
Normalizing Tumor Vasculature to Reduce Hypoxia, Enhance Perfusion, and Optimize Therapy Uptake




Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Sprouting Angiogenesis in Normal Physiology
1.2. Tumor Control of Angiogenesis
1.3. Factors Contributing to Tumor Vascular Dysfunction
1.4. Abnormal Vasculature Results in Limited Treatment Delivery
1.5. Hypoxia and Tumor Metabolism
1.6. Hypoxia and Drug Resistance
1.7. Hypoxia and the Immune Environment
1.8. Therapeutic Use of Vascular-Targeting Agents
2. Considerations for Success of Vascular Normalizing Agents
3. Vascular Normalizing Agents as Adjuvants to Traditional Cancer Therapeutics
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pereira, R.D.; de Long, N.E.; Wang, R.C.; Yazdi, F.T.; Holloway, A.C.; Raha, S. Angiogenesis in the Placenta: The Role of Reactive Oxygen Species Signaling. BioMed Res. Int. 2015, 2015, 814543. [Google Scholar] [CrossRef] [Green Version]
- Dangat, K.; Khaire, A.; Joshi, S. Cross Talk of Vascular Endothelial Growth Factor and Neurotrophins in Mammary Gland Development. Growth Factors 2020, 38, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kumar, S.; Udupa, E.P.; Kumar, U.; Rao, P.; Honnegowda, T. Role of Angiogenesis and Angiogenic Factors in Acute and Chronic Wound Healing. Plast. Aesthet. Res. 2015, 2, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Ucuzian, A.A.; Gassman, A.A.; East, A.T.; Greisler, H.P. Molecular Mediators of Angiogenesis. J. Burn Care Res. 2010, 31, 158–175. [Google Scholar] [CrossRef]
- Yokota, Y.; Nakajima, H.; Wakayama, Y.; Muto, A.; Kawakami, K.; Fukuhara, S.; Mochizuki, N. Endothelial Ca2+ Oscillations Reflect VEGFR Signaling-Regulated Angiogenic Capacity in Vivo. eLife 2015, 4, e08817. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; George, S.C.; Putnam, A.J. Matrix Metalloproteinase Control of Capillary Morphogenesis. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 251–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauteur, L.; Krudewig, A.; Herwig, L.; Ehrenfeuchter, N.; Lenard, A.; Affolter, M.; Belting, H.G. Cdh5/VE-Cadherin Promotes Endothelial Cell Interface Elongation via Cortical Actin Polymerization during Angiogenic Sprouting. Cell Rep. 2014, 9, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Franco, C.A.; Jones, M.L.; Bernabeu, M.O.; Vion, A.C.; Barbacena, P.; Fan, J.; Mathivet, T.; Fonseca, C.G.; Ragab, A.; Yamaguchi, T.P.; et al. Non-Canonical Wnt Signalling Modulates the Endothelial Shear Stress Flow Sensor in Vascular Remodelling. eLife 2016, 5, e07727. [Google Scholar] [CrossRef]
- Chen, Q.; Jiang, L.; Li, C.; Hu, D.; Bu, J.W.; Cai, D.; Du, J.L. Haemodynamics-Driven Developmental Pruning of Brain Vasculature in Zebrafish. PLoS Biol. 2012, 10, e1001374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M. Signaling Required for Blood Vessel Maintenance: Molecular Basis and Pathological Manifestations. Int. J. Vasc. Med. 2012, 2012, 293641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremnes, R.M.; Dønnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The Role of Tumor Stroma in Cancer Progression and Prognosis: Emphasis on Carcinoma-Associated Fibroblasts and Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.J.; Chambers, C.R.; Herrmann, D.; Timpson, P.; Pereira, B.A. Dynamic Stromal Alterations Influence Tumor-Stroma Crosstalk to Promote Pancreatic Cancer and Treatment Resistance. Cancers 2021, 13, 3481. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J.; Hanahan, D. Switch to the Angiogenic Phenotype during Tumorigenesis. Princess Takamatsu Symp. 1991, 22, 22. [Google Scholar]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-Inducible Factor-1-Dependent Regulation of the Multidrug Resistance (MDR1) Gene. Cancer Res. 2002, 62, 62. [Google Scholar]
- Wilkins, S.E.; Abboud, M.I.; Hancock, R.L.; Schofield, C.J. Targeting Protein-Protein Interactions in the HIF System. ChemMedChem 2016, 11, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.-W.; Bae, M.-K.; Ahn, M.-Y.; Kim, S.-H.; Sohn, T.-K.; Bae, M.-H.; Yoo, M.-A.; Song, E.J.; Lee, K.-J.; Kim, K.-W. Regulation and Destabilization of HIF-1 by ARD1-Mediated Acetylation Quitin-Proteasome Pathway (Salceda and Caro The Association of PVHL and HIF-1 under nor-Moxic Conditions Is Triggered by the Posttranslational. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Strowitzki, M.; Cummins, E.; Taylor, C. Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells 2019, 8, 384. [Google Scholar] [CrossRef] [Green Version]
- Yu, A.Y.; Frid, M.G.; Shimoda, L.A.; Wiener, C.M.; Stenmark, K.; Semenza, G.L. Temporal, Spatial, and Oxygen-Regulated Expression of Hypoxia-Inducible Factor-1 in the Lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 1998, 275, L818–L826. [Google Scholar] [CrossRef]
- McKeown, S.R. Defining Normoxia, Physoxia and Hypoxia in Tumours—Implications for Treatment Response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [Green Version]
- Artemov, A.V.; Zhigalova, N.; Zhenilo, S.; Mazur, A.M.; Prokhortchouk, E.B. VHL Inactivation without Hypoxia Is Sufficient to Achieve Genome Hypermethylation. Sci. Rep. 2018, 8, 10667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.A.; le Quesne, J.; Baena Acevedo, J.D.; et al. P53 Mutants Cooperate with HIF-1 in Transcriptional Regulation of Extracellular Matrix Components to Promote Tumor Progression. Proc. Natl. Acad. Sci. USA 2018, 115, E10869–E10878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of Tumor Angiogenesis by P53-Induced Degradation of Hypoxia- Inducible Factor 1α. Genes Dev. 2000, 14, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Mohlin, S.; Hamidian, A.; von Stedingk, K.; Bridges, E.; Wigerup, C.; Bexell, D.; Påhlman, S. PI3K-MTORC2 but Not PI3K-MTORC1 Regulates Transcription of HIF2A/EPAS1and Vascularization in Neuroblastoma. Cancer Res. 2015, 75, 4617–4628. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of Hypoxia-Inducible Factor 1α Expression by the Epidermal Growth Factor/Phosphatidylinositol 3-Kinase/PTEN/AKT/FRAP Pathway in Human Prostate Cancer Cells: Implications for Tumor Angiogenesis and Therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Lee, B.I.; Kim, W.H.; Jung, J.; Cho, S.J.; Park, J.W.; Kim, J.; Chung, H.Y.; Chang, M.S.; Nam, S.Y. A Hypoxia-Independent up-Regulation of Hypoxia-Inducible Factor-1 by AKT Contributes to Angiogenesis in Human Gastric Cancer. Carcinogenesis 2008, 29, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegeman, H.; Span, P.N.; Peeters, W.J.M.; Verheijen, M.M.G.; Grénman, R.; Meijer, T.W.H.; Kaanders, J.H.A.M.; Bussink, J. Interaction between Hypoxia, AKT and HIF-1 Signaling in HNSCC and NSCLC: Implications for Future Treatment Strategies. Future Sci. OA 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N. Vascular Endothelial Growth Factor: Basic Science and Clinical Progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Welsh, M. VEGFA and Tumour Angiogenesis. J. Intern. Med. 2013, 273, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Eriksson, U. Novel VEGF Family Members: VEGF-B, VEGF-C and VEGF-D. Int. J. Biochem. Cell Biol. 2001, 33, 421–426. [Google Scholar] [CrossRef]
- Liang, L.; Yue, Z.; Du, W.; Li, Y.; Tao, H.; Wang, D.; Wang, R.; Huang, Z.; He, N.; Xie, X.; et al. Molecular Imaging of Inducible VEGF Expression and Tumor Progression in a Breast Cancer Model. Cell. Physiol. Biochem. 2017, 42, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Chaudhuri, O. Beyond Proteases: Basement Membrane Mechanics and Cancer Invasion. J. Cell Biol. 2019, 218, 2456–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucker, S.; Vacirca, J. Role of Matrix Metalloproteinases (MMPs) in Colorectal Cancer. Cancer Metastasis Rev. 2004, 23, 101–117. [Google Scholar] [CrossRef]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between Defective Endothelial Cells Explain Tumor Vessel Leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef] [Green Version]
- Hida, K.; Hida, Y.; Amin, D.N.; Flint, A.F.; Panigrahy, D.; Morton, C.C.; Klagsbrun, M. Tumor-Associated Endothelial Cells with Cytogenetic Abnormalities. Cancer Res. 2004, 64, 8249–8255. [Google Scholar] [CrossRef] [Green Version]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the Role of the Tumor Vasculature in Antitumor Immunity and Immunotherapy Article. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, K.; Ohga, N.; Hida, Y.; Muraki, C.; Tsuchiya, K.; Kurosu, T.; Akino, T.; Shih, S.C.; Totsuka, Y.; Klagsbrun, M.; et al. Isolated Tumor Endothelial Cells Maintain Specific Character during Long-Term Culture. Biochem. Biophys. Res. Commun. 2010, 394, 947–954. [Google Scholar] [CrossRef]
- Sievert, W.; Tapio, S.; Breuninger, S.; Gaipl, U.; Andratschke, N.; Trott, K.R.; Multhoff, G. Adhesion Molecule Expression and Function of Primary Endothelial Cells in Benign and Malignant Tissues Correlates with Proliferation. PLoS ONE 2014, 9, e91808. [Google Scholar] [CrossRef] [Green Version]
- Ohmura-Kakutani, H.; Akiyama, K.; Maishi, N.; Ohga, N.; Hida, Y.; Kawamoto, T.; Iida, J.; Shindoh, M.; Tsuchiya, K.; Shinohara, N.; et al. Identification of Tumor Endothelial Cells with High Aldehyde Dehydrogenase Activity and a Highly Angiogenic Phenotype. PLoS ONE 2014, 9, e113910. [Google Scholar] [CrossRef] [PubMed]
- Mehran, R.; Nilsson, M.; Khajavi, M.; Du, Z.; Cascone, T.; Wu, H.K.; Cortes, A.; Xu, L.; Zurita, A.; Schier, R.; et al. Tumor Endothelial Markers Define Novel Subsets of Cancer-Specific Circulating Endothelial Cells Associated with Antitumor Efficacy. Cancer Res. 2014, 74, 2731–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatmaitan, Z.; Varticovski, L.; Ling, L.; Mikkelsen, R.; Steffan, A.M.; Arias, I.M. Studies on Fenestral Contraction in Rat Liver Endothelial Cells in Culture. Am. J. Pathol. 1996, 148, 2027–2041. [Google Scholar] [PubMed]
- Levick, J.R.; Smaje, L.H. An Analysis of the Permeability of a Fenestra. Microvasc. Res. 1987, 33, 233–256. [Google Scholar] [CrossRef]
- Suarez, S.; Ballmer-Hofer, K. VEGF Transiently Disrupts Gap Junctional Communication in Endothelial Cells. J. Cell Sci. 2001, 114, 1229–1235. [Google Scholar] [CrossRef]
- Nimlamool, W.; Andrews, R.M.K.; Falk, M.M. Connexin43 Phosphorylation by PKC and MAPK Signals VEGF-Mediated Gap Junction Internalization. Mol. Biol. Cell 2015, 26, 2755–2768. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D. The Vascular Endothelial Growth Factor-Induced Disruption of Gap Junctions is Relayed by an Autocrine Communication via ATP Release in Coronary Capillary Endothelium. Ann. N. Y. Acad. Sci. 2004, 1030, 14–27. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Senger, D.R.; Dvorak, A.M. Fibrin as a Component of the Tumor Stroma: Origins and Biological Significance. Cancer Metastasis Rev. 1983, 2, 41–73. [Google Scholar] [CrossRef]
- Xiang, D.; Feng, Y.; Wang, J.; Zhang, X.; Shen, J.; Zou, R.; Yuan, Y. Platelet-derived Growth Factor-BB Promotes Proliferation and Migration of Retinal Microvascular Pericytes by Up-regulating the Expression of C-X-C Chemokine Receptor Types 4. Exp. Ther. Med. 2019, 18, 4022–4030. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, P.; Johansson, B.R.; Levéen, P.; Betsholtz, C. Pericyte Loss and Microaneurysm Formation in PDGF-B-Deficient Mice. Science 1997, 277, 242–245. [Google Scholar] [CrossRef]
- Franco, M.; Roswall, P.; Cortez, E.; Hanahan, D.; Pietras, K. Pericytes Promote Endothelial Cell Survival through Induction of Autocrine VEGF-Asignaling and Bcl-w Expression. Blood 2011, 118, 2906–2917. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Flores, L.; Gutiérrez, R.; Madrid, J.F.; Varela, H.; Valladares, F.; Acosta, E.; Martín-Vasallo, P.; Díaz-Flores, J. Pericytes. Morphofunction, Interactions and Pathology in a Quiescent and Activated Mesenchymal Cell Niche. Histol. Histopathol. 2009, 24, 909–969. [Google Scholar] [PubMed]
- Jain, R.K. Transport of Molecules in the Tumor Interstitium: A Review. Cancer Res. 1987, 47, 3039–3051. [Google Scholar]
- Libutti, S.K.; Tamarkin, L.; Nilubol, N. Targeting the Invincible Barrier for Drug Delivery in Solid Cancers: Interstitial Fluid Pressure. Oncotarget 2018, 9, 35723–35725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffon-Etienne, G.; Boucher, Y.; Brekken, C.; Suit, H.D.; Jain, R.K. Taxane-Induced Apoptosis Decompresses Blood Vessels and Lowers Interstitial Fluid Pressure in Solid Tumors: Clinical Implications. Cancer Res. 1999, 59, 59. [Google Scholar]
- Padera, T.P.; Stoll, B.R.; Tooredman, J.B.; Capen, D.; di Tomaso, E.; Jain, R.K. Pathology: Cancer Cells Compress Intratumour Vessels 1 11672. Nature 2004, 427, 695. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Pathophysiological Consequences of VEGF-Induced Vascular Permeability. Nature 2005, 437, 497–504. [Google Scholar] [CrossRef]
- Nagy, J.A.; Dvorak, A.M.; Dvorak, H.F. Vascular Hyperpermeability, Angiogenesis, and Stroma Generation. Cold Spring Harb. Perspect. Med. 2012, 2, a006544. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, Consequences, and Remedies for Growth-Induced Solid Stress in Murine and Human Tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef] [Green Version]
- Helmlinger, G.; Yuan, F.; Dellian, M.; Jain, R.K. Interstitial PH and PO2 Gradients in Solid Tumors in Vivo: High-Resolution Measurements Reveal a Lack of Correlation. Nat. Med. 1997, 3, 177–182. [Google Scholar] [CrossRef]
- Simonsen, T.G.; Lund, K.V.; Hompland, T.; Kristensen, G.B.; Rofstad, E.K. DCE-MRI–Derived Measures of Tumor Hypoxia and Interstitial Fluid Pressure Predict Outcomes in Cervical Carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, 1193–1201. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, S.; Allegrini, P.R.; Becquet, M.M.; McSheehy, P.M.J. Tumor Interstitial Fluid Pressure as an Early-Response Marker for Anticancer Therapeutics. Neoplasia 2009, 11, 874–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baish, J.W.; Netti, P.A.; Jain, R.K. Transmural Coupling of Fluid Flow in Microcirculatory Network and Interstitium in Tumors. Microvasc. Res. 1997, 53, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Frieboes, H.B.; Chaplain, M.A.J.; McDougall, S.R.; Cristini, V.; Lowengrub, J.S. The Effect of Interstitial Pressure on Therapeutic Agent Transport: Coupling with the Tumor Blood and Lymphatic Vascular Systems. J. Theor. Biol. 2014, 355, 194–207. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K.; Stylianopoulos, T. Delivering Nanomedicine to Solid Tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Bender, L.H.; Abbate, F.; Walters, I.B. Intratumoral Administration of a Novel Cytotoxic Formulation with Strong Tissue Dispersive Properties Regresses Tumor Growth and Elicits Systemic Adaptive Immunity in in Vivo Models. Int. J. Mol. Sci. 2020, 21, 4493. [Google Scholar] [CrossRef]
- Sriraman, S.K.; Aryasomayajula, B.; Torchilin, V.P. Barriers to Drug Delivery in Solid Tumors. Tissue Barriers 2014, 2, e29528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of Glucose Metabolization in Cancer Cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Caswell, D.R.; Swanton, C. The Role of Tumour Heterogeneity and Clonal Cooperativity in Metastasis, Immune Evasion and Clinical Outcome. BMC Med. 2017, 15, 133. [Google Scholar] [CrossRef]
- Yang, T.; Wall, E.M.; Milne, K.; Theiss, P.; Watson, P.; Nelson, B.H. CD8+ T Cells Induce Complete Regression of Advanced Ovarian Cancers by an Interleukin (IL)-2/IL-15–Dependent Mechanism. Clin. Cancer Res. 2007, 13, 7172–7180. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, K.M.; Scarbrough, P.M.; Ribeiro, A.; Richardson, R.; Yuan, H.; Sonveaux, P.; Landon, C.D.; Chi, J.-T.; Pizzo, S.; Schroeder, T.; et al. Catabolism of Exogenous Lactate Reveals It as a Legitimate Metabolic Substrate in Breast Cancer. PLoS ONE 2013, 8, e75154. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Bai, C.; Ruan, Y.; Liu, M.; Chu, Q.; Qiu, L.; Yang, C.; Li, B. Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat. Commun. 2019, 10, 201. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; Van Hée, V.; De Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.-J.; Brisson, L.; et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 2016, 15, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatenby, R.A.; Gawlinski, E.T.; Gmitro, A.F.; Kaylor, B.; Gillies, R. Acid-Mediated Tumor Invasion: A Multidisciplinary Study. Cancer Res. 2006, 66, 5216–5223. [Google Scholar] [CrossRef] [Green Version]
- Rauschner, M.; Lange, L.; Hüsing, T.; Reime, S.; Nolze, A.; Maschek, M.; Thews, O.; Riemann, A. Impact of the acidic environment on gene expression and functional parameters of tumors in vitro and in vivo. J. Exp. Clin. Cancer Res. 2021, 40, 10. [Google Scholar] [CrossRef] [PubMed]
- Riemann, A.; Schneider, B.; Gündel, D.; Stock, C.; Thews, O.; Gekle, M. Acidic priming enhances metastatic potential of cancer cells. Pflügers Arch. Eur. J. Physiol. 2014, 466, 2127–2138. [Google Scholar] [CrossRef]
- Wu, H.; Estrella, V.; Beatty, M.; Abrahams, D.; El-Kenawi, A.; Russell, S.; Ibrahim-Hashim, A.; Longo, D.L.; Reshetnyak, Y.K.; Moshnikova, A.; et al. T-cells produce acidic niches in lymph nodes to suppress their own effector functions. Nat. Commun. 2020, 11, 4113. [Google Scholar] [CrossRef]
- Halcrow, P.W.; Geiger, J.D.; Chen, X. Overcoming Chemoresistance: Altering pH of Cellular Compartments by Chloroquine and Hydroxychloroquine. Front. Cell Dev. Biol. 2021, 9, 170. [Google Scholar] [CrossRef]
- Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C.A. The Concentration of Oxygen Dissolved in Tissues at the Time of Irradiation as a Factor in Radiotherapy. Br. J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef]
- Price, M.; Heilbrun, L.; Kessel, D. Effects of the oxygenation level on formation of different reactive oxygen species during photodynamic therapy. Photochem. Photobiol. 2012, 89, 683–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, I. Facing hypoxia: A must for photodynamic therapy. J. Photochem. Photobiol. B Biol. 1988, 2, 281–282. [Google Scholar] [CrossRef]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Y.; Zhao, S.; Han, J.; Zheng, L.; Yang, Z.; Zhao, L. Hypoxia-inducible factor-1α induces multidrug resistance protein in colon cancer. OncoTargets Ther. 2015, 8, 1941–1948. [Google Scholar] [CrossRef] [Green Version]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.-L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzaei, S.A.; Reiisi, S.; Tabari, P.G.; Shekari, A.; Aliakbari, F.; Azadfallah, E.; Elahian, F. Broad blocking of MDR efflux pumps by acetylshikonin and acetoxyisovalerylshikonin to generate hypersensitive phenotype of malignant carcinoma cells. Sci. Rep. 2018, 8, 3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ding, Z.; Peng, Y.; Pan, F.; Li, J.; Zou, L.; Zhang, Y.; Liang, H. HIF-1α Inhibition Reverses Multidrug Resistance in Colon Cancer Cells via Downregulation of MDR1/P-Glycoprotein. PLoS ONE 2014, 9, e98882. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.-J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Saggar, J.K.; Tannock, I.F. Chemotherapy Rescues Hypoxic Tumor Cells and Induces Their Reoxygenation and Repopulation—An Effect That Is Inhibited by the Hypoxia-Activated Prodrug TH-302. Clin. Cancer Res. 2015, 21, 2107–2114. [Google Scholar] [CrossRef] [Green Version]
- Iyikesici, M.S. Long-Term Survival Outcomes of Metabolically Supported Chemotherapy with Gemcitabine-Based or FOLFIRINOX Regimen Combined with Ketogenic Diet, Hyperthermia, and Hyperbaric Oxygen Therapy in Metastatic Pancreatic Cancer. Complement. Med. Res. 2020, 27, 31–39. [Google Scholar] [CrossRef]
- Alagoz, T.; Buller, R.E.; Anderson, B.; Terrell, K.L.; Squatrito, R.C.; Niemann, T.H.; Tatman, D.J.; Jebson, P. Evaluation of hyperbaric oxygen as a chemosensitizer in the treatment of epithelial ovarian cancer in xenografts in mice. Cancer 1995, 75, 2313–2322. [Google Scholar] [CrossRef]
- Takiguchi, N.; Saito, N.; Nunomura, M.; Kouda, K.; Oda, K.; Furuyama, N.; Nakajima, N. Use of 5-FU plus hyperbaric oxygen for treating malignant tumors: Evaluation of antitumor effect and measurement of 5-FU in individual organs. Cancer Chemother. Pharmacol. 2000, 47, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Khouzam, R.A.; Brodaczewska, K.; Filipiak, A.; Zeinelabdin, N.A.; Buart, S.; Szczylik, C.; Kieda, C.; Chouaib, S. Tumor Hypoxia Regulates Immune Escape/Invasion: Influence on Angiogenesis and Potential Impact of Hypoxic Biomarkers on Cancer Therapies. Front. Immunol. 2021, 11, 3479. [Google Scholar] [CrossRef]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P.; et al. Hypoxia-Inducible Factor-1 Alpha-Dependent Induction of FoxP3 Drives Regulatory T-Cell Abundance and Function during Inflammatory Hypoxia of the Mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A Mechanism of Hypoxia-Mediated Escape from Adaptive Immunity in Cancer Cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Buart, S.; van Pelt, J.; Richon, C.; Hasmim, M.; Leleu, N.; Suchorska, W.M.; Jalil, A.; Lecluse, Y.; el Hage, F.; et al. The Cooperative Induction of Hypoxia-Inducible Factor-1 Alpha and STAT3 during Hypoxia Induced an Impairment of Tumor Susceptibility to CTL-Mediated Cell Lysis. J. Immunol. (Baltim. Md. 1950) 2009, 182, 3510–3521. [Google Scholar] [CrossRef]
- Damgaci, S.; Ibrahim-Hashim, A.; Enriquez-Navas, P.M.; Pilon-Thomas, S.; Guvenis, A.; Gillies, R.J. Hypoxia and Acidosis: Immune Suppressors and Therapeutic Targets. Immunology 2018, 154, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Choukèr, A.; Thiel, M.; Lukashev, D.; Ward, J.M.; Kaufmann, I.; Apasov, S.; Sitkovsky, M.V.; Ohta, A. Critical Role of Hypoxia and A2A Adenosine Receptors in Liver Tissue-Protecting Physiological Anti-Inflammatory Pathway. Mol. Med. 2008, 14, 116–123. [Google Scholar] [CrossRef]
- Mpekris, F.; Voutouri, C.; Baish, J.W.; Duda, D.G.; Munn, L.L.; Stylianopoulos, T.; Jain, R.K. Combining Microenvironment Normalization Strategies to Improve Cancer Immunotherapy. Proc. Natl. Acad. Sci. USA 2020, 117, 3728–3737. [Google Scholar] [CrossRef] [Green Version]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic Stress: Obstacles and Opportunities for Innovative Immunotherapy of Cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Torres, N.; Regge, M.V.; Secchiari, F.; Friedrich, A.D.; Spallanzani, R.G.; Raffo Iraolagoitia, X.L.; Núñez, S.Y.; Sierra, J.M.; Ziblat, A.; Santilli, M.C.; et al. Restoration of Antitumor Immunity through Anti-MICA Antibodies Elicited with a Chimeric Protein. J. ImmunoTherapy Cancer 2020, 8, e000233. [Google Scholar] [CrossRef] [PubMed]
- Labiano, S.; Palazon, A.; Melero, I. Immune Response Regulation in the Tumor Microenvironment by Hypoxia. Semin. Oncol. 2015, 42, 378–386. [Google Scholar] [CrossRef]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-β Signaling and Epithelial-Mesenchymal Transition in Cancer Progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Borriello, L.; Nakata, R.; Sheard, M.A.; Fernandez, G.E.; Sposto, R.; Malvar, J.; Blavier, L.; Shimada, H.; Asgharzadeh, S.; Seeger, R.C.; et al. Cancer-Associated Fibroblasts Share Characteristics and Protumorigenic Activity with Mesenchymal Stromal Cells. Cancer Res. 2017, 77, 5142–5157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, C.W.; Gold, M.J.; Garcia-Batres, C.; Tai, K.; Elford, A.R.; Himmel, M.E.; Elia, A.J.; Ohashi, P.S. Hypoxia-Inducible Factor 1 Alpha Limits Dendritic Cell Stimulation of CD8 T Cell Immunity. PLoS ONE 2020, 15, e0244366. [Google Scholar] [CrossRef]
- Weigert, A.; Weichand, B.; Sekar, D.; Sha, W.; Hahn, C.; Mora, J.; Ley, S.; Essler, S.; Dehne, N.; Brüne, B. HIF-1α Is a Negative Regulator of Plasmacytoid DC Development in Vitro and in Vivo. Blood 2012, 120, 3001–3006. [Google Scholar] [CrossRef] [Green Version]
- Colliez, F.; Gallez, B.; Jordan, B.F. Assessing Tumor Oxygenation for Predicting Outcome in Radiation Oncology: A Review of Studies Correlating Tumor Hypoxic Status and Outcome in the Preclinical and Clinical Settings. Front. Oncol. 2017, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Yeh, J.J.; Kim, W.Y. Targeting Tumor Hypoxia with Hypoxia-Activated Prodrugs. J. Clin. Oncol. 2015, 33, 1505–1508. [Google Scholar] [CrossRef]
- Thews, O.; Vaupel, P. Spatial Oxygenation Profiles in Tumors during Normo- and Hyperbaric Hyperoxia. Strahlenther. Und Onkol. 2015, 191, 875–882. [Google Scholar] [CrossRef]
- Gainer, J.L. Increasing Oxygen in Hypoxic Tumors. Clin. Exp. Pharmacol. 2012, 1. [Google Scholar] [CrossRef]
- Eisenbrey, J.R.; Shraim, R.; Liu, J.B.; Li, J.; Stanczak, M.; Oeffinger, B.; Leeper, D.B.; Keith, S.W.; Jablonowski, L.J.; Forsberg, F.; et al. Sensitization of Hypoxic Tumors to Radiation Therapy Using Ultrasound-Sensitive Oxygen Microbubbles. Int. J. Radiat. Oncol. Biol. Phys. 2018, 101, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Cheng, H.; Jiang, C.; Qiu, X.; Wang, K.; Huan, W.; Yuan, A.; Wu, J.; Hu, Y. Perfluorocarbon Nanoparticles Enhance Reactive Oxygen Levels and Tumour Growth Inhibition in Photodynamic Therapy. Nat. Commun. 2015, 6, 8785. [Google Scholar] [CrossRef]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of Vascular Endothelial Growth Factor-Induced Angiogenesis Suppresses Tumour Growth in Vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef]
- Van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.M.; Thijssen, V.L.; Griffioen, A.W. The Great Escape; the Hallmarks of Resistance to Antiangiogenic Therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Eales, K.L.; Hollinshead, K.E.R.; Tennant, D.A. Hypoxia and Metabolic Adaptation of Cancer Cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [Green Version]
- Godet, I.; Shin, Y.J.; Ju, J.A.; Ye, I.C.; Wang, G.; Gilkes, D.M. Fate-Mapping Post-Hypoxic Tumor Cells Reveals a ROS-Resistant Phenotype That Promotes Metastasis. Nat. Commun. 2019, 10, 4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verduzco, D.; Lloyd, M.; Xu, L.; Ibrahim-Hashim, A.; Balagurunathan, Y.; Gatenby, R.A.; Gillies, R.J. Intermittent Hypoxia Selects for Genotypes and Phenotypes That Increase Survival, Invasion, and Therapy Resistance. PLoS ONE 2015, 10, e0120958. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Dor, Y.; Porat, R.; Keshet, E. Vascular Endothelial Growth Factor and Vascular Adjustments to Perturbations in Oxygen Homeostasis. Am. J. Physiol. Cell Physiol. 2001, 280, C1367–C1374. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; Seano, G.; Jain, R.K. Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu. Rev. Physiol. 2019, 81, 505–534. [Google Scholar] [CrossRef] [PubMed]
- Tewari, K.S.; Sill, M.W.; Long, H.J.; Penson, R.T.; Huang, H.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; Leitao, M.M.; et al. Improved Survival with Bevacizumab in Advanced Cervical Cancer. N. Engl. J. Med. 2014, 370, 734–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabbinavar, F.F.; Hambleton, J.; Mass, R.D.; Hurwitz, H.I.; Bergsland, E.; Sarkar, S. Combined Analysis of Efficacy: The Addition of Bevacizumab to Fluorouracil/Leucovorin Improves Survival for Patients with Metastatic Colorectal Cancer. J. Clin. Oncol. 2005, 23, 3706–3712. [Google Scholar] [CrossRef]
- Sandler, A.; Gray, R.; Perry, M.C.; Brahmer, J.; Schiller, J.H.; Dowlati, A.; Lilenbaum, R.; Johnson, D.H. Paclitaxel-Carboplatin Alone or with Bevacizumab for Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2006, 355, 2542–2550. [Google Scholar] [CrossRef] [Green Version]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A Phase 3 Trial of Bevacizumab in Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Pluzanska, A.; Koralewski, P.; Ravaud, A.; Bracarda, S.; Szczylik, C.; Chevreau, C.; Filipek, M.; Melichar, B.; Bajetta, E.; et al. Bevacizumab plus Interferon Alfa-2a for Treatment of Metastatic Renal Cell Carcinoma: A Randomised, Double-Blind Phase III Trial. Lancet 2007, 370, 2103–2111. [Google Scholar] [CrossRef]
- Escudier, B.; Bellmunt, J.; Négrier, S.; Bajetta, E.; Melichar, B.; Bracarda, S.; Ravaud, A.; Golding, S.; Jethwa, S.; Sneller, V. Phase III Trial of Bevacizumab plus Interferon Alfa-2a in Patients with Metastatic Renal Cell Carcinoma (AVOREN): Final Analysis of Overall Survival. J. Clin. Oncol. 2010, 28, 2144–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.H.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Elisei, R.; Schlumberger, M.J.; Müller, S.P.; Schöffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in Progressive Medullary Thyroid Cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef] [Green Version]
- Beaver, J.; Park, B.H. The BOLERO-2 Trial: The Addition of Everolimus to Exemestane in the Treatment of Postmenopausal Hormone Receptor-Positive Advanced Breast Cancer. Future Oncol. 2013, 8, 651–657. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, Everolimus, and the Combination in Patients with Metastatic Renal Cell Carcinoma: A Randomised, Phase 2, Open-Label, Multicentre Trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in Locally Advanced or Metastatic Renal Cell Carcinoma: Results of a Randomized Phase III Trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef]
- van der Graaf, W.T.A.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for Metastatic Soft-Tissue Sarcoma (PALETTE): A Randomised, Double-Blind, Placebo-Controlled Phase 3 Trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Grothey, A.; van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib Monotherapy for Previously Treated Metastatic Colorectal Cancer (CORRECT): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Effi Cacy and Safety of Regorafenib for Advanced Gastrointestinal Stromal Tumours after Failure of Imatinib and Sunitinib (GRID): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for Patients with Hepatocellular Carcinoma Who Progressed on Sorafenib Treatment (RESORCE): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in Advanced Clear-Cell Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; Cosme de Oliveira, A.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 2498. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de La Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in Radioactive Iodine-Refractory, Locally Advanced or Metastatic Diff Erentiated Thyroid Cancer: A Randomised, Double-Blind, Phase 3 Trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Faivre, S.; Niccoli, P.; Castellano, D.; Valle, J.W.; Hammel, P.; Raoul, J.L.; Vinik, A.; van Cutsem, E.; Bang, Y.J.; Lee, S.H.; et al. Sunitinib in Pancreatic Neuroendocrine Tumors: Updated Progression-Free Survival and Final Overall Survival from a Phase III Randomized Study. Ann. Oncol. 2017, 28, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in Patients with Locally Advanced or Metastatic Medullary Thyroid Cancer: A Randomized, Double-Blind Phase III Trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausová, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of Aflibercept to Fluorouracil, Leucovorin, and Irinotecan Improves Survival in a Phase III Randomized Trial in Patients with Metastatic Colorectal Cancer Previously Treated with an Oxaliplatin-Based Regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilke, H.; Muro, K.; van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab plus Paclitaxel versus Placebo plus Paclitaxel in Patients with Previously Treated Advanced Gastric or Gastro-Oesophageal Junction Adenocarcinoma (RAINBOW): A Double-Blind, Randomised Phase 3 Trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Garon, E.B.; Ciuleanu, T.E.; Arrieta, O.; Prabhash, K.; Syrigos, K.N.; Goksel, T.; Park, K.; Gorbunova, V.; Kowalyszyn, R.D.; Pikiel, J.; et al. Ramucirumab plus Docetaxel versus Placebo plus Docetaxel for Second-Line Treatment of Stage IV Non-Small-Cell Lung Cancer after Disease Progression on Platinum-Based Therapy (REVEL): A Multicentre, Double-Blind, Randomised Phase 3 Trial. Lancet 2014, 384, 665–673. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after Sorafenib in Patients with Advanced Hepatocellular Carcinoma and Increased α-Fetoprotein Concentrations (REACH-2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Bais, C.; Mueller, B.; Brady, M.F.; Mannel, R.S.; Burger, R.A.; Wei, W.; Marien, K.M.; Kockx, M.M.; Husain, A.; Birrer, M.J. Tumor Microvessel Density as a Potential Predictive Marker for Bevacizumab Benefit: GOG-0218 Biomarker Analyses. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Martin, J.D.; Fukumura, D.; Duda, D.G.; Boucher, Y.; Jain, R.K. Reengineering the Tumor Microenvironment to Alleviate Hypoxia and Overcome Cancer Heterogeneity. Cold Spring Harb. Perspect. Med. 2016, 6, a027094. [Google Scholar] [CrossRef] [Green Version]
- Tolaney, S.M.; Boucher, Y.; Duda, D.G.; Martin, J.D.; Seano, G.; Ancukiewicz, M.; Barry, W.T.; Goel, S.; Lahdenrata, J.; Isakoff, S.J.; et al. Role of Vascular Density and Normalization in Response to Neoadjuvant Bevacizumab and Chemotherapy in Breast Cancer Patients. Proc. Natl. Acad. Sci. USA 2015, 112, 14325–14330. [Google Scholar] [CrossRef] [Green Version]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic Therapy in Oncology: Current Status and Future Directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Lawler, J. The Structural and Functional Properties of Thrombospondin. Blood 1986, 67, 1197–1209. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals Leading to Apoptosis-Dependent Inhibition of Neovascularization by Thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Bocci, G.; Francia, G.; Man, S.; Lawler, J.; Kerbel, R.S. Thrombospondin 1, a Mediator of the Antiangiogenic Effects of Low-Dose Metronomic Chemotherapy. Proc. Natl. Acad. Sci. USA 2003, 100, 12917–12922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; Gupta, N.; Walcott, B.P.; Snuderl, M.; Kesler, C.T.; Kirkpatrick, N.D.; Heishi, T.; Huang, Y.; Martin, J.D.; Ager, E.; et al. Effects of Vascular-Endothelial Protein Tyrosine Phosphatase Inhibition on Breast Cancer Vasculature and Metastatic Progression. J. Natl. Cancer Inst. 2013, 105, 1188–1201. [Google Scholar] [CrossRef]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual Inhibition of Ang-2 and VEGF Receptors Normalizes Tumor Vasculature and Prolongs Survival in Glioblastoma by Altering Macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Goldstein, A.; Wang, H.; Lo, H.C.; Kim, I.S.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual Regulation of Tumour Vessel Normalization and Immunostimulatory Reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef]
- Kłosowska-Wardȩga, A.; Hasumi, Y.; Burmakin, M.; Åhgren, A.; Stuhr, L.; Moen, I.; Reed, R.K.; Rubin, K.; Hellberg, C.; Heldin, C.H. Combined Anti-Angiogenic Therapy Targeting PDGF and Vegf Receptors Lowers the Interstitial Fluid Pressure in a Murine Experimental Carcinoma. PLoS ONE 2009, 4, e8149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matuszewska, K.; Santry, L.A.; van Vloten, J.P.; AuYeung, A.W.K.; Major, P.P.; Lawler, J.; Wootton, S.K.; Bridle, B.W.; Petrik, J. Combining Vascular Normalization with an Oncolytic Virus Enhances Immunotherapy in a Preclinical Model of Advanced-Stage Ovarian Cancer. Clin. Cancer Res. 2019, 25, 1624–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic Agents Can Increase Lymphocyte Infiltration into Tumor and Enhance the Effectiveness of Adoptive Immunotherapy of Cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; Wong, A.H.K.; Jain, R.K. Vascular Normalization as a Therapeutic Strategy for Malignant and Nonmalignant Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006486. [Google Scholar] [CrossRef]



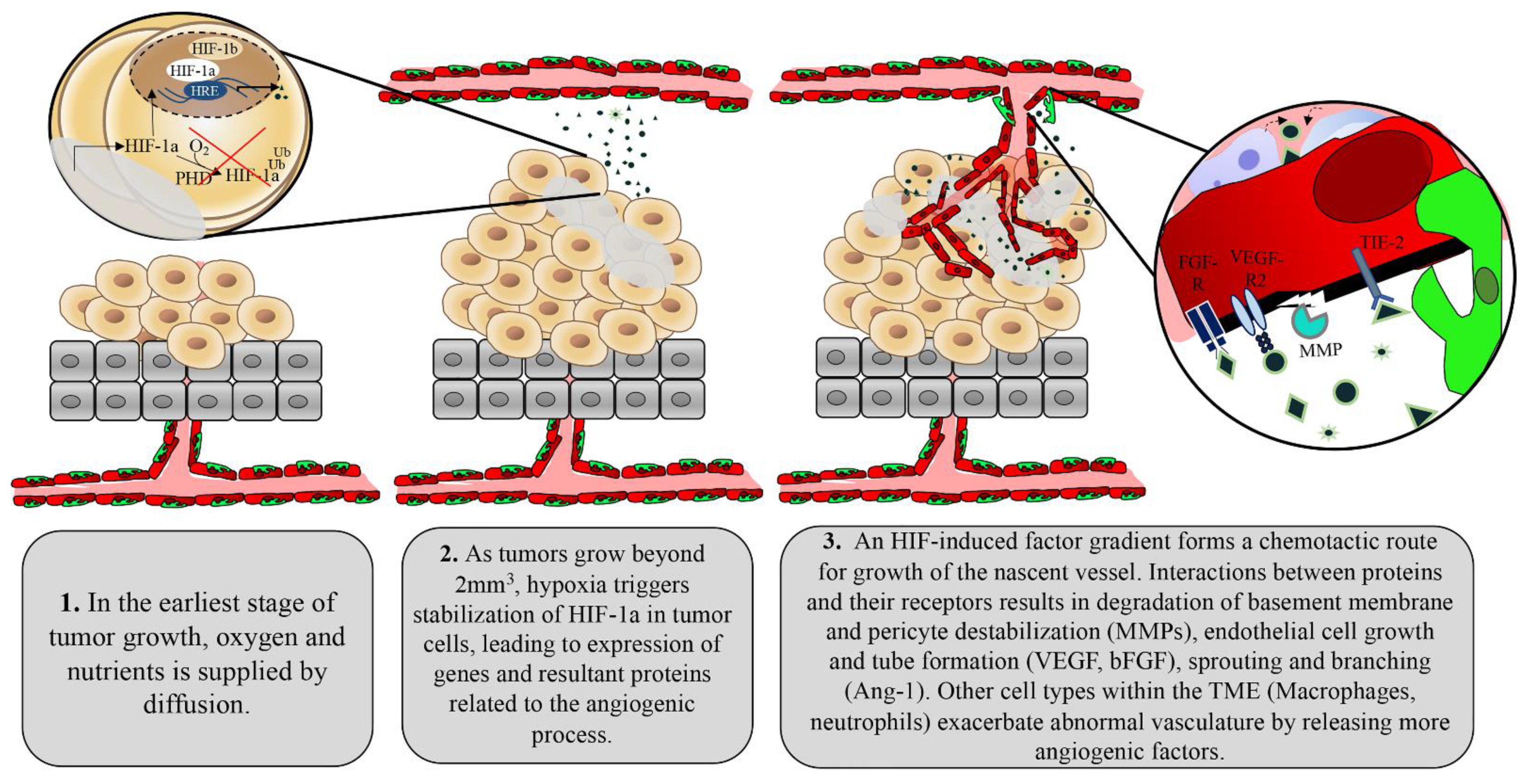{kind=link}
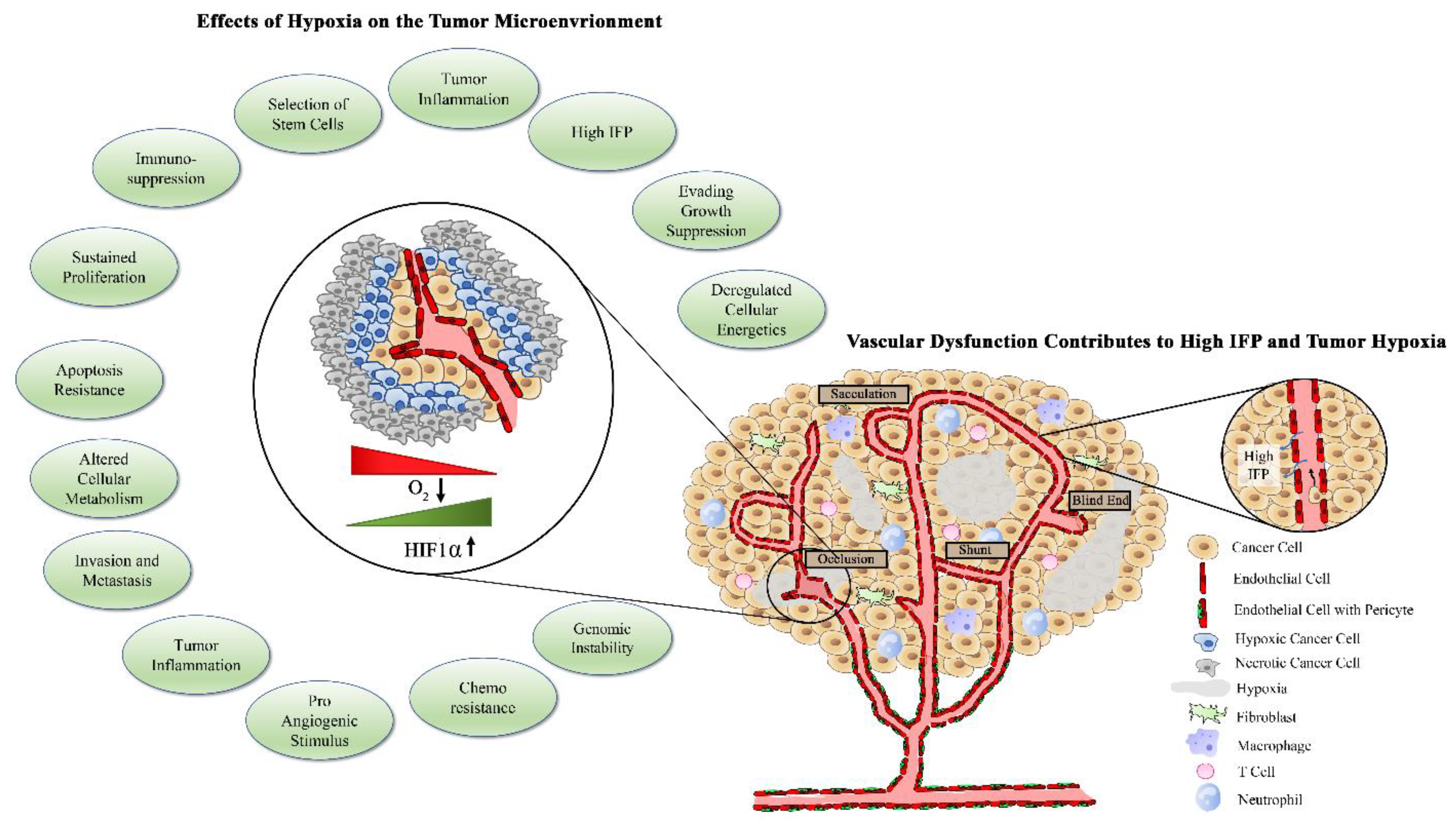{kind=link}
| Anti-Angiogenic Drug | FDA Approval | Mechanism | Indication | Combination Agent | Anti-angiogenic Drug + Combination Agent vs. Combination Agent Alone (*) | Ref. | |
|---|---|---|---|---|---|---|---|
| PFS (mts) | OS (mts) | ||||||
| Bevacizumab (Avastin®) | 2004 | Humanized monoclonal antibody that binds to and inhibits the activity of VEGF-A | Cervical | Paclitaxel + Cisplatin | 9.63 (* 6.67) | 17.51 (* 12.68) | [122] |
| Paclitaxel + Topotecan | 7.36 (* 5.29) | 16.20 (* 12.68) | [122] | ||||
| Colorectal (metastatic) | 5-Fluorouracil | 8.8 (* 5.6) | 17.9 (* 14.6) | [123] | |||
| NSCLC | Carboplatin + Paclitaxel | 6.2 (* 4.5) | 12.3 (* 10.3) | [124] | |||
| Ovarian, Fallopian, primary peritoneal | Carboplatin + Paclitaxel | 18.1 (* 14.5) | 36.6 (* 28.8) | [125] | |||
| Renal Cell | Interferon alfa | 10.2 (* 5.4) | 23.3 (* 21.3) | [126,127] | |||
| Axitinib (Inlyta®) | 2012 | Tyrosine Kinase Inhibitor (VEGFR-1, VEGFR-2, VEGFR-3) | Renal Cell | Pembrolizu mab | 15.1 (* 11.1) | - | [128] |
| Cabozantinib (Cometriq ®) | 2012 | Tyrosine Kinase Inhibitor (VEGF, MET, AXL) | Hepatocellular | Placebo | 5.2 (* 1.9) | 10.2 (* 8.0) | [129] |
| Medullary Thyroid | Placebo | 11.2 (* 4.0) | - | [130] | |||
| Everolimus (Afinitor®, Zortress ®) | 2009 | mTOR inhibitor | Breast | Exemestane | 10.6 (* 4.1) | - | [131] |
| Advanced Kidney | Lenvatinib | 14.6 (* 7.4) | - | [132] | |||
| Pazopanib (votrient®) | 2009 | Tyrosine Kinase Inhibitor (VEGFR-1,-2, -3, PDGFR-a, -b, c-KIT, FGFR-1, -3) | Renal Cell | placebo | 9.2 (* 4.2) | 22.9 (* 20.5) | [133] |
| Soft Tissue Sarcoma | placebo | 4.6 (* 1.6) | 12.5 (* 10.7) | [134,135] | |||
| Regorafenib (Stivarga®) | 2012 | Tyrosine Kinase Inhibitor (VEGFR-1, -2, -3, TIE-2, PDGRF, FGFR, KIT, RET, RAF-1, BRAF) | Colorectal Cancer | Placebo | - | 6.4 (5.0) | [135] |
| Gastrointestinal | Placebo | 4.8 (* 0.9) | - | [136] | |||
| Hepatocellular | Placebo | - | 10.6 (* 7.8) | [137] | |||
| Sorafenib (Nexavar®) | 2005 | Tyrosine Kinase Inhibitor (Raf, PDGF, VEGFR-2, -3, c-KIT) | Renal Cell | placebo | 5.5 (* 2.8) | 19.3 (* 15.9) | [138] |
| Hepatocellular | placebo | 5.5 (* 2.8) | 10.7 (* 7.9) | [139] | |||
| Advanced thyroid | placebo | 10.8 (* 5.8) | - | [140,141] | |||
| Sunitinib (Sutent®) | 2006 | Tyrosine Kinase Inhibitor (PDGF-a, b, VEGFR-1, -2, -3, KIT, FLT-3, CSF-1R) | Pancreatic Neuo-endocrine | Placebo | 12.6 (* 5.8) | 38.6 (* 29.1) | [141] |
| Gastrointestinal Stromal | Placebo | - | 18.5 (* 8.9) | [136] | |||
| Vandetanib | 2011 | Tyrosine Kinase Inhibitor (VEGFR-2, EGFR, RET) | Medullary Thyroid | Placebo | 30.5 (* 19.2) | - | [142] |
| Ziv-aflibercept | 2012 | Fusion protein (two human VEGF receptors connected by Fc domain) | Colorectal | FOLFIRI chemo (Folinic Acid, Fluorouracil, irinotecan) | 6.9 (* 4.7) | 13.5 (* 12.0) | [143] |
| Ramucirumab | 2014 | Human monoclonal antibody against VEGFR-2 | Gastric | Paclitaxel | - | 9.6 (* 7.4) | [144,145] |
| NSCLC | Docetaxel | 4.5 (* 3.0) | 10.5 (* 9.1) | [145] | |||
| Colorectal | Placebo | 2.8 (* 1.6) | 8.5 (* 7.3) | [146] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matuszewska, K.; Pereira, M.; Petrik, D.; Lawler, J.; Petrik, J. Normalizing Tumor Vasculature to Reduce Hypoxia, Enhance Perfusion, and Optimize Therapy Uptake. Cancers 2021, 13, 4444. https://doi.org/10.3390/cancers13174444
Matuszewska K, Pereira M, Petrik D, Lawler J, Petrik J. Normalizing Tumor Vasculature to Reduce Hypoxia, Enhance Perfusion, and Optimize Therapy Uptake. Cancers. 2021; 13(17):4444. https://doi.org/10.3390/cancers13174444
Chicago/Turabian StyleMatuszewska, Kathy, Madison Pereira, Duncan Petrik, Jack Lawler, and Jim Petrik. 2021. "Normalizing Tumor Vasculature to Reduce Hypoxia, Enhance Perfusion, and Optimize Therapy Uptake" Cancers 13, no. 17: 4444. https://doi.org/10.3390/cancers13174444
APA StyleMatuszewska, K., Pereira, M., Petrik, D., Lawler, J., & Petrik, J. (2021). Normalizing Tumor Vasculature to Reduce Hypoxia, Enhance Perfusion, and Optimize Therapy Uptake. Cancers, 13(17), 4444. https://doi.org/10.3390/cancers13174444