
The CDH1 c.1901C>T Variant: A Founder Variant in the Portuguese Population with Severe Impact in mRNA Splicing

 , , , , , , , , add
Show full author list
, , , , , , , , add
Show full author list
Abstract
:Simple Summary
Abstract
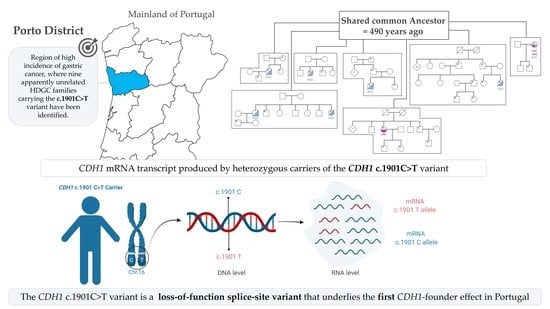
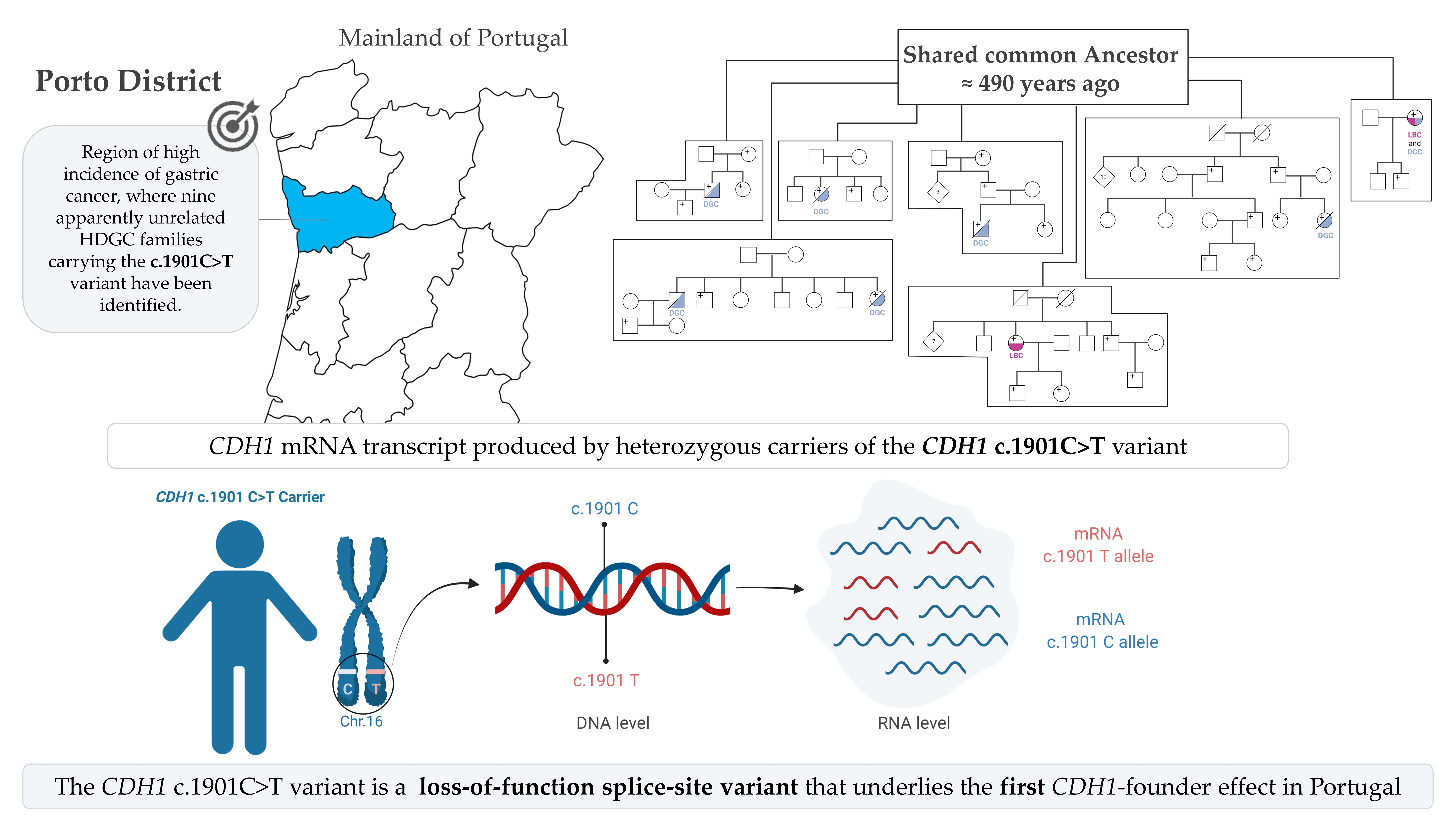
1. Introduction
2. Materials and Methods
2.1. Study Design and Data Collection
2.2. Biological Material (DNA and RNA) Collection from CDH1 c.1901C>T Variant Bearing Families
2.3. cDNA Synthesis and PCR Amplification
2.4. Cloning and Colony PCR
2.5. CDH1 Allelic Specific Expression (ASE) by SNaPshot
2.6. Histopathological Analysis
2.7. CDH1 c.1901C>T Variant Classification According to ACMG Guidelines
2.8. Identification of the Recombination Point in 16q22.1 Chromosome’s Region, Haplotyping of Different Polymorphic Markers
2.9. Characterization and Estimation of Age of the CDH1 c.1901C>T Variant
3. Results
3.1. The CDH1 c.1901C>T Variant Generates Cryptic Splicing within Exon 12, Leading to Premature Truncation and Decreased CDH1 RNA Levels
3.2. Age Estimation of CDH1 c.1901C>T Variant through Haplotyping and Identification of the Recombination Point in 16q22.1 Chromosome
3.3. CDH1 c.1901C>T a Founder Variant with Approximately 500 Years
3.4. Clinical Presentations in HDGC Families Carrying CDH1 c.1901C>T
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oliveira, C.; Pinheiro, H.; Figueiredo, J.; Seruca, R.; Carneiro, F. Familial gastric cancer: Genetic susceptibility, pathology, and implications for management. Lancet Oncol. 2015, 16, e60–e70. [Google Scholar] [CrossRef]
- Guilford, P.; Hopkins, J.; Harraway, J.; McLeod, M.; McLeod, N.; Harawira, P.; Taite, H.; Scoular, R.; Miller, A.; Reeve, A.E. E-cadherin germline mutations in familial gastric cancer. Nature 1998, 392, 402–405. [Google Scholar] [CrossRef]
- Caldas, C.; Carneiro, F.; Lynch, H.T.; Yokota, J.; Wiesner, G.L.; Powell, S.M.; Lewis, F.R.; Huntsman, D.G.; Pharoah, P.D.; Jankowski, J.A.; et al. Familial gastric cancer: Overview and guidelines for management. J. Med. Genet. 1999, 36, 873–880. [Google Scholar]
- Oliveira, C.; Senz, J.; Kaurah, P.; Pinheiro, H.; Sanges, R.; Haegert, A.; Corso, G.; Schouten, J.; Fitzgerald, R.; Vogelsang, H.; et al. Germline CDH1 deletions in hereditary diffuse gastric cancer families. Hum. Mol. Genet. 2009, 18, 1545–1555. [Google Scholar] [CrossRef]
- Blair, V.R.; McLeod, M.; Carneiro, F.; Coit, D.G.; D’Addario, J.L.; van Dieren, J.M.; Harris, K.L.; Hoogerbrugge, N.; Oliveira, C.; van der Post, R.S.; et al. Hereditary diffuse gastric cancer: Updated clinical practice guidelines. Lancet Oncol. 2020, 21, e386–e397. [Google Scholar] [CrossRef]
- Fitzgerald, R.C.; Hardwick, R.; Huntsman, D.; Carneiro, F.; Guilford, P.; Blair, V.; Chung, D.C.; Norton, J.; Ragunath, K.; Van Krieken, J.H.; et al. Hereditary diffuse gastric cancer: Updated consensus guidelines for clinical management and directions for future research. J. Med. Genet. 2010, 47, 436–444. [Google Scholar] [CrossRef] [Green Version]
- van der Post, R.S.; Vogelaar, I.P.; Carneiro, F.; Guilford, P.; Huntsman, D.; Hoogerbrugge, N.; Caldas, C.; Schreiber, K.E.; Hardwick, R.H.; Ausems, M.G.; et al. Hereditary diffuse gastric cancer: Updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J. Med. Genet. 2015, 52, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, M.E.; Ranola, J.M.O.; Marshall, M.L.; Susswein, L.R.; Graceffo, S.; Bohnert, K.; Tsai, G.; Klein, R.T.; Hruska, K.S.; Shirts, B.H. Comparison of CDH1 Penetrance Estimates in Clinically Ascertained Families vs Families Ascertained for Multiple Gastric Cancers. JAMA Oncol. 2019, 5, 1325–1331. [Google Scholar] [CrossRef]
- Pharoah, P.D.; Guilford, P.; Caldas, C.; International Gastric Cancer Linkage Consortium. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001, 121, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Xicola, R.M.; Li, S. Clinical features and cancer risk in families with pathogenic CDH1 variants irrespective of clinical criteria. J. Med. Genet. 2019, 56, 838–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Krempely, K. Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum. Mutat. 2018, 39, 1553–1568. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.; Figueiredo, J. Predicting the Functional Impact of CDH1 Missense Mutations in Hereditary Diffuse Gastric Cancer. Int. J. Mol. Sci. 2017, 18, 2687. [Google Scholar] [CrossRef] [Green Version]
- Kaurah, P.; MacMillan, A.; Boyd, N.; Senz, J.; De Luca, A.; Chun, N.; Suriano, G.; Zaor, S.; Van Manen, L.; Gilpin, C.; et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 2007, 297, 2360–2372. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.A. Old meets new: Identifying founder mutations in genetic disease. Can. Med. Assoc. J. 2015, 187, 93–94. [Google Scholar] [CrossRef] [Green Version]
- Kivisild, T. Founder Effect. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Academic Press: Massachusetts, MA, USA, 2013; pp. 100–101. [Google Scholar]
- Keinan, A.; Clark, A.G. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science 2012, 336, 740–743. [Google Scholar] [CrossRef] [Green Version]
- Castro, C.; Antunes, L.; Lunet, N.; Bento, M.J. Cancer incidence predictions in the North of Portugal: Keeping population-based cancer registration up to date. Eur. J. Cancer Prev. 2016, 25, 472–480. [Google Scholar] [CrossRef]
- Castro, C.; Peleteiro, B.; Bento, M.J.; Lunet, N. Trends in gastric and esophageal cancer incidence in northern Portugal (1994-2009) by subsite and histology, and predictions for 2015. Tumori 2017, 103, 155–163. [Google Scholar] [CrossRef]
- Ernst, M. Systematics and the Origin of Species; Columbia University Press: New York City, NY, USA, 1942; p. 237. [Google Scholar]
- Vecsey-Semjen, B.; Becker, K.F.; Sinski, A.; Blennow, E.; Vietor, I.; Zatloukal, K.; Beug, H.; Wagner, E.; Huber, L.A. Novel colon cancer cell lines leading to better understanding of the diversity of respective primary cancers. Oncogene 2002, 21, 4646–4662. [Google Scholar] [CrossRef] [Green Version]
- Corso, G.; Corso, F. Geographical Distribution of E-cadherin Germline Mutations in the Context of Diffuse Gastric Cancer: A Systematic Review. Cancers 2021, 13, 1269. [Google Scholar] [CrossRef]
- McPeek, M.S.; Strahs, A. Assessment of linkage disequilibrium by the decay of haplotype sharing, with application to fine-scale genetic mapping. Am. J. Hum. Genet. 1999, 65, 858–875. [Google Scholar] [CrossRef] [Green Version]
- Vaz Rodrigues, L.; Costa, F.; Marques, P.; Mendonça, C.; Rocha, J.; Seixas, S. Severe α-1 antitrypsin deficiency caused by Q0(Ourém) allele: Clinical features, haplotype characterization and history. Clin. Genet. 2012, 81, 462–469. [Google Scholar] [CrossRef]
- Weber, J.L.; Wong, C. Mutation of human short tandem repeats. Hum. Mol. Genet. 1993, 2, 1123–1128. [Google Scholar] [CrossRef]
- Nachman, M.W.; Crowell, S.L. Estimate of the mutation rate per nucleotide in humans. Genetics 2000, 156, 297–304. [Google Scholar] [CrossRef]
- Chang, Y.F.; Imam, J.S.; Wilkinson, M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007, 76, 51–74. [Google Scholar] [CrossRef] [Green Version]
- Karam, R.; Carvalho, J.; Bruno, I.; Graziadio, C.; Senz, J.; Huntsman, D.; Carneiro, F.; Seruca, R.; Wilkinson, M.F.; Oliveira, C. The NMD mRNA surveillance pathway downregulates aberrant E-cadherin transcripts in gastric cancer cells and in CDH1 mutation carriers. Oncogene 2008, 27, 4255–4260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo, J.; Soderberg, O.; Simoes-Correia, J.; Grannas, K.; Suriano, G.; Seruca, R. The importance of E-cadherin binding partners to evaluate the pathogenicity of E-cadherin missense mutations associated to HDGC. Eur. J. Hum. Genet. 2013, 21, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, H.; Bordeira-Carrico, R.; Seixas, S.; Carvalho, J.; Senz, J.; Oliveira, P.; Inacio, P.; Gusmao, L.; Rocha, J.; Huntsman, D.; et al. Allele-specific CDH1 downregulation and hereditary diffuse gastric cancer. Hum. Mol. Genet. 2010, 19, 943–952. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.; Sato, H.; Tang, Y.; Matsuda, D.; Maquat, L.E. UPF1 association with the cap-binding protein, CBP80, promotes nonsense-mediated mRNA decay at two distinct steps. Mol. Cell 2010, 39, 396–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borràs, N.; Orriols, G.; Batlle, J.; Pérez-Rodríguez, A.; Fidalgo, T.; Martinho, P.; López-Fernández, M.F.; Rodríguez-Trillo, Á.; Lourés, E.; Parra, R.; et al. Unraveling the effect of silent, intronic and missense mutations on VWF splicing: Contribution of next generation sequencing in the study of mRNA. Haematologica 2019, 104, 587–598. [Google Scholar] [CrossRef]
- Pereira, P.S.; Teixeira, A.; Pinho, S.; Ferreira, P.; Fernandes, J.; Oliveira, C.; Seruca, R.; Suriano, G.; Casares, F. E-cadherin missense mutations, associated with hereditary diffuse gastric cancer (HDGC) syndrome, display distinct invasive behaviors and genetic interactions with the Wnt and Notch pathways in Drosophila epithelia. Hum. Mol. Genet. 2006, 15, 1704–1712. [Google Scholar] [CrossRef]
- Stella, A.; Lastella, P.; Loconte, D.C.; Bukvic, N. Accurate Classification of NF1 Gene Variants in 84 Italian Patients with Neurofibromatosis Type 1. Genes 2018, 9, 216. [Google Scholar] [CrossRef] [Green Version]
- Gergics, P.; Smith, C.; Bando, H.; Jorge, A.A.L.; Rockstroh-Lippold, D.; Vishnopolska, S.A.; Castinetti, F.; Maksutova, M.; Carvalho, L.R.S.; Hoppmann, J.; et al. High-throughput splicing assays identify missense and silent splice-disruptive POU1F1 variants underlying pituitary hormone deficiency. Am. J. Hum. Genet. 2021, 108, 1526–1539. [Google Scholar] [CrossRef]
- Suriano, G.; Oliveira, C.; Ferreira, P.; Machado, J.C.; Bordin, M.C.; De Wever, O.; Bruyneel, E.A.; Moguilevsky, N.; Grehan, N.; Porter, T.R.; et al. Identification of CDH1 germline missense mutations associated with functional inactivation of the E-cadherin protein in young gastric cancer probands. Hum. Mol. Genet. 2003, 12, 575–582. [Google Scholar] [CrossRef] [PubMed]
- More, H.; Humar, B.; Weber, W.; Ward, R.; Christian, A.; Lintott, C.; Graziano, F.; Ruzzo, A.M.; Acosta, E.; Boman, B.; et al. Identification of seven novel germline mutations in the human E-cadherin (CDH1) gene. Hum. Mutat. 2007, 28, 203. [Google Scholar] [CrossRef]
- Oliveira, C.; Ferreira, P.; Nabais, S.; Campos, L.; Ferreira, A.; Cirnes, L.; Alves, C.C.; Veiga, I.; Fragoso, M.; Regateiro, F.; et al. E-Cadherin (CDH1) and p53 rather than SMAD4 and Caspase-10 germline mutations contribute to genetic predisposition in Portuguese gastric cancer patients. Eur. J. Cancer 2004, 40, 1897–1903. [Google Scholar] [CrossRef] [Green Version]
- Shriver, M.D.; Jin, L.; Chakraborty, R.; Boerwinkle, E. VNTR allele frequency distributions under the stepwise mutation model: A computer simulation approach. Genetics 1993, 134, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.P.; Gullo, I.; Wen, X.; Devezas, V.; Baptista, M.; Oliveira, C.; Carneiro, F. Pathological features of total gastrectomy specimens from asymptomatic hereditary diffuse gastric cancer patients and implications for clinical management. Histopathology 2018, 73, 878–886. [Google Scholar] [CrossRef]
- Barber, M.E.; Save, V.; Carneiro, F.; Dwerryhouse, S.; Lao-Sirieix, P.; Hardwick, R.H.; Caldas, C.; Fitzgerald, R.C. Histopathological and molecular analysis of gastrectomy specimens from hereditary diffuse gastric cancer patients has implications for endoscopic surveillance of individuals at risk. J. Pathol. 2008, 216, 286–294. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total nº of Tested Individuals | Total nº of Carriers | DGC Cases | Mean Age of DGC Diagnosis (±SD) | RRG | RRG with Lesions | |
|---|---|---|---|---|---|---|
| Males | 60 | 27 | 4 | 27 ± 7 | 10 | 8 |
| Females | 74 | 31 | 7 * | 37 ± 13 | 16 | 13 # |
| Total | 134 | 58 | 11 | 33 ± 12 | 26 | 21 |
| Number of Females Tested | Total nº of Carriers | LBC Cases | Mean Age of LBC Diagnosis (± SD) | RRM | RRM with Lesions | Lesions in both RRG and RRM |
|---|---|---|---|---|---|---|
| 74 | 31 | 6 | 50 ± 8 | 8 | 4 * | 3 # |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbosa-Matos, R.; Leal Silva, R.; Garrido, L.; Aguiar, A.C.; Garcia-Pelaez, J.; André, A.; Seixas, S.; Sousa, S.P.; Ferro, L.; Vilarinho, L.; et al. The CDH1 c.1901C>T Variant: A Founder Variant in the Portuguese Population with Severe Impact in mRNA Splicing. Cancers 2021, 13, 4464. https://doi.org/10.3390/cancers13174464
Barbosa-Matos R, Leal Silva R, Garrido L, Aguiar AC, Garcia-Pelaez J, André A, Seixas S, Sousa SP, Ferro L, Vilarinho L, et al. The CDH1 c.1901C>T Variant: A Founder Variant in the Portuguese Population with Severe Impact in mRNA Splicing. Cancers. 2021; 13(17):4464. https://doi.org/10.3390/cancers13174464
Chicago/Turabian StyleBarbosa-Matos, Rita, Rafaela Leal Silva, Luzia Garrido, Ana Cerqueira Aguiar, José Garcia-Pelaez, Ana André, Susana Seixas, Sónia Passos Sousa, Luísa Ferro, Lúcia Vilarinho, and et al. 2021. "The CDH1 c.1901C>T Variant: A Founder Variant in the Portuguese Population with Severe Impact in mRNA Splicing" Cancers 13, no. 17: 4464. https://doi.org/10.3390/cancers13174464
APA StyleBarbosa-Matos, R., Leal Silva, R., Garrido, L., Aguiar, A. C., Garcia-Pelaez, J., André, A., Seixas, S., Sousa, S. P., Ferro, L., Vilarinho, L., Gullo, I., Devezas, V., Oliveira, R., Fernandes, S., Costa, S. C., Magalhães, A., Baptista, M., Carneiro, F., Pinheiro, H., ... Oliveira, C. (2021). The CDH1 c.1901C>T Variant: A Founder Variant in the Portuguese Population with Severe Impact in mRNA Splicing. Cancers, 13(17), 4464. https://doi.org/10.3390/cancers13174464