
Deletion of NEMO Inhibits EMT and Reduces Metastasis in KPC Mice

 , ,
, ,  ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Mice
2.2. RNA Isolation, cDNA Synthesis and qRT-PCR
2.3. Protein Isolation and Western Blot
2.4. Histology
2.5. Cancer Grading, Differentiation Status and Gross Anatomy
2.6. Evaluation of Ascites Development
2.7. Quantification of Aspartate Transaminase (AST) and Alanine Transaminase (ALT) Levels
2.8. Cell Isolation from Ascites and Immunofluorescence Staining
2.9. Evaluation of Macro- and Micro-Metastasis
2.10. Isolation of Primary Cancer Cells and Primary Cell Culture Establishment
2.11. Tumor Necrosis Factor a (TNF-α) Treatment and Nuclear Extraction
2.12. Cell Migration and Invasion Assays
2.13. Statistics
3. Results
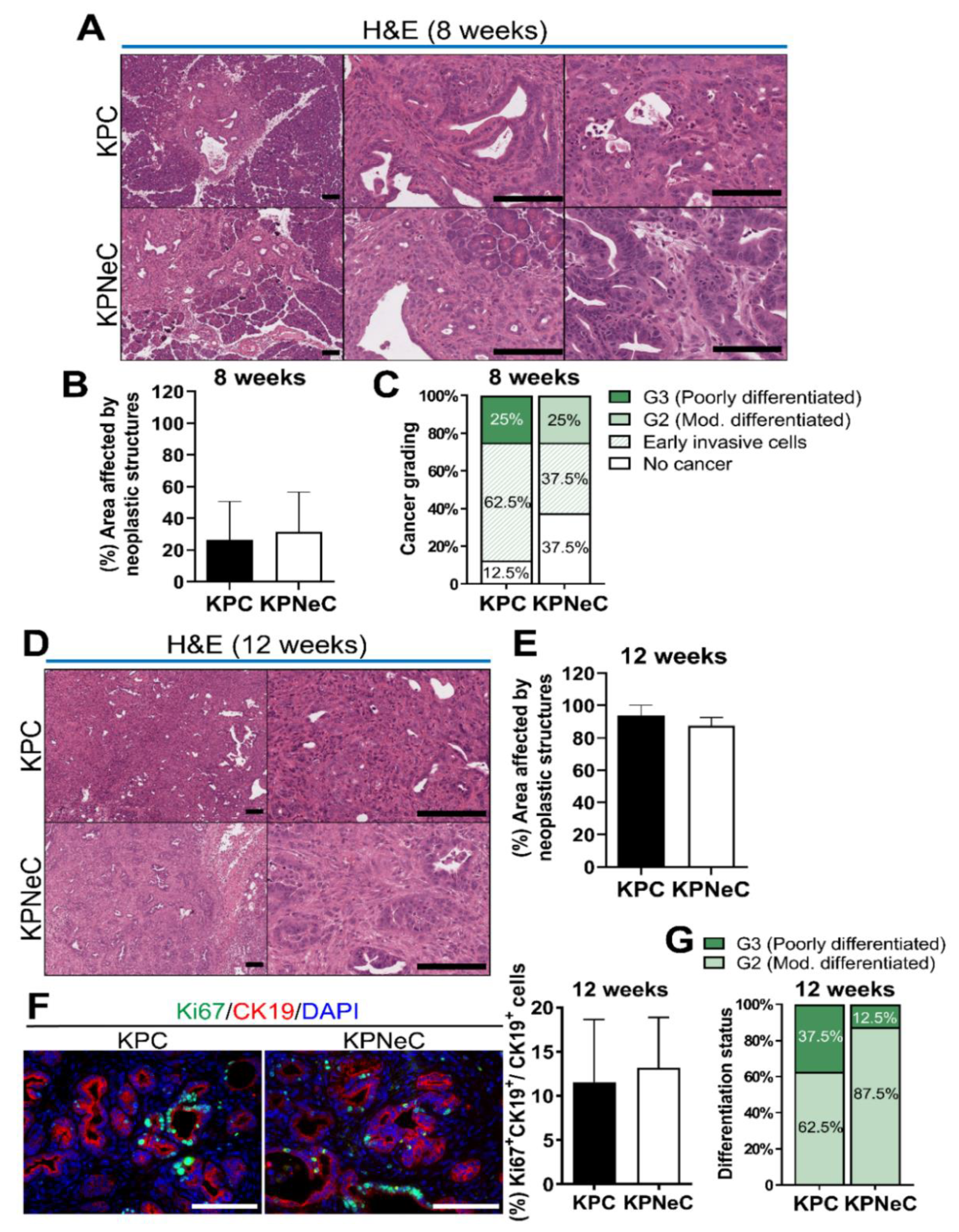
3.1. NEMO Is Dispensable for the Development of PDAC but Modulates Progression of Tumors in KPC Mice
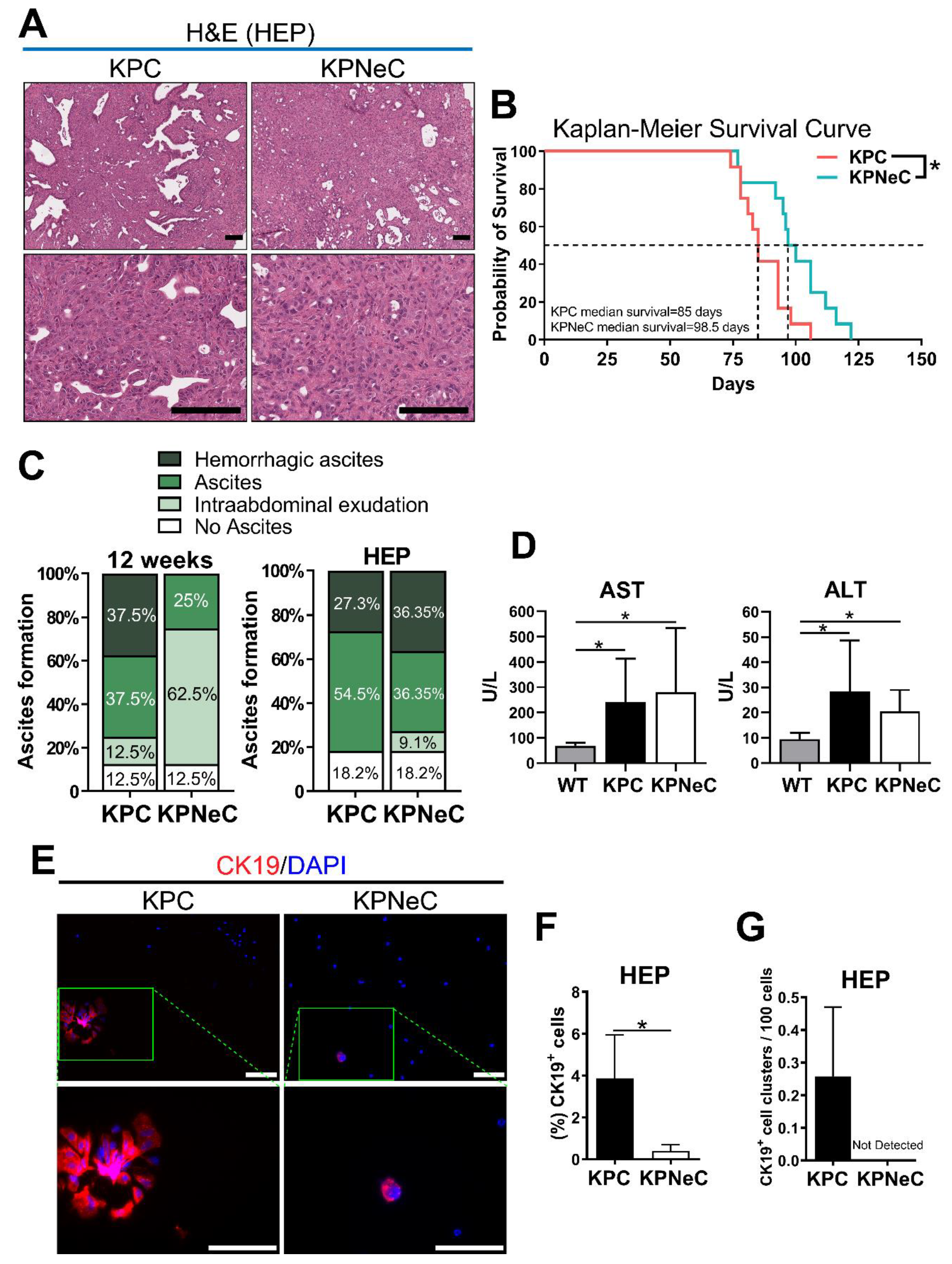
3.2. Pancreas-Specific NEMO Ablation Improves Survival in KPC Mice
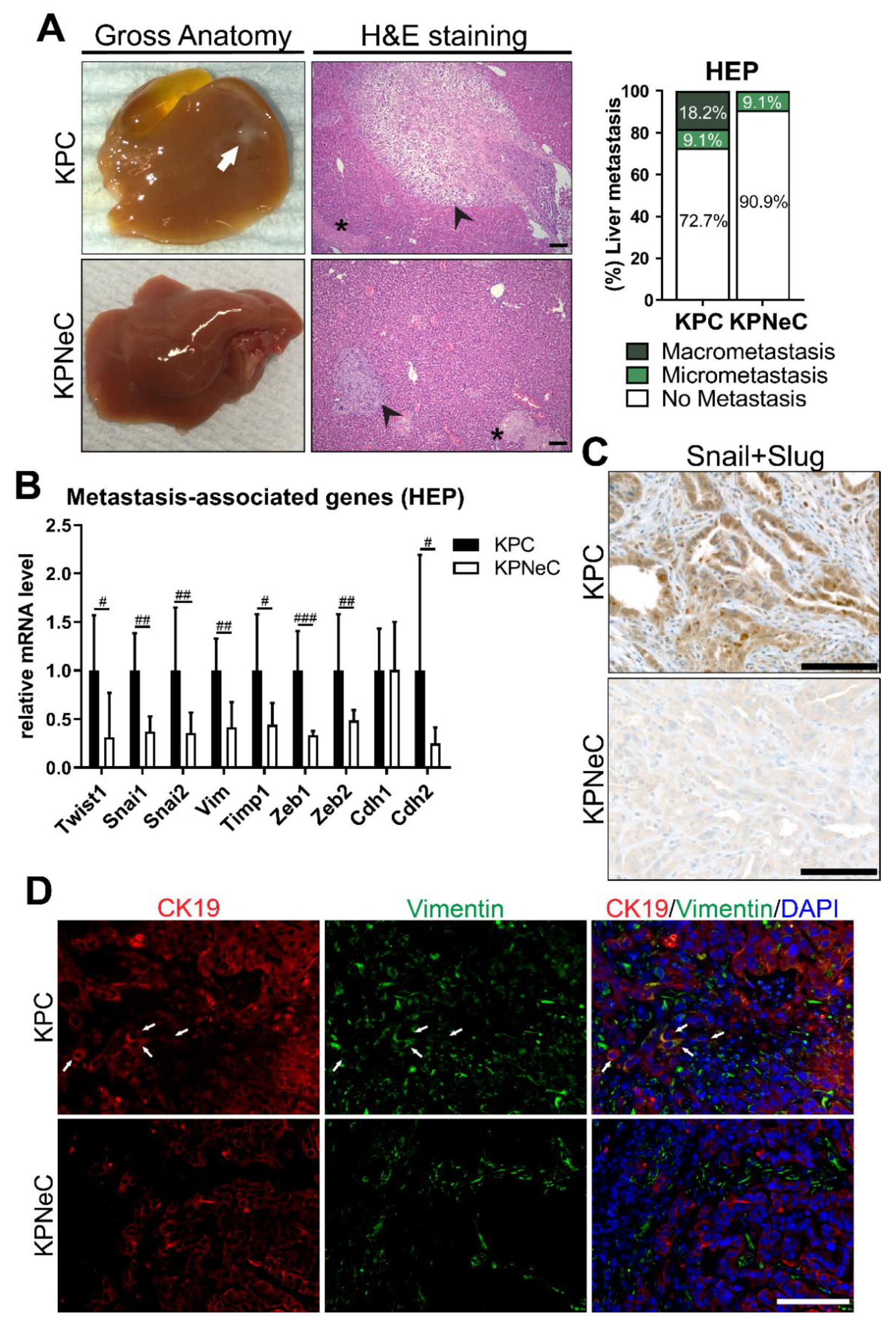
3.3. Pancreas-Specific NEMO Ablation Reduces the Metastasis Rate in KPC Mice and Blocks EMT
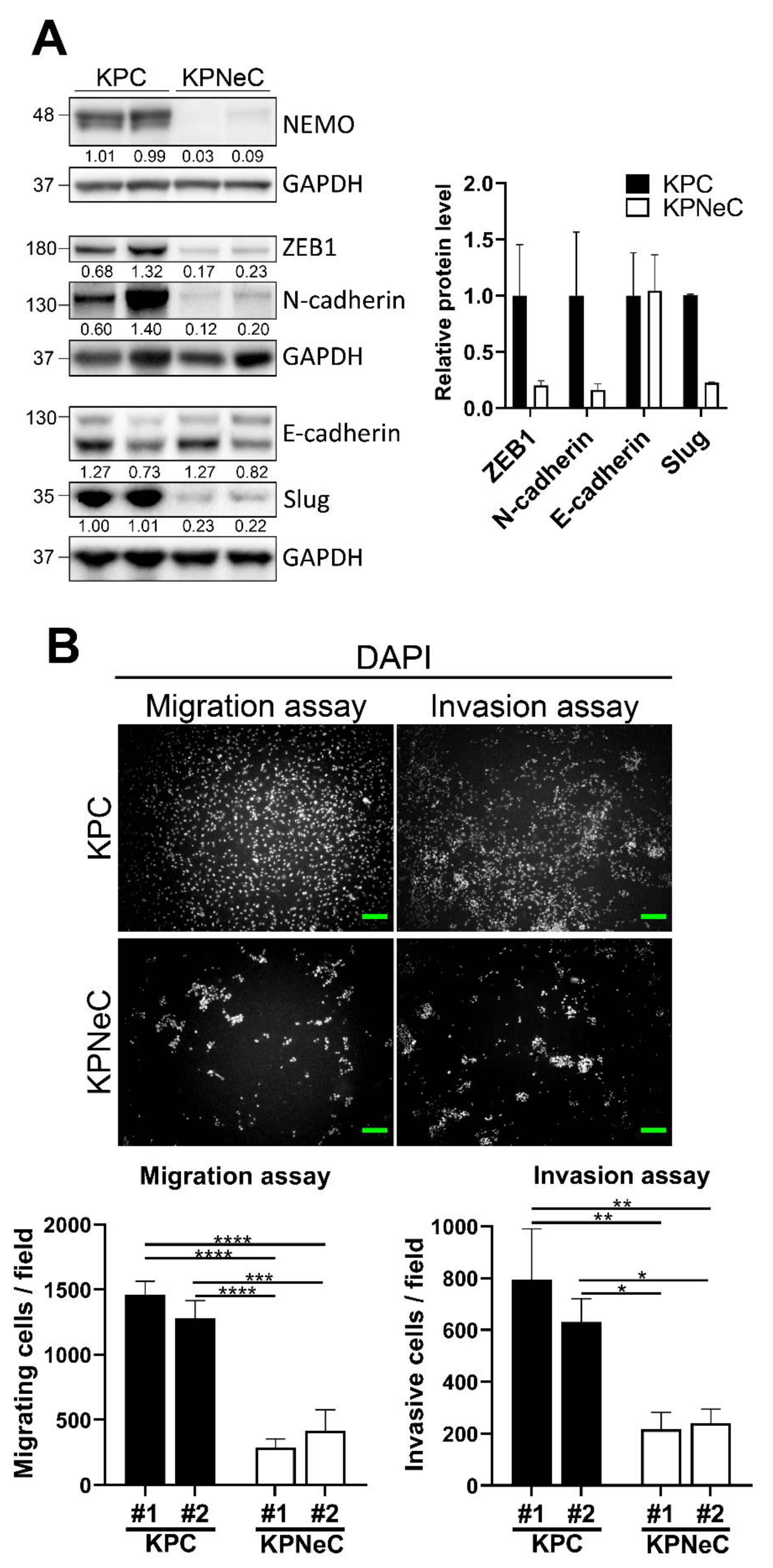
3.4. NEMO Ablation Diminishes the Migrating and Invasive Properties of KPC Cells Ex Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.X.; Liu, Z.W.; You, L.; Wu, W.M.; Zhao, Y.P. Advances in understanding the molecular mechanism of pancreatic cancer metastasis. Hepatob. Pancreat. Dis. Int. 2016, 15, 361–370. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Sipos, B.; Frank, S.; Gress, T.; Hahn, S.; Kloppel, G. Pancreatic intraepithelial neoplasia revisited and updated. Pancreatology 2009, 9, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ottenhof, N.A.; de Wilde, R.F.; Maitra, A.; Hruban, R.H.; Offerhaus, G.J. Molecular characteristics of pancreatic ductal adenocarcinoma. Pathol. Res. Int. 2011, 2011, 620601. [Google Scholar] [CrossRef]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of Somatic Mutations in Most Early-Stage Pancreatic Intraepithelial Neoplasia. Gastroenterology 2012, 142, 730–733. [Google Scholar] [CrossRef]
- Caldwell, M.E.; DeNicola, G.M.; Martins, C.P.; Jacobetz, M.A.; Maitra, A.; Hruban, R.H.; Tuveson, D.A. Cellular features of senescence during the evolution of human and murine ductal pancreatic cancer. Oncogene 2012, 31, 1599–1608. [Google Scholar] [CrossRef]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef]
- Deeb, A.; Haque, S.-U.; Olowokure, O. Pulmonary metastases in pancreatic cancer, is there a survival influence? J. Gastrointest. Oncol. 2015, 6, E48. [Google Scholar]
- Hass, R.; von der Ohe, J.; Ungefroren, H. The Intimate Relationship among EMT, MET and TME: A T(ransdifferentiation) E(nhancing) M(ix) to Be Exploited for Therapeutic Purposes. Cancers 2020, 12, 3674. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, S.; Sun, Y.L. Epithelial-Mesenchymal Transition in Pancreatic Cancer: A Review. Biomed. Res. Int. 2017, 2017, 2646148. [Google Scholar] [CrossRef] [PubMed]
- Fujii-Nishimura, Y.; Yamazaki, K.; Masugi, Y.; Douguchi, J.; Kurebayashi, Y.; Kubota, N.; Ojima, H.; Kitago, M.; Shinoda, M.; Hashiguchi, A.; et al. Mesenchymal-epithelial transition of pancreatic cancer cells at perineural invasion sites is induced by Schwann cells. Pathol. Int. 2018, 68, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Smith, S.M.; Lyu, Y.L.; Cai, L. NF-kappaB affects proliferation and invasiveness of breast cancer cells by regulating CD44 expression. PLoS ONE 2014, 9, e106966. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.L.; Kamata, H.; Karin, M. IKK/NF-kappaB signaling: Balancing life and death—A new approach to cancer therapy. J. Clin. Invest. 2005, 115, 2625–2632. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Prabhu, L.; Mundade, R.; Korc, M.; Loehrer, P.J.; Lu, T. Critical role of NF-kappaB in pancreatic cancer. Oncotarget 2014, 5, 10969–10975. [Google Scholar] [CrossRef]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef]
- Wang, W.X.; Abbruzzese, J.L.; Evans, D.B.; Larry, L.; Cleary, K.R.; Chiao, P.J. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin. Cancer Res. 1999, 5, 119–127. [Google Scholar]
- Ling, J.; Kang, Y.a.; Zhao, R.; Xia, Q.; Lee, D.-F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-Induced IKK2/ß/NF-κB Activation by IL-1a and p62 Feedforward Loops Is Required for Development of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef]
- Maier, H.J.; Wagner, M.; Schips, T.G.; Salem, H.H.; Baumann, B.; Wirth, T. Requirement of NEMO/IKKgamma for effective expansion of KRAS-induced precancerous lesions in the pancreas. Oncogene 2013, 32, 2690–2695. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.K.; Gerstenlauer, M.; Konukiewitz, B.; Steiger, K.; Weichert, W.; Wirth, T.; Maier, H.J. Epithelial NEMO/IKKgamma limits fibrosis and promotes regeneration during pancreatitis. Gut 2017, 66, 1995–2007. [Google Scholar] [CrossRef]
- Maniati, E.; Bossard, M.; Cook, N.; Candido, J.B.; Emami-Shahri, N.; Nedospasov, S.A.; Balkwill, F.R.; Tuveson, D.A.; Hagemann, T. Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. J. Clin. Investig. 2011, 121, 4685–4699. [Google Scholar] [CrossRef]
- Gannon, M.; Herrera, P.L.; Wright, C.V.E. Mosaic Cre-mediated recombination in pancreas using the pdx-1 enhancer/promoter. Genesis 2000, 26, 143–144. [Google Scholar] [CrossRef]
- Johnson, L.; Mercer, K.; Greenbaum, D.; Bronson, R.T.; Crowley, D.; Tuveson, D.A.; Jacks, T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001, 410, 1111–1116. [Google Scholar] [CrossRef]
- Marino, S.; Vooijs, M.; van Der Gulden, H.; Jonkers, J.; Berns, A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000, 14, 994–1004. [Google Scholar] [PubMed]
- Schmidt-Supprian, M.; Bloch, W.; Courtois, G.; Addicks, K.; Israel, A.; Rajewsky, K.; Pasparakis, M. Nemo/ikkg-deficient mice model incontinentia pigmenti. J. Mol. Med. 2000, 78, B41. [Google Scholar]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed]
- Proteinatlas.org. The Human Protein Atlas. Available online: http://www.proteinatlas.org (accessed on 7 September 2021).
- Jogi, A.; Vaapil, M.; Johansson, M.; Pahlman, S. Cancer cell differentiation heterogeneity and aggressive behavior in solid tumors. Ups. J. Med. Sci. 2012, 117, 217–224. [Google Scholar] [CrossRef]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated Pancreatic Stellate Cells Sequester CD8(+) T Cells to Reduce Their Infiltration of the Juxtatumoral Compartment of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef]
- Hicks, A.M.; Chou, J.; Capanu, M.; Lowery, M.A.; Yu, K.H.; O’Reilly, E.M. Pancreas Adenocarcinoma: Ascites, Clinical Manifestations, and Management Implications. Clin. Colorectal. Cancer 2016, 15, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Baretti, M.; Pulluri, B.; Tsai, H.L.; Blackford, A.L.; Wolfgang, C.L.; Laheru, D.; Zheng, L.; Herman, J.; Le, D.T.; Narang, A.K.; et al. The Significance of Ascites in Patients With Pancreatic Ductal Adenocarcinoma: A Case-Control Study. Pancreas 2019, 48, 585–589. [Google Scholar] [CrossRef]
- Gabriel, A.N.A.; Jiao, Q.L.; Yvette, U.; Yang, X.M.; Al-Ameri, S.A.; Du, L.T.; Wang, Y.S.; Wang, C.X. Differences between KC and KPC pancreatic ductal adenocarcinoma mice models, in terms of their modeling biology and their clinical relevance. Pancreatology 2020, 20, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e684. [Google Scholar] [CrossRef]
- Kumar, M.; Allison, D.F.; Baranova, N.N.; Wamsley, J.J.; Katz, A.J.; Bekiranov, S.; Jones, D.R.; Mayo, M.W. NF-kappaB regulates mesenchymal transition for the induction of non-small cell lung cancer initiating cells. PLoS ONE 2013, 8, e68597. [Google Scholar] [CrossRef]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef]
- Huber, M.A.; Beug, H.; Wirth, T. Epithelial-mesenchymal transition: NF-kappaB takes center stage. Cell Cycle 2004, 3, 1477–1480. [Google Scholar] [CrossRef]
- Maier, H.J.; Schmidt-Strassburger, U.; Huber, M.A.; Wiedemann, E.M.; Beug, H.; Wirth, T. NF-kappaB promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010, 295, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, B.; Harant, V.; Schaten, S.; Frühschütz, M.; Spallek, R.; Höchst, B.; Stutzer, K.; Berchtold, S.; Erkan, M.; Prokopchuk, O.; et al. Pancreatic Premalignant Lesions Secrete Tissue Inhibitor of Metalloproteinases-1, Which Activates Hepatic Stellate Cells Via CD63 Signaling to Create a Premetastatic Niche in the Liver. Gastroenterology 2016, 151, 1011–1024.e1017. [Google Scholar] [CrossRef]
- Tanaka, T.; Nakayama, H.; Yoshitake, Y.; Irie, A.; Nagata, M.; Kawahara, K.; Takamune, Y.; Yoshida, R.; Nakagawa, Y.; Ogi, H.; et al. Selective inhibition of nuclear factor-kappaB by nuclear factor-kappaB essential modulator-binding domain peptide suppresses the metastasis of highly metastatic oral squamous cell carcinoma. Cancer Sci. 2012, 103, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Czauderna, C.; Castven, D.; Mahn, F.L.; Marquardt, J.U. Context-Dependent Role of NF-kappaB Signaling in Primary Liver Cancer-from Tumor Development to Therapeutic Implications. Cancers 2019, 11, 1053. [Google Scholar] [CrossRef]
- He, J.; Gerstenlauer, M.; Chan, L.K.; Leithauser, F.; Yeh, M.M.; Wirth, T.; Maier, H.J. Block of NF-kB signaling accelerates MYC-driven hepatocellular carcinogenesis and modifies the tumor phenotype towards combined hepatocellular cholangiocarcinoma. Cancer Lett. 2019, 458, 113–122. [Google Scholar] [CrossRef]
- Lesina, M.; Wormann, S.M.; Morton, J.; Diakopoulos, K.N.; Korneeva, O.; Wimmer, M.; Einwachter, H.; Sperveslage, J.; Demir, I.E.; Kehl, T.; et al. RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis. J. Clin. Investig. 2016, 126, 2919–2932. [Google Scholar] [CrossRef]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Qin, Y.; Zhao, D.; Zhou, H.G.; Wang, X.H.; Zhong, W.L.; Chen, S.; Gu, W.G.; Wang, W.; Zhang, C.H.; Liu, Y.R.; et al. Apigenin inhibits NF-kappaB and snail signaling, EMT and metastasis in human hepatocellular carcinoma. Oncotarget 2016, 7, 41421–41431. [Google Scholar] [CrossRef]
- Huber, M.A.; Maier, H.J.; Alacakaptan, M.; Wiedemann, E.; Braunger, J.; Boehmelt, G.; Madwed, J.B.; Young, E.R.; Marshall, D.R.; Pehamberger, H.; et al. BI 5700, a Selective Chemical Inhibitor of IkappaB Kinase 2, Specifically Suppresses Epithelial-Mesenchymal Transition and Metastasis in Mouse Models of Tumor Progression. Genes Cancer 2010, 1, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Peeters, J.K.; Smid, M.; Timmermans, M.; Verhoog, L.C.; Westenend, P.J.; Heine, A.A.J.; Chan, A.; Sieuwerts, A.M.; Wiemer, E.A.C.; et al. Loss of E-cadherin is not a necessity for epithelial to mesenchymal transition in human breast cancer. Breast Cancer Res. Treat. 2013, 138, 47–57. [Google Scholar]
- Nilsson, G.M.; Akhtar, N.; Kannius-Janson, M.; Baeckström, D. Loss of E-cadherin expression is not a prerequisite for c-erbB2-induced epithelial-mesenchymal transition. Int. J. Oncol. 2014, 45, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Sommariva, M.; Gagliano, N.A.-O. E-Cadherin in Pancreatic Ductal Adenocarcinoma: A Multifaceted Actor during EMT. Cells 2020, 9, 1040. [Google Scholar] [CrossRef]
- Zheng, X.F.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525. [Google Scholar] [CrossRef]
- Fujioka, S.; Sclabas, G.M.; Schmidt, C.; Frederick, W.A.; Dong, Q.G.; Abbruzzese, J.L.; Evans, D.B.; Baker, C.; Chiao, P.J. Function of nuclear factor kappaB in pancreatic cancer metastasis. Clin. Cancer Res. 2003, 9, 346–354. [Google Scholar]
- Guo, X.; Zheng, L.; Jiang, J.; Zhao, Y.; Wang, X.; Shen, M.; Zhu, F.; Tian, R.; Shi, C.; Xu, M.; et al. Blocking NF-κB Is Essential for the Immunotherapeutic Effect of Recombinant IL18 in Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 5939–5950. [Google Scholar] [CrossRef]
- Cheng, Z.-X.; Sun, B.; Wang, S.-J.; Gao, Y.; Zhang, Y.-M.; Zhou, H.-X.; Jia, G.; Wang, Y.-W.; Kong, R.; Pan, S.-H.; et al. Nuclear factor-κB-dependent epithelial to mesenchymal transition induced by HIF-1a activation in pancreatic cancer cells under hypoxic conditions. PLoS ONE 2011, 6, e23752. [Google Scholar] [CrossRef] [PubMed]
- Omar, O.M.; Soutto, M.; Bhat, N.S.; Bhat, A.A.; Lu, H.; Chen, Z.; El-Rifai, W. TFF1 antagonizes TIMP-1 mediated proliferative functions in gastric cancer. Mol. Carcinog. 2018, 57, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Fan, X.; Hao, M.; Wang, J.; Zhou, X.; Sun, X. Higher levels of TIMP-1 expression are associated with a poor prognosis in triple-negative breast cancer. Mol. Cancer 2016, 15, 30. [Google Scholar] [CrossRef]
- Bommarito, A.; Richiusa, P.; Carissimi, E.; Pizzolanti, G.; Rodolico, V.; Zito, G.; Criscimanna, A.; Di Blasi, F.; Pitrone, M.; Zerilli, M.; et al. BRAFV600E mutation, TIMP-1 upregulation, and NF-kappaB activation: Closing the loop on the papillary thyroid cancer trilogy. Endocr. Relat. Cancer 2011, 18, 669–685. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Model | Genotype |
|---|---|
| WT | --- |
| NeC | Pdx1-Cre; NEMOfl/fl |
| PC | Pdx1-Cre; p53fl/fl |
| PNeC | Pdx1-Cre; p53fl/fl; NEMOfl/fl |
| KC | Pdx1-Cre; LSL-KRASG12D |
| KNeC | Pdx1-Cre; LSL-KRASG12D; NEMOfl/fl |
| KPC | Pdx1-Cre; LSL-KRASG12D; p53fl/fl |
| KPNeC | Pdx1-Cre; LSL-KRASG12D; p53fl/fl; NEMOfl/fl |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsesmelis, M.; Tiwary, K.; Steiger, K.; Sperb, N.; Gerstenlauer, M.; Manfras, U.; Maier, H.J.; Hermann, P.C.; Chan, L.K.; Wirth, T. Deletion of NEMO Inhibits EMT and Reduces Metastasis in KPC Mice. Cancers 2021, 13, 4541. https://doi.org/10.3390/cancers13184541
Tsesmelis M, Tiwary K, Steiger K, Sperb N, Gerstenlauer M, Manfras U, Maier HJ, Hermann PC, Chan LK, Wirth T. Deletion of NEMO Inhibits EMT and Reduces Metastasis in KPC Mice. Cancers. 2021; 13(18):4541. https://doi.org/10.3390/cancers13184541
Chicago/Turabian StyleTsesmelis, Miltiadis, Kanishka Tiwary, Katja Steiger, Nadine Sperb, Melanie Gerstenlauer, Uta Manfras, Harald J. Maier, Patrick C. Hermann, Lap Kwan Chan, and Thomas Wirth. 2021. "Deletion of NEMO Inhibits EMT and Reduces Metastasis in KPC Mice" Cancers 13, no. 18: 4541. https://doi.org/10.3390/cancers13184541
APA StyleTsesmelis, M., Tiwary, K., Steiger, K., Sperb, N., Gerstenlauer, M., Manfras, U., Maier, H. J., Hermann, P. C., Chan, L. K., & Wirth, T. (2021). Deletion of NEMO Inhibits EMT and Reduces Metastasis in KPC Mice. Cancers, 13(18), 4541. https://doi.org/10.3390/cancers13184541