
Inhibitors Targeting CDK9 Show High Efficacy against Osimertinib and AMG510 Resistant Lung Adenocarcinoma Cells


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. K-Ras and EGFR-Mutant Lung Cancer Cells Are Sensitive to CDK9 Inhibitors
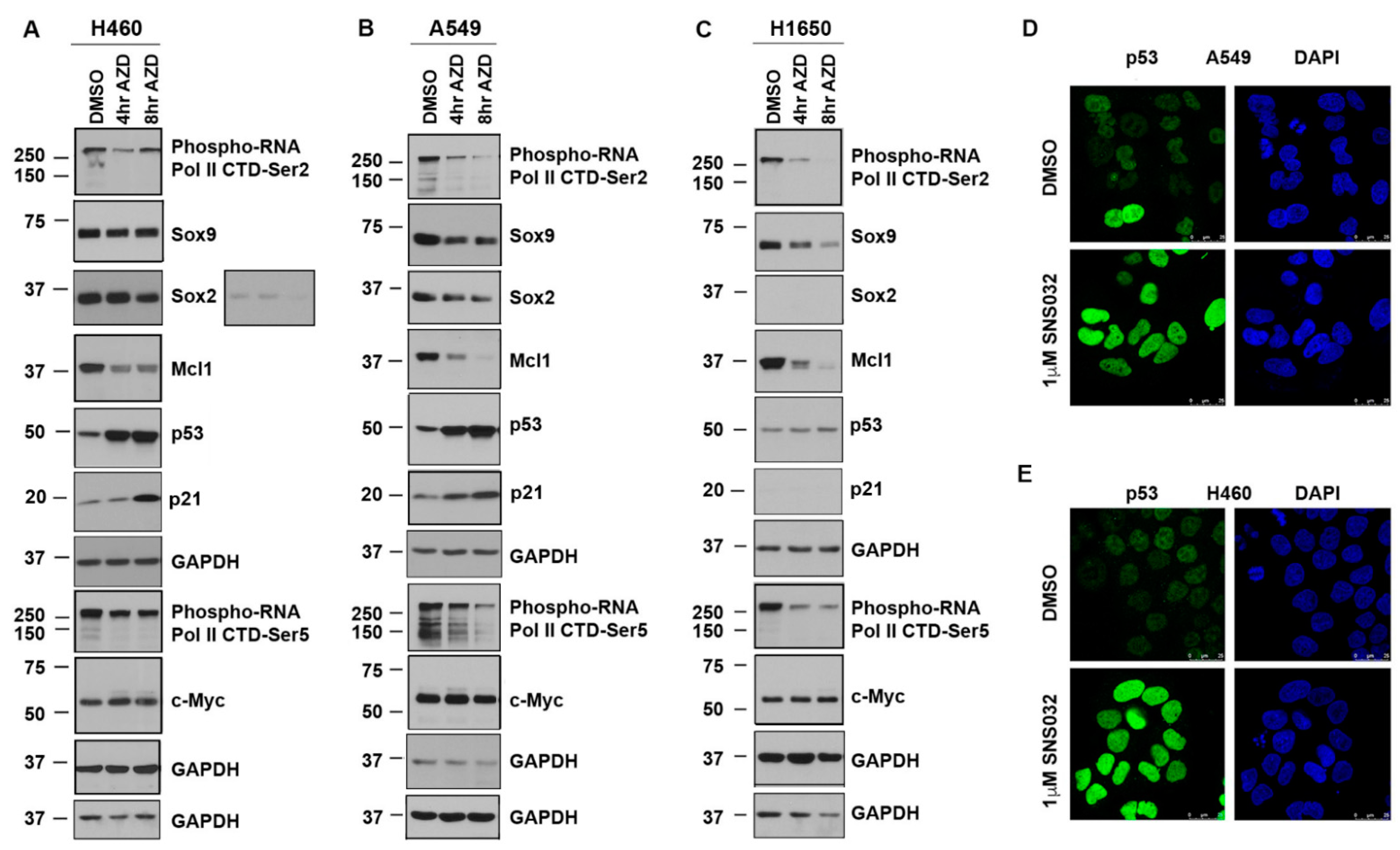
3.2. CDK9 Inhibition Suppresses Sox2, Sox9, and Mcl1 and Elevates p53 Protein Expression
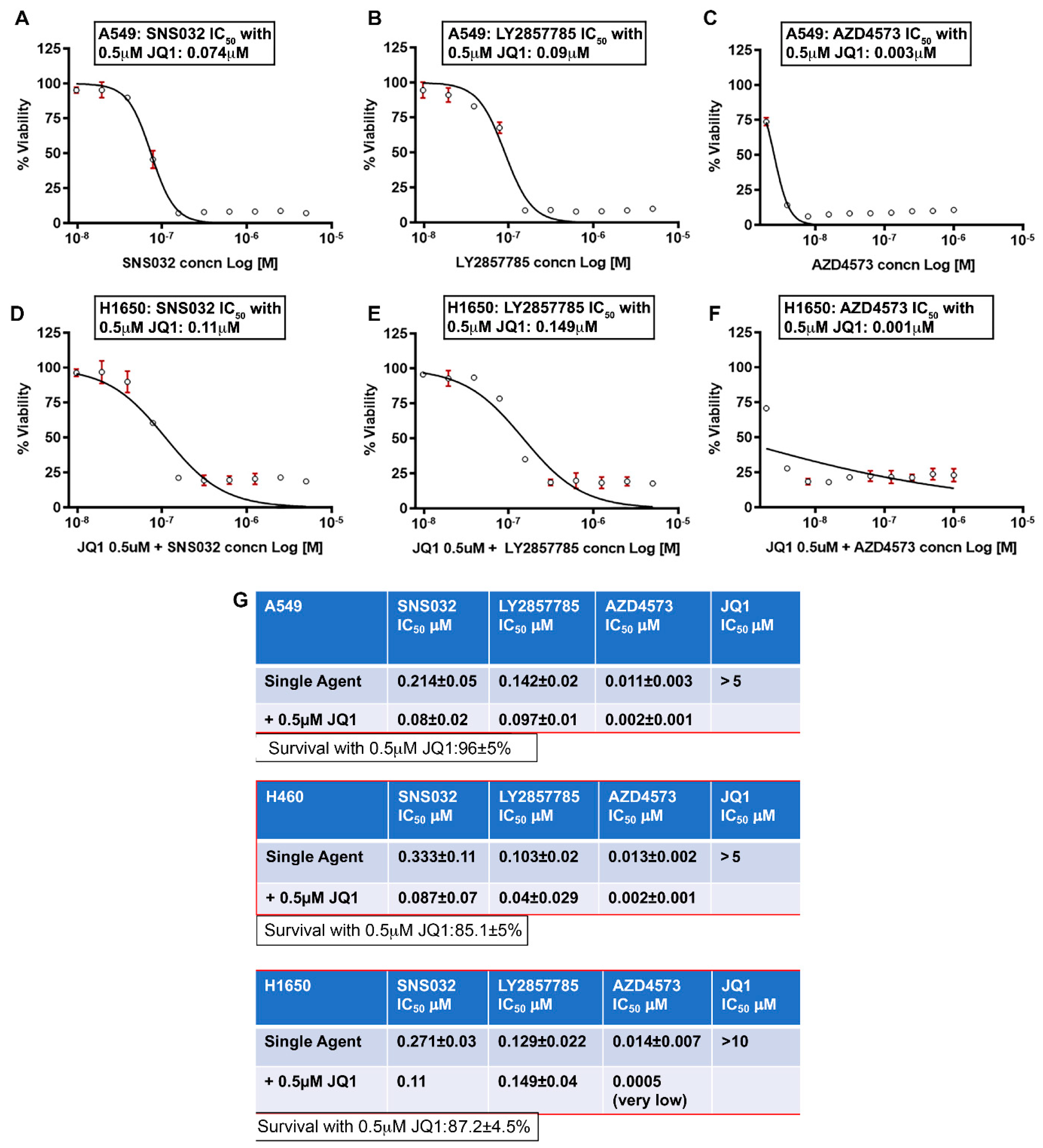
3.3. Combined Inhibition of CDK9 and BRD4 Markedly Reduces Cell Viability
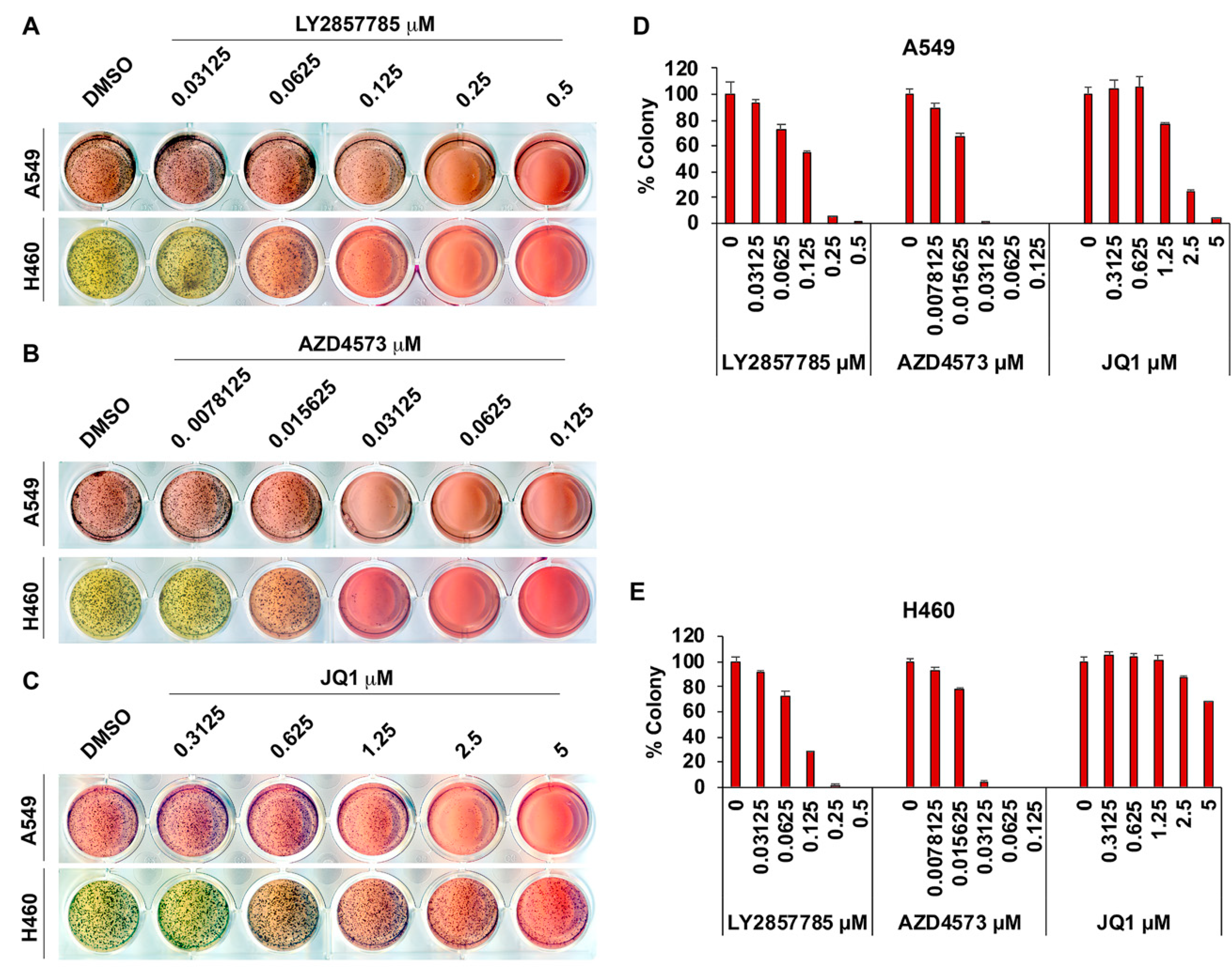
3.4. Adherence-Independent Growth of Lung Cancer Cells Is Abrogated upon CDK9 Inhibition
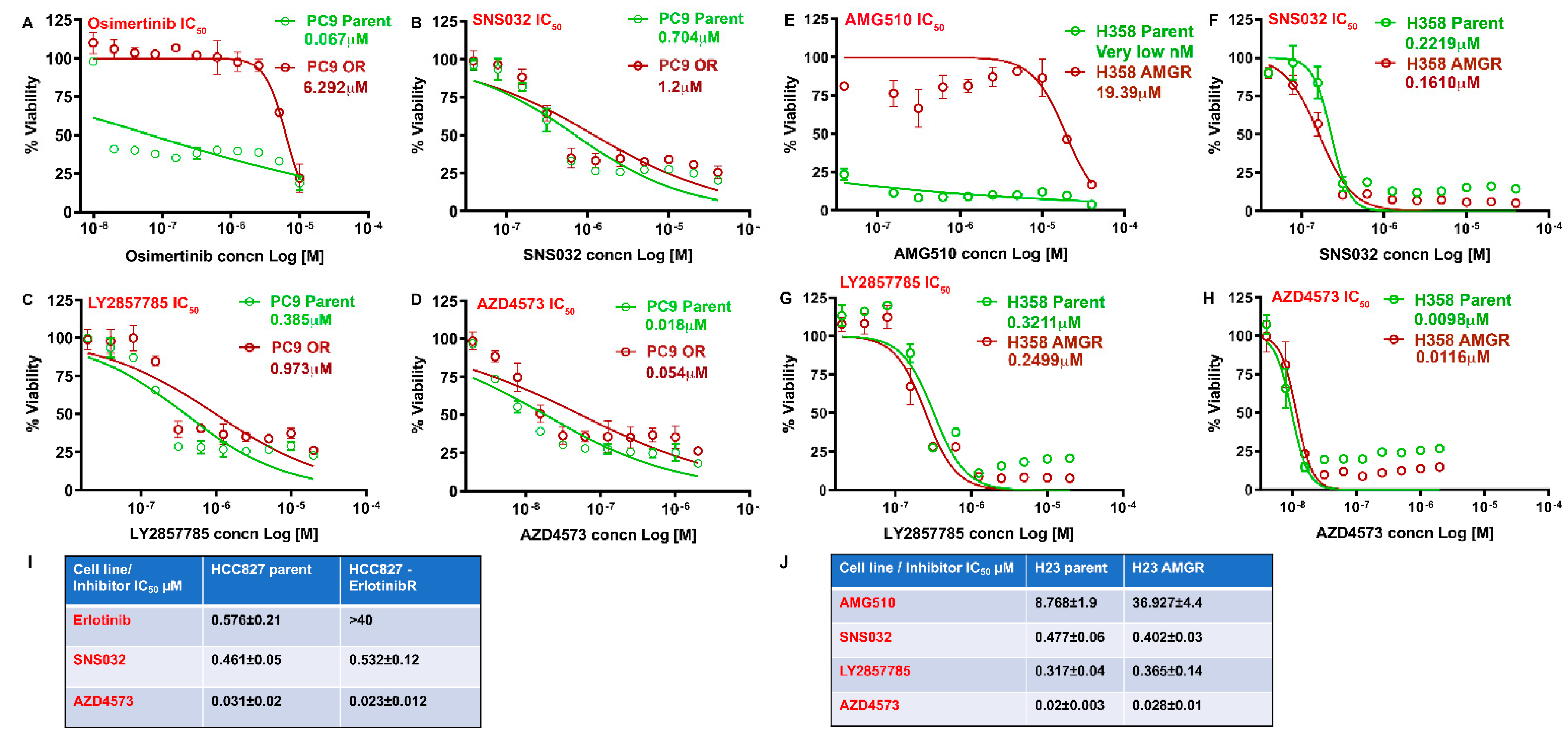
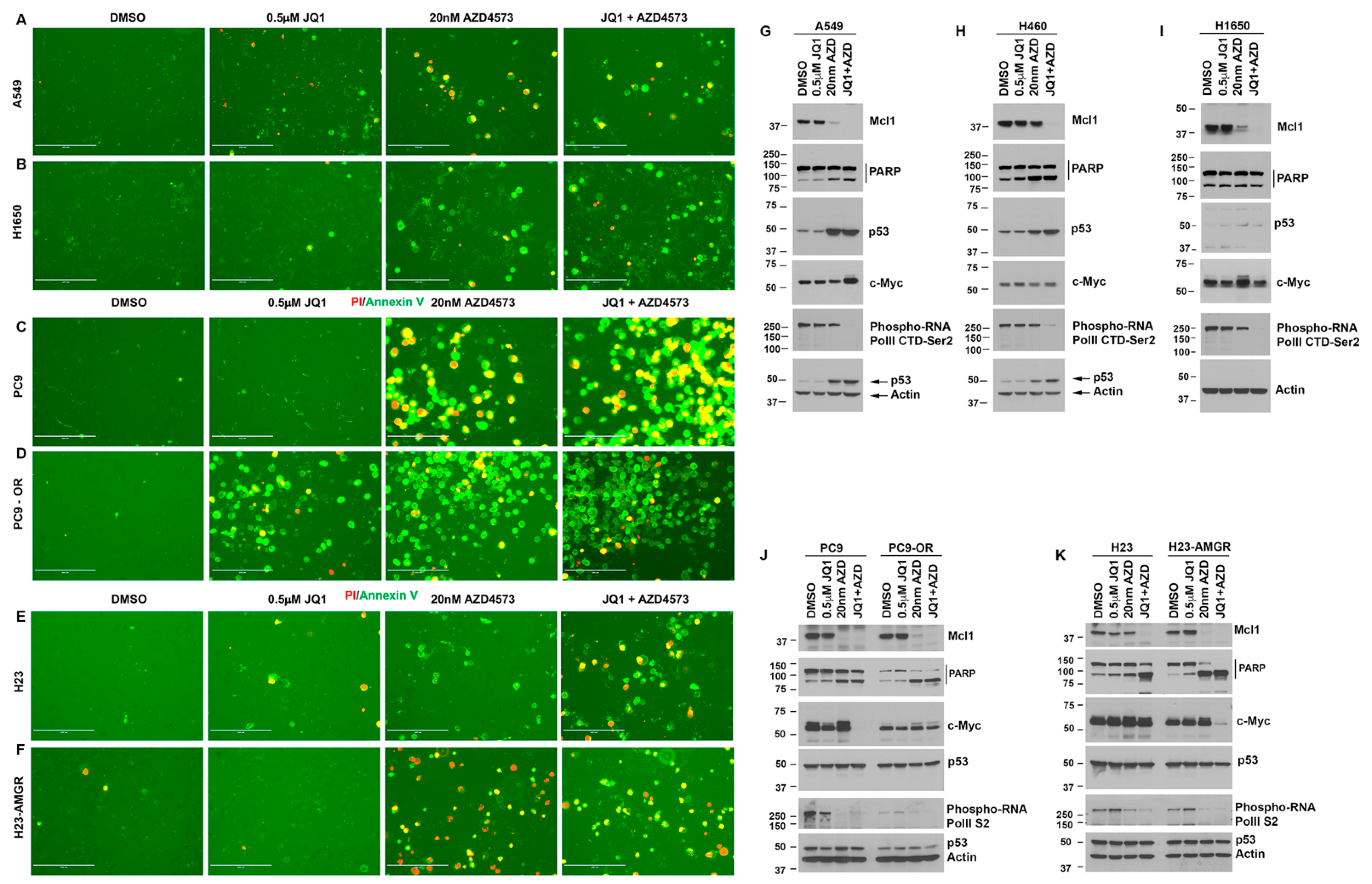
3.5. CDK9 Inhibitors Can Eliminate Drug Resistant NSCLC Cells
3.6. Drug-Sensitive and Resistant Lung Cancer Cells Treated with AZD4573 and JQ1 Show Increased Apoptosis and Reduced Mcl1 Expression
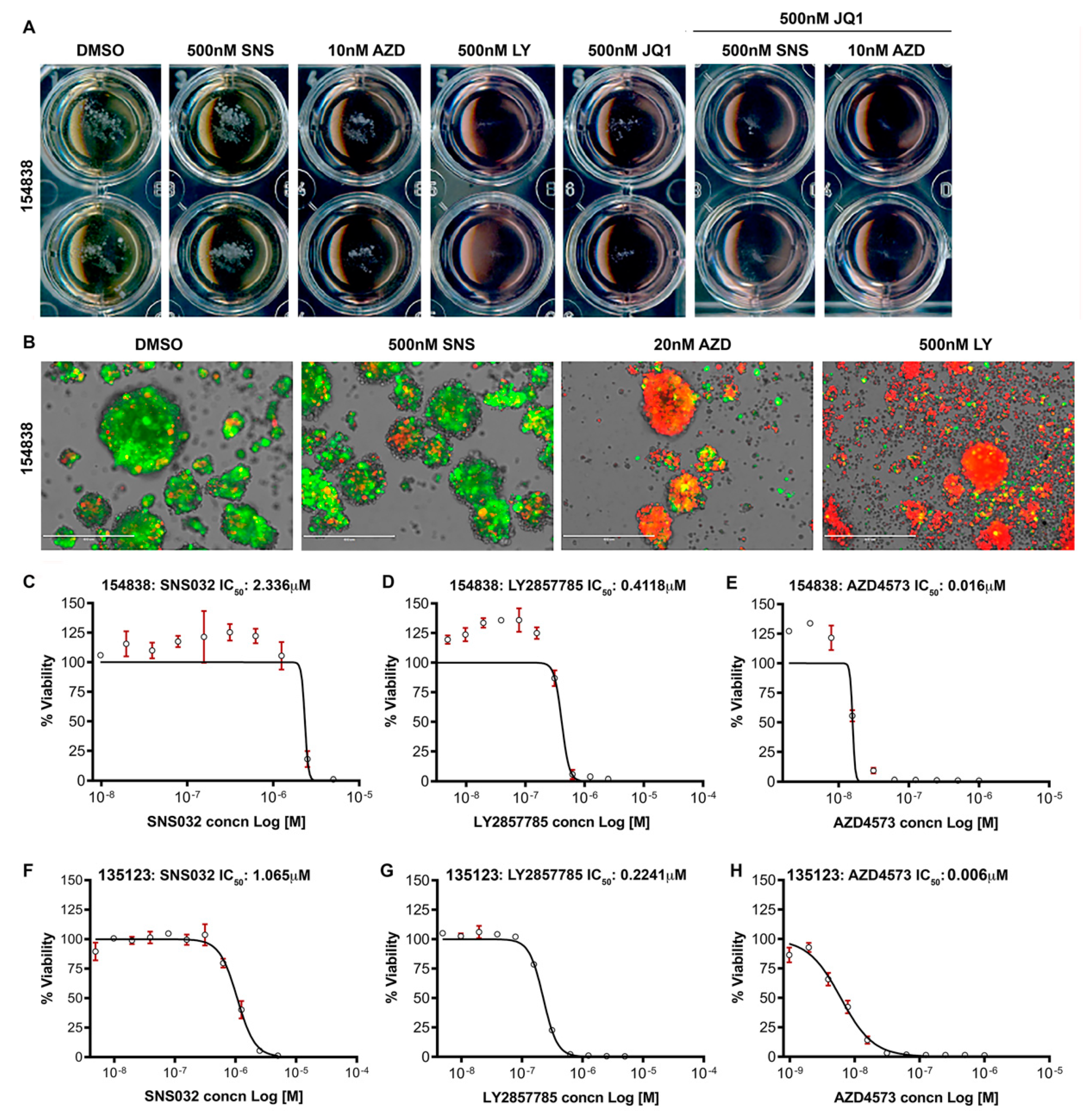
3.7. CDK9 Inhibitors Reduce the Survival of Lung Cancer Organoids
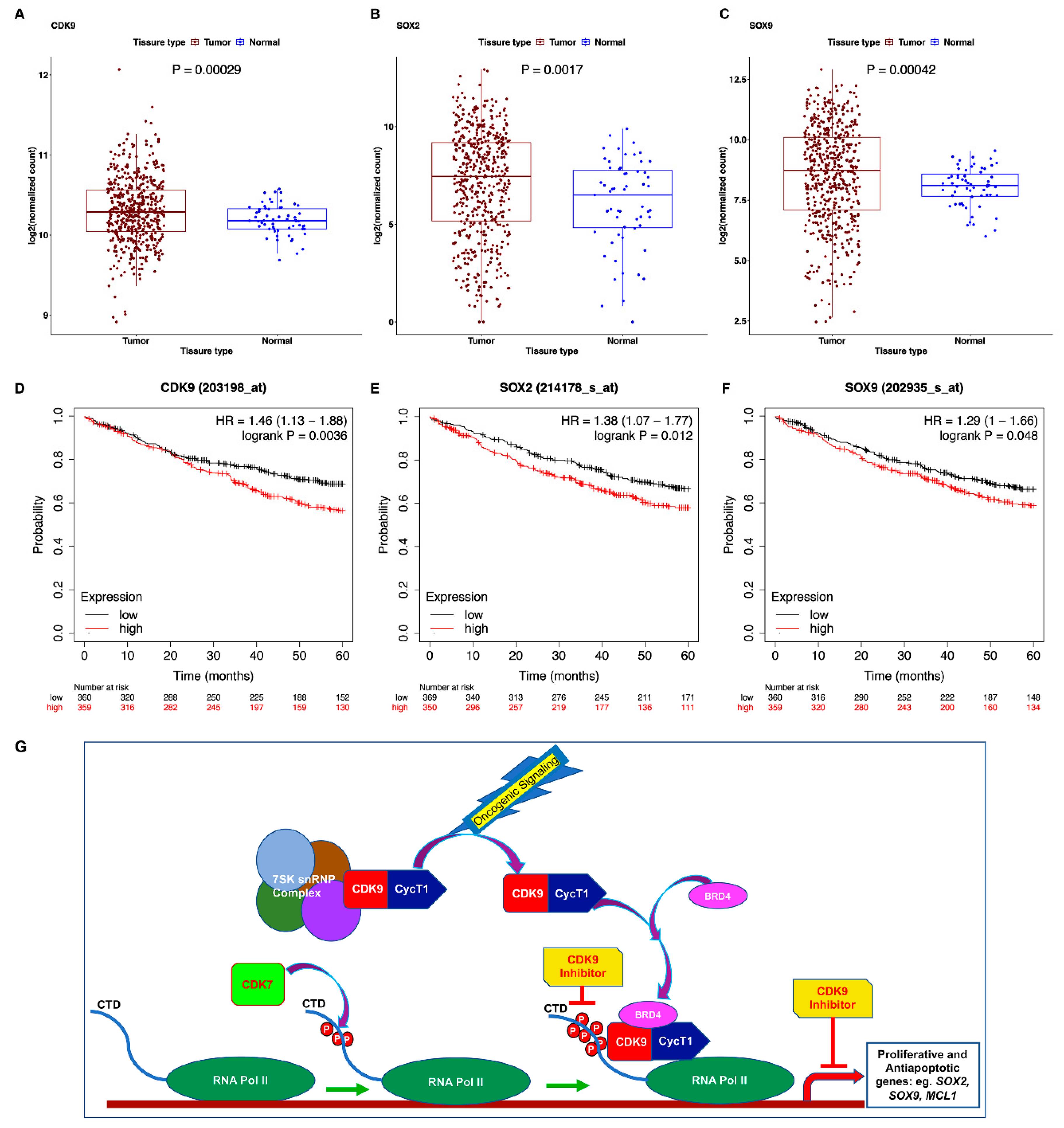
3.8. TCGA Dataset Shows Alterations in CDK9 and Its Targets
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Pillai, S.; Chellappan, S.P. Genetic and biochemical alterations in non-small cell lung cancer. Biochem. Res. Int. 2012, 2012, 940405. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, L.; Eskiocak, B.; Kohn, R.; Dang, C.; Joshi, N.S.; DuPage, M.; Lee, D.Y.; Jacks, T. Enhanced adaptive immune responses in lung adenocarcinoma through natural killer cell stimulation. Proc. Natl. Acad. Sci. USA 2019, 116, 17460–17469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naylor, E.C.; Desani, J.K.; Chung, P.K. Targeted Therapy and Immunotherapy for Lung Cancer. Surg. Oncol. Clin. N. Am. 2016, 25, 601–609. [Google Scholar] [CrossRef]
- Proto, C.; Ferrara, R.; Signorelli, D.; Lo Russo, G.; Galli, G.; Imbimbo, M.; Prelaj, A.; Zilembo, N.; Ganzinelli, M.; Pallavicini, L.M.; et al. Choosing wisely first line immunotherapy in non-small cell lung cancer (NSCLC): What to add and what to leave out. Cancer Treat Rev. 2019, 75, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Stinchcombe, T.E.; Borghaei, H.; Barker, S.S.; Treat, J.A.; Obasaju, C. Pemetrexed with Platinum Combination as a Backbone for Targeted Therapy in Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2016, 17, 1–9. [Google Scholar] [CrossRef]
- Horn, L.; Spigel, D.R.; Vokes, E.E.; Holgado, E.; Ready, N.; Steins, M.; Poddubskaya, E.; Borghaei, H.; Felip, E.; Paz-Ares, L.; et al. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). J. Clin. Oncol. 2017, 35, 3924–3933. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Jeanson, A.; Tomasini, P.; Souquet-Bressand, M.; Brandone, N.; Boucekine, M.; Grangeon, M.; Chaleat, S.; Khobta, N.; Milia, J.; Mhanna, L.; et al. Efficacy of Immune Checkpoint Inhibitors in KRAS-Mutant Non-Small Cell Lung Cancer (NSCLC). J. Thorac. Oncol. 2019, 14, 1095–1101. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Bellosillo, B.; Caux, C.; de Langen, A.; Mazieres, J.; Normanno, N.; Preusser, M.; Provencio, M.; Rojo, F.; Wolf, J.; et al. Immune checkpoint inhibitor treatment in patients with oncogene- addicted non-small cell lung cancer (NSCLC): Summary of a multidisciplinary round-table discussion. ESMO Open 2019, 4, e000498. [Google Scholar] [CrossRef] [Green Version]
- Kazandjian, D.; Khozin, S.; Blumenthal, G.; Zhang, L.; Tang, S.; Libeg, M.; Kluetz, P.; Sridhara, R.; Keegan, P.; Pazdur, R. Benefit-Risk Summary of Nivolumab for Patients With Metastatic Squamous Cell Lung Cancer After Platinum-Based Chemotherapy: A Report From the US Food and Drug Administration. JAMA Oncol. 2016, 2, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Antonia, S.; Goldberg, S.B.; Balmanoukian, A.; Chaft, J.E.; Sanborn, R.E.; Gupta, A.; Narwal, R.; Steele, K.; Gu, Y.; Karakunnel, J.J.; et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: A multicentre, phase 1b study. Lancet Oncol. 2016, 17, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Mei, Q.; Liu, L.; Cheng, T.; Wang, P.; Chen, L.; Zhou, J. The progress and challenge of anti-PD-1/PD-L1 immunotherapy in treating non-small cell lung cancer. Ther. Adv. Med. Oncol. 2021, 13, 1758835921992968. [Google Scholar] [CrossRef] [PubMed]
- Papadimitrakopoulou, V.A.; Mok, T.S.; Han, J.Y.; Ahn, M.J.; Delmonte, A.; Ramalingam, S.S.; Kim, S.W.; Shepherd, F.A.; Laskin, J.; He, Y.; et al. Osimertinib versus platinum-pemetrexed for patients with EGFR T790M advanced NSCLC and progression on a prior EGFR-tyrosine kinase inhibitor: AURA3 overall survival analysis. Ann. Oncol. 2020, 31, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Vendrell, J.A.; Mazieres, J.; Senal, R.; Rouquette, I.; Quantin, X.; Pujol, J.L.; Roch, B.; Bouidioua, A.; Godreuil, S.; Coyaud, E.; et al. Ultra-sensitive EGFR (T790M) Detection as an Independent Prognostic Marker for Lung Cancer Patients Harboring EGFR (del19) Mutations and Treated with First-generation TKIs. Clin. Cancer Res. 2019, 25, 4280–4289. [Google Scholar] [CrossRef] [Green Version]
- Stewart, E.L.; Tan, S.Z.; Liu, G.; Tsao, M.S. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations-a review. Transl. Lung Cancer Res. 2015, 4, 67–81. [Google Scholar] [CrossRef]
- Goss, G.; Tsai, C.M.; Shepherd, F.A.; Bazhenova, L.; Lee, J.S.; Chang, G.C.; Crino, L.; Satouchi, M.; Chu, Q.; Hida, T.; et al. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2016, 17, 1643–1652. [Google Scholar] [CrossRef]
- Wangari-Talbot, J.; Hopper-Borge, E. Drug Resistance Mechanisms in Non-Small Cell Lung Carcinoma. J. Can. Res. Updat. 2013, 2, 265–282. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Chen, L.; Liu, L.; Niu, X. EMT-Mediated Acquired EGFR-TKI Resistance in NSCLC: Mechanisms and Strategies. Front. Oncol. 2019, 9, 1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Bora-Singhal, N.; Nguyen, J.; Schaal, C.; Perumal, D.; Singh, S.; Coppola, D.; Chellappan, S. YAP1 Regulates OCT4 Activity and SOX2 Expression to Facilitate Self-Renewal and Vascular Mimicry of Stem-Like Cells. Stem Cells 2015, 33, 1705–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchinetti, F.; Rossi, G.; Bria, E.; Soria, J.C.; Besse, B.; Minari, R.; Friboulet, L.; Tiseo, M. Oncogene addiction in non-small cell lung cancer: Focus on ROS1 inhibition. Cancer Treat. Rev. 2017, 55, 83–95. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Smolle, E.; Leithner, K.; Olschewski, H. Oncogene addiction and tumor mutational burden in non-small-cell lung cancer: Clinical significance and limitations. Thorac. Cancer 2020, 11, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef]
- Sundar, V.; Vimal, S.; Sai Mithlesh, M.S.; Dutta, A.; Tamizhselvi, R.; Manickam, V. Transcriptional cyclin-dependent kinases as the mediators of inflammation-a review. Gene 2021, 769, 145200. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Quigley, D.A.; Robinson, T.M.; Feng, F.Y.; Ashworth, A. Transcription-Associated Cyclin-Dependent Kinases as Targets and Biomarkers for Cancer Therapy. Cancer Discov. 2020, 10, 351–370. [Google Scholar] [CrossRef] [Green Version]
- Paculova, H.; Kohoutek, J. The emerging roles of CDK12 in tumorigenesis. Cell Div. 2017, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Galbraith, M.D.; Bender, H.; Espinosa, J.M. Therapeutic targeting of transcriptional cyclin-dependent kinases. Transcription 2019, 10, 118–136. [Google Scholar] [CrossRef]
- Martin, R.D.; Hebert, T.E.; Tanny, J.C. Therapeutic Targeting of the General RNA Polymerase II Transcription Machinery. Int. J. Mol. Sci. 2020, 21, 3354. [Google Scholar] [CrossRef]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R.; Duckett, D.R. Therapeutic Targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell 2019, 36, 545–558.e547. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; George, R.E. Super-Enhancer-Driven Transcriptional Dependencies in Cancer. Trends Cancer 2017, 3, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Bacon, C.W.; D’Orso, I. CDK9: A signaling hub for transcriptional control. Transcription 2019, 10, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Price, D.H. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell Biol. 2000, 20, 2629–2634. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Yoon, H.G.; Qin, J.; Wong, J. Regulation of P-TEFb elongation complex activity by CDK9 acetylation. Mol. Cell Biol. 2007, 27, 4641–4651. [Google Scholar] [CrossRef] [Green Version]
- Bowman, E.A.; Kelly, W.G. RNA polymerase II transcription elongation and Pol II CTD Ser2 phosphorylation: A tail of two kinases. Nucleus 2014, 5, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cidado, J.; Boiko, S.; Proia, T.; Ferguson, D.; Criscione, S.W.; San Martin, M.; Pop-Damkov, P.; Su, N.; Roamio Franklin, V.N.; Sekhar Reddy Chilamakuri, C.; et al. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin. Cancer Res. 2020, 26, 922–934. [Google Scholar] [CrossRef] [Green Version]
- Barlaam, B.; Casella, R.; Cidado, J.; Cook, C.; De Savi, C.; Dishington, A.; Donald, C.S.; Drew, L.; Ferguson, A.D.; Ferguson, D.; et al. Discovery of AZD4573, a Potent and Selective Inhibitor of CDK9 That Enables Short Duration of Target Engagement for the Treatment of Hematological Malignancies. J. Med. Chem. 2020, 63, 15564–15590. [Google Scholar] [CrossRef]
- Gargano, B.; Amente, S.; Majello, B.; Lania, L. P-TEFb is a crucial co-factor for Myc transactivation. Cell Cycle 2007, 6, 2031–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashiguchi, T.; Bruss, N.; Best, S.; Lam, V.; Danilova, O.; Paiva, C.J.; Wolf, J.; Gilbert, E.W.; Okada, C.Y.; Kaur, P.; et al. Cyclin-Dependent Kinase-9 Is a Therapeutic Target in MYC-Expressing Diffuse Large B-Cell Lymphoma. Mol. Cancer Ther. 2019, 18, 1520–1532. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Lujambio, A.; Zuber, J.; Tschaharganeh, D.F.; Doran, M.G.; Evans, M.J.; Kitzing, T.; Zhu, N.; de Stanchina, E.; Sawyers, C.L.; et al. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 2014, 28, 1800–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.L.; Abudureheman, T.; Xia, J.; Chu, L.; Zhou, H.; Zheng, W.W.; Zhou, N.; Shi, R.Y.; Li, M.H.; Zhu, J.M.; et al. CDK9 Inhibitor Induces the Apoptosis of B-Cell Acute Lymphocytic Leukemia by Inhibiting c-Myc-Mediated Glycolytic Metabolism. Front. Cell Dev. Biol. 2021, 9, 641271. [Google Scholar] [CrossRef]
- Blake, D.R.; Vaseva, A.V.; Hodge, R.G.; Kline, M.P.; Gilbert, T.S.K.; Tyagi, V.; Huang, D.; Whiten, G.C.; Larson, J.E.; Wang, X.; et al. Application of a MYC degradation screen identifies sensitivity to CDK9 inhibitors in KRAS-mutant pancreatic cancer. Sci. Signal. 2019, 12, eaav7259. [Google Scholar] [CrossRef] [PubMed]
- Gregory, G.P.; Hogg, S.J.; Kats, L.M.; Vidacs, E.; Baker, A.J.; Gilan, O.; Lefebure, M.; Martin, B.P.; Dawson, M.A.; Johnstone, R.W.; et al. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia 2015, 29, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Tang, H.; Wu, S.; Chen, J.; Liu, J.; Liao, C. The cyclin-dependent kinase inhibitor SNS-032 induces apoptosis in breast cancer cells via depletion of Mcl-1 and X-linked inhibitor of apoptosis protein and displays antitumor activity in vivo. Int. J. Oncol. 2014, 45, 804–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Finlay, M.R.; Anderton, M.; Ashton, S.; Ballard, P.; Bethel, P.A.; Box, M.R.; Bradbury, R.H.; Brown, S.J.; Butterworth, S.; Campbell, A.; et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 2014, 57, 8249–8267. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018, 175, 1244–1258.e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Bottinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef]
- Woods, N.; Trevino, J.; Coppola, D.; Chellappan, S.; Yang, S.; Padmanabhan, J. Fendiline inhibits proliferation and invasion of pancreatic cancer cells by interfering with ADAM10 activation and beta-catenin signaling. Oncotarget 2015, 6, 35931–35948. [Google Scholar] [CrossRef] [Green Version]
- Woods, N.K.; Padmanabhan, J. Inhibition of amyloid precursor protein processing enhances gemcitabine-mediated cytotoxicity in pancreatic cancer cells. J. Biol. Chem. 2013, 288, 30114–30124. [Google Scholar] [CrossRef] [Green Version]
- Padmanabhan, J. Detection of histone H3 phosphorylation in cultured cells and tissue sections by immunostaining. Methods Mol. Biol. 2009, 523, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, J. Immunostaining analysis of tissue cultured cells and tissue sections using phospho-Histone H3 (Serine 10) antibody. Methods Mol. Biol. 2015, 1288, 231–244. [Google Scholar] [CrossRef]
- Kroesen, B.J.; Mesander, G.; ter Haar, J.G.; The, T.H.; de Leij, L. Direct visualisation and quantification of cellular cytotoxicity using two colour flourescence. J. Immunol. Methods 1992, 156, 47–54. [Google Scholar] [CrossRef]
- Chang, L.; Gusewitch, G.A.; Chritton, D.B.; Folz, J.C.; Lebeck, L.K.; Nehlsen-Cannarella, S.L. Rapid flow cytometric assay for the assessment of natural killer cell activity. J. Immunol. Methods 1993, 166, 45–54. [Google Scholar] [CrossRef]
- Shimamura, T.; Chen, Z.; Soucheray, M.; Carretero, J.; Kikuchi, E.; Tchaicha, J.H.; Gao, Y.; Cheng, K.A.; Cohoon, T.J.; Qi, J.; et al. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 6183–6192. [Google Scholar] [CrossRef] [Green Version]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; He, N.; Zhou, Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell Biol. 2008, 28, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.F.; Wu, Y.B.; Long, X.; Zhu, S.Q.; Jin, C.; Xu, J.J.; Ding, J.Y. High level of BRD4 promotes non-small cell lung cancer progression. Oncotarget 2016, 7, 9491–9500. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Gyorffy, B.; Surowiak, P.; Budczies, J.; Lanczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [Green Version]
- Ji, W.; Choi, Y.J.; Kang, M.H.; Sung, K.J.; Kim, D.H.; Jung, S.; Choi, C.M.; Lee, J.C.; Rho, J.K. Efficacy of the CDK7 Inhibitor on EMT-Associated Resistance to 3rd Generation EGFR-TKIs in Non-Small Cell Lung Cancer Cell Lines. Cells 2020, 9, 2596. [Google Scholar] [CrossRef]
- La Monica, S.; Fumarola, C.; Cretella, D.; Bonelli, M.; Minari, R.; Cavazzoni, A.; Digiacomo, G.; Galetti, M.; Volta, F.; Mancini, M.; et al. Efficacy of the CDK4/6 Dual Inhibitor Abemaciclib in EGFR-Mutated NSCLC Cell Lines with Different Resistance Mechanisms to Osimertinib. Cancers 2020, 13, 6. [Google Scholar] [CrossRef]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. CDK4/6 inhibitor palbociclib overcomes acquired resistance to third-generation EGFR inhibitor osimertinib in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 2389–2397. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y.; Jiao, L.; Lin, C.; Lu, C.; Zhang, K.; Hu, C.; Ye, J.; Zhang, D.; Wu, H.; et al. Protective autophagy decreases osimertinib cytotoxicity through regulation of stem cell-like properties in lung cancer. Cancer Lett. 2019, 452, 191–202. [Google Scholar] [CrossRef]
- Akhave, N.S.; Biter, A.B.; Hong, D.S. Mechanisms of Resistance to KRAS(G12C)-Targeted Therapy. Cancer Discov. 2021, 11, 1345–1352. [Google Scholar] [CrossRef]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef] [Green Version]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Munkhbaatar, E.; Dietzen, M.; Agrawal, D.; Anton, M.; Jesinghaus, M.; Boxberg, M.; Pfarr, N.; Bidola, P.; Uhrig, S.; Hockendorf, U.; et al. MCL-1 gains occur with high frequency in lung adenocarcinoma and can be targeted therapeutically. Nat. Commun. 2020, 11, 4527. [Google Scholar] [CrossRef]
- Tong, W.G.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.D.; Badros, A.Z.; Popplewell, L.; Coutre, S.; Fox, J.A.; et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J. Clin. Oncol. 2010, 28, 3015–3022. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padmanabhan, J.; Saha, B.; Powell, C.; Mo, Q.; Perez, B.A.; Chellappan, S. Inhibitors Targeting CDK9 Show High Efficacy against Osimertinib and AMG510 Resistant Lung Adenocarcinoma Cells. Cancers 2021, 13, 3906. https://doi.org/10.3390/cancers13153906
Padmanabhan J, Saha B, Powell C, Mo Q, Perez BA, Chellappan S. Inhibitors Targeting CDK9 Show High Efficacy against Osimertinib and AMG510 Resistant Lung Adenocarcinoma Cells. Cancers. 2021; 13(15):3906. https://doi.org/10.3390/cancers13153906
Chicago/Turabian StylePadmanabhan, Jaya, Biswarup Saha, Chase Powell, Qianxing Mo, Bradford A. Perez, and Srikumar Chellappan. 2021. "Inhibitors Targeting CDK9 Show High Efficacy against Osimertinib and AMG510 Resistant Lung Adenocarcinoma Cells" Cancers 13, no. 15: 3906. https://doi.org/10.3390/cancers13153906
APA StylePadmanabhan, J., Saha, B., Powell, C., Mo, Q., Perez, B. A., & Chellappan, S. (2021). Inhibitors Targeting CDK9 Show High Efficacy against Osimertinib and AMG510 Resistant Lung Adenocarcinoma Cells. Cancers, 13(15), 3906. https://doi.org/10.3390/cancers13153906