
A New Player in Neuroblastoma: YAP and Its Role in the Neuroblastoma Microenvironment


Abstract
:Simple Summary
Abstract
1. Introduction
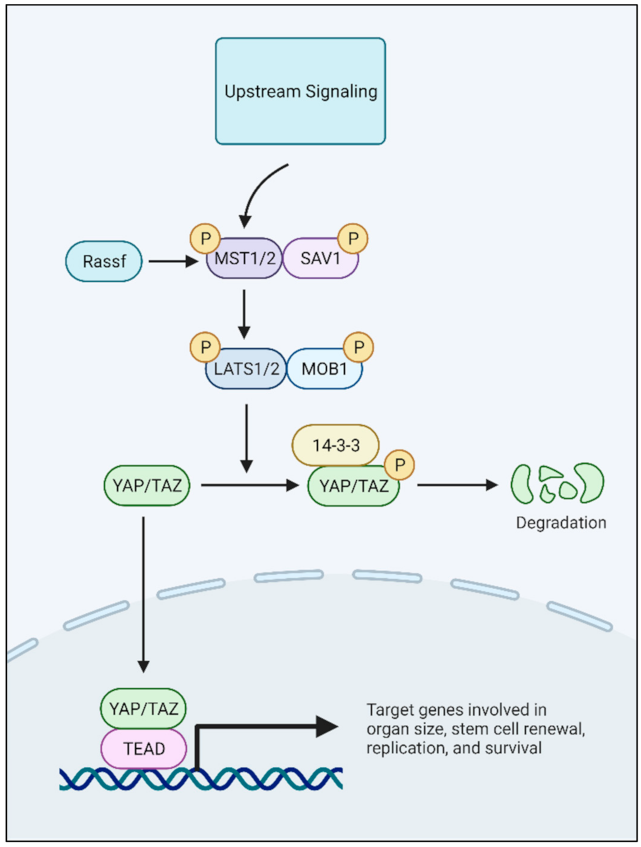
2. YAP and Its Role in High-Risk Neuroblastoma
2.1. Tumorigenesis
2.1.1. Cell Proliferation
2.1.2. Tumor Growth
2.2. Metastasis
2.3. Therapy Resistance
2.3.1. Cytotoxic Therapy Resistance Imparted by YAP Differs In Vitro Versus In Vivo
2.3.2. Tyrosine Kinase Inhibitor Resistance Due to YAP
2.4. Mesenchymal Properties
2.4.1. Neurosphere Formation
2.4.2. Mesenchymal Phenotype
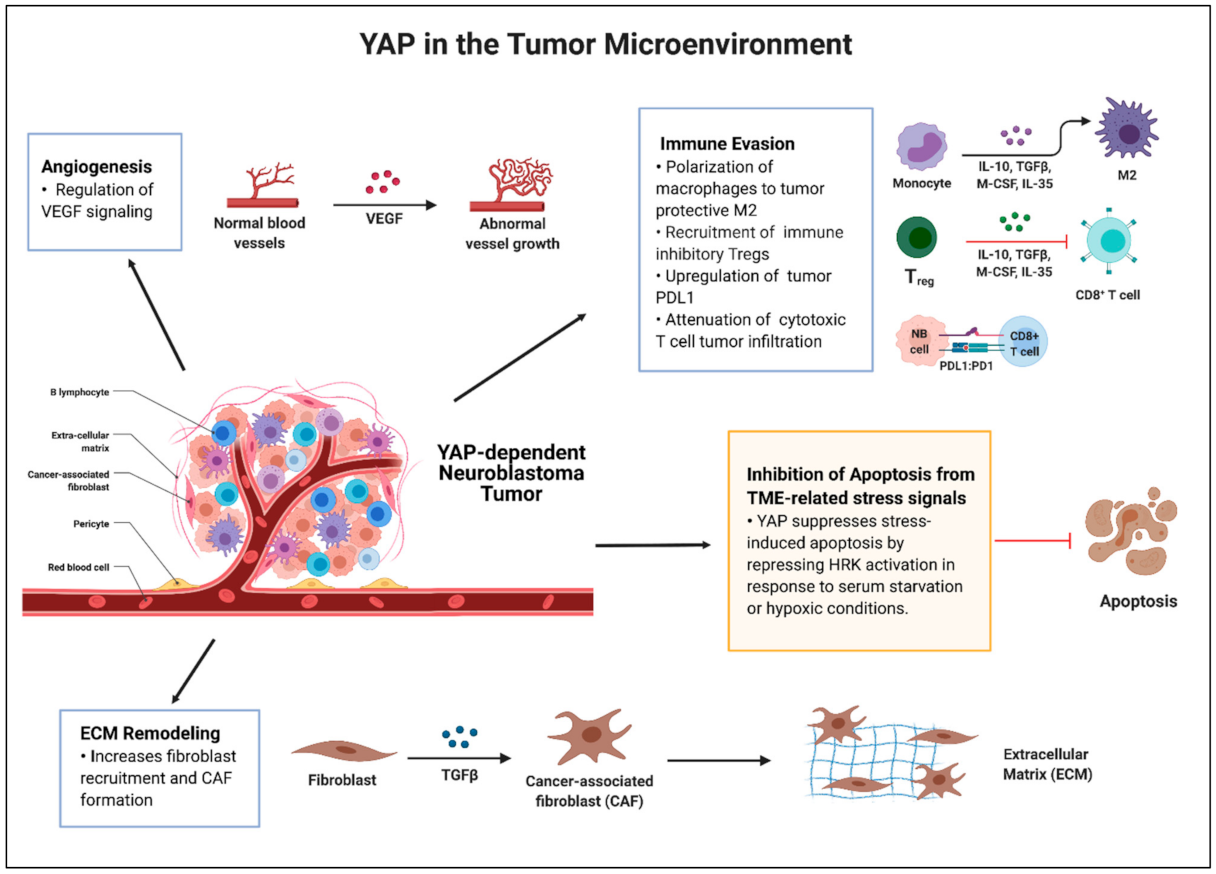
3. YAP and the Tumor Microenvironment in Neuroblastoma
3.1. Current Knowledge and Potential Contributions
3.1.1. Tumor Environmental Stress-Induced Apoptosis
3.1.2. Hypoxia and Angiogenesis
3.1.3. Extracellular Matrix Remodeling
3.1.4. Immune Milieu
3.2. Future Investigations
4. Therapeutic Targeting of YAP in Neuroblastoma
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Park, J.R.; Bagatell, R.; London, W.B.; Maris, J.M.; Cohn, S.L.; Mattay, K.K.; Hogarty, M.; Committee, C.O.G.N. Children’s Oncology Group’s 2013 blueprint for research: Neuroblastoma. Pediatr. Blood Cancer 2013, 60, 985–993. [Google Scholar] [CrossRef]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [Green Version]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S. Targeting the Tumor Microenvironment in Neuroblastoma: Recent Advances and Future Directions. Cancers 2020, 12, 57. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Padovan-Merhar, O.M.; Raman, P.; Ostrovnaya, I.; Kalletla, K.; Rubnitz, K.R.; Sanford, E.M.; Ali, S.M.; Miller, V.A.; Mosse, Y.P.; Granger, M.P.; et al. Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet. 2016, 12, e1006501. [Google Scholar] [CrossRef]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Colmet Daage, L.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Koster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, E.L.; Mosse, Y.P. Targeting ALK in neuroblastoma--preclinical and clinical advancements. Nat. Rev. Clin. Oncol. 2012, 9, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.H.; Voss, S.D.; Hall, D.C.; Minard, C.G.; Balis, F.M.; Wilner, K.; Berg, S.L.; Fox, E.; Adamson, P.C.; Blaney, S.; et al. Activity of Crizotinib in Patients with ALK-aberrant Relapsed/Refractory Neuroblastoma: A Children’s Oncology Group Study (ADVL0912). Clin. Cancer Res. 2021, 27, 3543–3548. [Google Scholar] [CrossRef] [PubMed]
- Krytska, K.; Ryles, H.T.; Sano, R.; Raman, P.; Infarinato, N.R.; Hansel, T.D.; Makena, M.R.; Song, M.M.; Reynolds, C.P.; Mosse, Y.P. Crizotinib Synergizes with Chemotherapy in Preclinical Models of Neuroblastoma. Clin. Cancer Res. 2016, 22, 948–960. [Google Scholar] [CrossRef] [Green Version]
- Trigg, R.M.; Turner, S.D. ALK in Neuroblastoma: Biological and Therapeutic Implications. Cancers 2018, 10, 113. [Google Scholar] [CrossRef] [Green Version]
- Borriello, L.; Seeger, R.C.; Asgharzadeh, S.; DeClerck, Y.A. More than the genes, the tumor microenvironment in neuroblastoma. Cancer Lett. 2016, 380, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Blavier, L.; Yang, R.M.; DeClerck, Y.A. The Tumor Microenvironment in Neuroblastoma: New Players, New Mechanisms of Interaction and New Perspectives. Cancers 2020, 12, 2912. [Google Scholar] [CrossRef]
- Castellan, M.; Guarnieri, A.; Fujimura, A.; Zanconato, F.; Battilana, G.; Panciera, T.; Sladitschek, H.L.; Contessotto, P.; Citron, A.; Grilli, A.; et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in Glioblastoma. Nat. Cancer 2021, 2, 174–188. [Google Scholar] [CrossRef]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Ni, X.; Tao, J.; Barbi, J.; Chen, Q.; Park, B.V.; Li, Z.; Zhang, N.; Lebid, A.; Ramaswamy, A.; Wei, P.; et al. YAP Is Essential for Treg-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2018, 8, 1026–1043. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Tian, Y.; Cao, C.; Niu, G. The Emerging Role of YAP/TAZ in Tumor Immunity. Mol. Cancer Res. 2019, 17, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, H.; Li, Y.; Xia, D.; Yang, L.; Ma, Y.; Li, H. The role of YAP/TAZ activity in cancer metabolic reprogramming. Mol. Cancer 2018, 17, 134. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, T.; Zhang, J.; Cheng, A.S.L.; Yu, J.; Kang, W.; To, K.F. Emerging roles of Hippo signaling in inflammation and YAP-driven tumor immunity. Cancer Lett. 2018, 426, 73–79. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Ma, Y.; Yang, L.; Wang, T.; Meng, X.; Zong, Z.; Sun, X.; Hua, X.; Li, H. Yes-associated protein (YAP) binds to HIF-1alpha and sustains HIF-1alpha protein stability to promote hepatocellular carcinoma cell glycolysis under hypoxic stress. J. Exp. Clin. Cancer Res. 2018, 37, 216. [Google Scholar] [CrossRef]
- Gumbiner, B.M.; Kim, N.G. The Hippo-YAP signaling pathway and contact inhibition of growth. J. Cell Sci. 2014, 127, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moya, I.M.; Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2019, 20, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Li, L.; Zhao, B. The regulation and function of YAP transcription co-activator. Acta Biochim. Biophys. Sin. 2015, 47, 16–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Zhang, W.; Pan, Y.; Gao, Y.; Deng, L.; Li, F.; Li, F.; Ma, X.; Hou, S.; Xu, J.; et al. YAP Suppresses Lung Squamous Cell Carcinoma Progression via Deregulation of the DNp63-GPX2 Axis and ROS Accumulation. Cancer Res. 2017, 77, 5769–5781. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Eisenbarth, D.; Choi, W.; Kim, H.; Choi, C.; Lee, D.; Lim, D.S. YAP and AP-1 Cooperate to Initiate Pancreatic Cancer Development from Ductal Cells in Mice. Cancer Res. 2020, 80, 4768–4779. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, L.; Wang, J.; Chen, P.; Jia, Y.; Wang, C.; Yang, W.; Wen, Z.; Song, Q.; Tan, B.; et al. Yes-associated protein (YAP) predicts poor prognosis and regulates progression of esophageal squamous cell cancer through epithelial-mesenchymal transition. Exp. Med. 2019, 18, 2993–3001. [Google Scholar] [CrossRef] [Green Version]
- Azar, W.J.; Christie, E.L.; Mitchell, C.; Liu, D.S.; Au-Yeung, G.; Bowtell, D.D.L. Noncanonical IL6 Signaling-Mediated Activation of YAP Regulates Cell Migration and Invasion in Ovarian Clear Cell Cancer. Cancer Res. 2020, 80, 4960–4971. [Google Scholar] [CrossRef]
- Gregorieff, A.; Liu, Y.; Inanlou, M.R.; Khomchuk, Y.; Wrana, J.L. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 2015, 526, 715–718. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122 e112. [Google Scholar] [CrossRef]
- Ling, H.H.; Kuo, C.C.; Lin, B.X.; Huang, Y.H.; Lin, C.W. Elevation of YAP promotes the epithelial-mesenchymal transition and tumor aggressiveness in colorectal cancer. Exp. Cell Res. 2017, 350, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. Bioessays 2020, 42, e1900162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.A.; Mohamed, A.D.; Gener, M.; Li, W.; Taboada, E. YAP and the Hippo pathway in pediatric cancer. Mol. Cell. Oncol. 2017, 4, e1295127. [Google Scholar] [CrossRef] [Green Version]
- Morice, S.; Danieau, G.; Redini, F.; Brounais-Le-Royer, B.; Verrecchia, F. Hippo/YAP Signaling Pathway: A Promising Therapeutic Target in Bone Paediatric Cancers? Cancers 2020, 12, 645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.H.; Lawlor, E.R. BMI-1 suppresses contact inhibition and stabilizes YAP in Ewing sarcoma. Oncogene 2011, 30, 2077–2085. [Google Scholar] [CrossRef] [Green Version]
- Slemmons, K.K.; Crose, L.E.S.; Riedel, S.; Sushnitha, M.; Belyea, B.; Linardic, C.M. A Novel Notch-YAP Circuit Drives Stemness and Tumorigenesis in Embryonal Rhabdomyosarcoma. Mol. Cancer Res. 2017, 15, 1777–1791. [Google Scholar] [CrossRef] [Green Version]
- Slemmons, K.K.; Yeung, C.; Baumgart, J.T.; Juarez, J.O.M.; McCalla, A.; Helman, L.J. Targeting Hippo-Dependent and Hippo-Independent YAP1 Signaling for the Treatment of Childhood Rhabdomyosarcoma. Cancer Res. 2020, 80, 3046–3056. [Google Scholar] [CrossRef]
- LaQuaglia, M.J.; Grijalva, J.L.; Mueller, K.A.; Perez-Atayde, A.R.; Kim, H.B.; Sadri-Vakili, G.; Vakili, K. YAP Subcellular Localization and Hippo Pathway Transcriptome Analysis in Pediatric Hepatocellular Carcinoma. Sci. Rep. 2016, 6, 30238. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, S.W.; Lin, K.C.; Moore, J.L., 3rd; Tan, F.E.; Ma, S.; Ye, Z.; Qiu, Y.; Ren, B.; Guan, K.L. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J. Biol. Chem. 2018, 293, 11230–11240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finch-Edmondson, M.L.; Strauss, R.P.; Passman, A.M.; Sudol, M.; Yeoh, G.C.; Callus, B.A. TAZ Protein Accumulation Is Negatively Regulated by YAP Abundance in Mammalian Cells. J. Biol. Chem. 2015, 290, 27928–27938. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Liu, Y.; Zou, J.; Yang, R.; Xuan, F.; Wang, Y.; Gao, N.; Cui, H. Transcriptional co-activator TAZ sustains proliferation and tumorigenicity of neuroblastoma by targeting CTGF and PDGF-beta. Oncotarget 2015, 6, 9517–9530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Huang, M.; Tan, J.; Hou, J.; He, J.; Wang, F.; Cui, H.; Yi, L. Transcriptional co-activator with PDZ-binding motif overexpression promotes cell proliferation and transcriptional co-activator with PDZ-binding motif deficiency induces cell cycle arrest in neuroblastoma. Oncol. Lett. 2017, 13, 4295–4301. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.; Lee, J.Y.; Jonus, H.C.; Arnold, A.; Schnepp, R.W.; Janssen, K.M.; Maximov, V.; Goldsmith, K.C. YAP-Mediated Repression of HRK Regulates Tumor Growth, Therapy Response, and Survival Under Tumor Environmental Stress in Neuroblastoma. Cancer Res. 2020, 80, 4741–4753. [Google Scholar] [CrossRef]
- Coggins, G.E.; Farrel, A.; Rathi, K.S.; Hayes, C.M.; Scolaro, L.; Rokita, J.L.; Maris, J.M. YAP1 Mediates Resistance to MEK1/2 Inhibition in Neuroblastomas with Hyperactivated RAS Signaling. Cancer Res. 2019, 79, 6204–6214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Tan, J.; Zhu, J.; Wang, S.; Wei, G. YAP promotes tumorigenesis and cisplatin resistance in neuroblastoma. Oncotarget 2017, 8, 37154–37163. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Xu, X.; Xie, C.; Liu, H.; Yang, D.; Zhang, J.; Wu, Q.; Feng, W.; Wang, L.; Du, L.; et al. YAP promotes the proliferation of neuroblastoma cells through decreasing the nuclear location of p27(Kip1) mediated by Akt. Cell Prolif. 2020, 53, e12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, B.K.; Fathers, K.E.; Hallett, R.; Yung, C.K.; Stein, L.D.; Mouaaz, S.; Kee, L.; Hawkins, C.E.; Irwin, M.S.; Kaplan, D.R. A Metastatic Mouse Model Identifies Genes That Regulate Neuroblastoma Metastasis. Cancer Res. 2017, 77, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Chen, K.; Cheng, C.; Xu, Y.; Cheng, Q.; Xu, G.; Wu, Y.; Wu, Z. Prp19 Is an Independent Prognostic Marker and Promotes Neuroblastoma Metastasis by Regulating the Hippo-YAP Signaling Pathway. Front. Oncol. 2020, 10, 575366. [Google Scholar] [CrossRef] [PubMed]
- van Groningen, T.; Akogul, N.; Westerhout, E.M.; Chan, A.; Hasselt, N.E.; Zwijnenburg, D.A.; Broekmans, M.; Stroeken, P.; Haneveld, F.; Hooijer, G.K.J.; et al. A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat. Commun. 2019, 10, 1530. [Google Scholar] [CrossRef]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Hindley, C.J.; Condurat, A.L.; Menon, V.; Thomas, R.; Azmitia, L.M.; Davis, J.A.; Pruszak, J. The Hippo pathway member YAP enhances human neural crest cell fate and migration. Sci. Rep. 2016, 6, 23208. [Google Scholar] [CrossRef] [Green Version]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Piccolo, S.; Cordenonsi, M.; Dupont, S. Molecular pathways: YAP and TAZ take center stage in organ growth and tumorigenesis. Clin. Cancer Res. 2013, 19, 4925–4930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, A.P. Anoikis. Cell Death Differ. 2005, 12 (Suppl. 2), 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Guadamillas, M.C.; Cerezo, A.; Del Pozo, M.A. Overcoming anoikis—Pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011, 124, 3189–3197. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.Y.; Yu, J.; Guan, K.L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yang, L.; Szeto, P.; Abali, G.K.; Zhang, Y.; Kulkarni, A.; Amarasinghe, K.; Li, J.; Vergara, I.A.; Molania, R.; et al. The Hippo pathway oncoprotein YAP promotes melanoma cell invasion and spontaneous metastasis. Oncogene 2020, 39, 5267–5281. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Sun, Y.; Wei, Y.; Zhang, P.; Rezaeian, A.H.; Teruya-Feldstein, J.; Gupta, S.; Liang, H.; Lin, H.K.; Hung, M.C.; et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat. Med. 2012, 18, 1511–1517. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Yang, X.; Huang, X.; Yao, Y.; Wei, X.; Yang, S.; Zhou, D.; Zhang, W.; Long, Z.; Xu, X.; et al. 2-Hydroxylation of Fatty Acids Represses Colorectal Tumorigenesis and Metastasis via the YAP Transcriptional Axis. Cancer Res. 2021, 81, 289–302. [Google Scholar] [CrossRef]
- Zhao, B.; Xie, J.; Zhou, X.; Zhang, L.; Cheng, X.; Liang, C. YAP activation in melanoma contributes to anoikis resistance and metastasis. Exp. Biol. Med. (Maywood) 2020, 246, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Reggiani, F.; Gobbi, G.; Ciarrocchi, A.; Ambrosetti, D.C.; Sancisi, V. Multiple roles and context-specific mechanisms underlying YAP and TAZ-mediated resistance to anti-cancer therapy. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188341. [Google Scholar] [CrossRef]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, J. Role of YAP/TAZ transcriptional regulators in resistance to anti-cancer therapies. Cell. Mol. Life Sci. 2017, 74, 1457–1474. [Google Scholar] [CrossRef]
- Zeng, R.; Dong, J. The Hippo Signaling Pathway in Drug Resistance in Cancer. Cancers 2021, 13, 318. [Google Scholar] [CrossRef]
- Gujral, T.S.; Kirschner, M.W. Hippo pathway mediates resistance to cytotoxic drugs. Proc. Natl. Acad. Sci. USA 2017, 114, E3729–E3738. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wu, L.; Tu, J.; Zhao, Z.; Fan, X.; Mao, J.; Weng, Q.; Wu, X.; Huang, L.; Xu, M.; et al. miR-590-5p suppresses hepatocellular carcinoma chemoresistance by targeting YAP1 expression. EBioMedicine 2018, 35, 142–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wu, B.; Zhong, Z. Downregulation of YAP inhibits proliferation, invasion and increases cisplatin sensitivity in human hepatocellular carcinoma cells. Oncol. Lett. 2018, 16, 585–593. [Google Scholar] [CrossRef]
- Chen, X.; Gu, W.; Wang, Q.; Fu, X.; Wang, Y.; Xu, X.; Wen, Y. C-MYC and BCL-2 mediate YAP-regulated tumorigenesis in OSCC. Oncotarget 2018, 9, 668–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; He, F.; Zhao, X.; Zhang, Y.; Chu, X.; Hua, C.; Qu, Y.; Duan, Y.; Ming, L. YAP Inhibits the Apoptosis and Migration of Human Rectal Cancer Cells via Suppression of JNK-Drp1-Mitochondrial Fission-HtrA2/Omi Pathways. Cell. Physiol. Biochem. 2017, 44, 2073–2089. [Google Scholar] [CrossRef]
- Goldsmith, K.C.; Gross, M.; Peirce, S.; Luyindula, D.; Liu, X.; Vu, A.; Sliozberg, M.; Guo, R.; Zhao, H.; Reynolds, C.P.; et al. Mitochondrial Bcl-2 family dynamics define therapy response and resistance in neuroblastoma. Cancer Res. 2012, 72, 2565–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.F.; Tseng, Y.C.; Chang, W.C.; Chen, Y.C.; Kao, Y.R.; Chou, T.Y.; Ho, C.C.; Wu, C.W. YAP1 is essential for tumor growth and is a potential therapeutic target for EGFR-dependent lung adenocarcinomas. Oncotarget 2017, 8, 89539–89551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.E.; Park, H.S.; Lee, D.; Yoo, G.; Kim, T.; Jeon, H.; Yeo, M.K.; Lee, C.S.; Moon, J.Y.; Jung, S.S.; et al. Hippo pathway effector YAP inhibition restores the sensitivity of EGFR-TKI in lung adenocarcinoma having primary or acquired EGFR-TKI resistance. Biochem. Biophys. Res. Commun. 2016, 474, 154–160. [Google Scholar] [CrossRef]
- Ghiso, E.; Migliore, C.; Ciciriello, V.; Morando, E.; Petrelli, A.; Corso, S.; De Luca, E.; Gatti, G.; Volante, M.; Giordano, S. YAP-Dependent AXL Overexpression Mediates Resistance to EGFR Inhibitors in NSCLC. Neoplasia 2017, 19, 1012–1021. [Google Scholar] [CrossRef]
- Hsu, P.C.; You, B.; Yang, Y.L.; Zhang, W.Q.; Wang, Y.C.; Xu, Z.; Dai, Y.; Liu, S.; Yang, C.T.; Li, H.; et al. YAP promotes erlotinib resistance in human non-small cell lung cancer cells. Oncotarget 2016, 7, 51922–51933. [Google Scholar] [CrossRef] [Green Version]
- Ney, G.M.; McKay, L.; Koschmann, C.; Mody, R.; Li, Q. The Emerging Role of Ras Pathway Signaling in Pediatric Cancer. Cancer Res. 2020, 80, 5155–5163. [Google Scholar] [CrossRef]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef] [Green Version]
- Schreck, K.C.; Grossman, S.A.; Pratilas, C.A. BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers 2019, 11, 1262. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Slemmons, K.K.; Crose, L.E.; Rudzinski, E.; Bentley, R.C.; Linardic, C.M. Role of the YAP Oncoprotein in Priming Ras-Driven Rhabdomyosarcoma. PLoS ONE 2015, 10, e0140781. [Google Scholar] [CrossRef]
- Hong, X.; Nguyen, H.T.; Chen, Q.; Zhang, R.; Hagman, Z.; Voorhoeve, P.M.; Cohen, S.M. Opposing activities of the Ras and Hippo pathways converge on regulation of YAP protein turnover. EMBO J. 2014, 33, 2447–2457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, J.Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell Biol. 2009, 11, 1444–1450. [Google Scholar] [CrossRef]
- Woodfield, S.E.; Zhang, L.; Scorsone, K.A.; Liu, Y.; Zage, P.E. Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression. BMC Cancer 2016, 16, 172. [Google Scholar] [CrossRef] [Green Version]
- Umapathy, G.; Guan, J.; Gustafsson, D.E.; Javanmardi, N.; Cervantes-Madrid, D.; Djos, A.; Martinsson, T.; Palmer, R.H.; Hallberg, B. MEK inhibitor trametinib does not prevent the growth of anaplastic lymphoma kinase (ALK)-addicted neuroblastomas. Sci. Signal. 2017, 10, eaam7550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misiak, D.; Hagemann, S.; Bell, J.L.; Busch, B.; Lederer, M.; Bley, N.; Schulte, J.H.; Huttelmaier, S. The MicroRNA Landscape of MYCN-Amplified Neuroblastoma. Front. Oncol. 2021, 11, 647737. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Healy, J.R.; Hart, L.S.; Shazad, A.L.; Gagliardi, M.E.; Tsang, M.; Elias, J.; Ruden, J.; Farrel, A.; Rokita, J.L.; Li, Y.; et al. Limited antitumor activity of combined BET and MEK inhibition in neuroblastoma. Pediatr. Blood Cancer 2020, 67, e28267. [Google Scholar] [CrossRef]
- Hart, L.S.; Rader, J.; Raman, P.; Batra, V.; Russell, M.R.; Tsang, M.; Gagliardi, M.; Chen, L.; Martinez, D.; Li, Y.; et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clin. Cancer Res. 2017, 23, 1785–1796. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- Santamaria, P.G.; Moreno-Bueno, G.; Cano, A. Contribution of Epithelial Plasticity to Therapy Resistance. J. Clin. Med. 2019, 8, 676. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.A.; Lu, C.Y.; Cheng, T.Y.; Pan, S.H.; Chen, H.F.; Chang, N.S. WW Domain-Containing Proteins YAP and TAZ in the Hippo Pathway as Key Regulators in Stemness Maintenance, Tissue Homeostasis, and Tumorigenesis. Front. Oncol. 2019, 9, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Jiang, D.; Chi, F.; Zhao, B. The Hippo pathway regulates stem cell proliferation, self-renewal, and differentiation. Protein Cell 2012, 3, 291–304. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, K.; Mishima, T.; Takano, S.; Yoshitomi, H.; Furukawa, K.; Takayashiki, T.; Kuboki, S.; Takada, M.; Miyazaki, M.; Ohtsuka, M. The Expression of Yes-Associated Protein (YAP) Maintains Putative Cancer Stemness and Is Associated with Poor Prognosis in Intrahepatic Cholangiocarcinoma. Am. J. Pathol. 2019, 189, 1863–1877. [Google Scholar] [CrossRef] [Green Version]
- Lavado, A.; Park, J.Y.; Pare, J.; Finkelstein, D.; Pan, H.; Xu, B.; Fan, Y.; Kumar, R.P.; Neale, G.; Kwak, Y.D.; et al. The Hippo Pathway Prevents YAP/TAZ-Driven Hypertranscription and Controls Neural Progenitor Number. Dev. Cell 2018, 47, 576–591 e578. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Pfaff, S.L.; Gage, F.H. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 2008, 22, 3320–3334. [Google Scholar] [CrossRef] [Green Version]
- Maguire, L.H.; Thomas, A.R.; Goldstein, A.M. Tumors of the neural crest: Common themes in development and cancer. Dev. Dyn. 2015, 244, 311–322. [Google Scholar] [CrossRef]
- Bate-Eya, L.T.; Ebus, M.E.; Koster, J.; den Hartog, I.J.; Zwijnenburg, D.A.; Schild, L.; van der Ploeg, I.; Dolman, M.E.; Caron, H.N.; Versteeg, R.; et al. Newly-derived neuroblastoma cell lines propagated in serum-free media recapitulate the genotype and phenotype of primary neuroblastoma tumours. Eur. J. Cancer 2014, 50, 628–637. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, F.; Li, G.; Li, G.; Yang, X.; Liu, L.; Zhang, R.; Zhang, B.; Feng, Y. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2018, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Lyu, X.; Faleti, O.D.; He, M.L. The special stemness functions of Tbx3 in stem cells and cancer development. Semin. Cancer Biol. 2019, 57, 105–110. [Google Scholar] [CrossRef]
- Chen, X.; Yang, W.; Deng, X.; Ye, S.; Xiao, W. Interleukin-6 promotes proliferative vitreoretinopathy by inducing epithelial-mesenchymal transition via the JAK1/STAT3 signaling pathway. Mol. Vis. 2020, 26, 517–529. [Google Scholar]
- Gkretsi, V.; Stylianou, A.; Papageorgis, P.; Polydorou, C.; Stylianopoulos, T. Remodeling Components of the Tumor Microenvironment to Enhance Cancer Therapy. Front. Oncol. 2015, 5, 214. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Pike, L.R.; Phadwal, K.; Simon, A.K.; Harris, A.L. ATF4 orchestrates a program of BH3-only protein expression in severe hypoxia. Mol. Biol. Rep. 2012, 39, 10811–10822. [Google Scholar] [CrossRef]
- Nakamura, M.; Shimada, K.; Konishi, N. The role of HRK gene in human cancer. Oncogene 2008, 27 (Suppl. 1), S105–S113. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Zeng, C.; Ye, S.; Dai, X.; He, Q.; Yang, B.; Zhu, H. Yes-associated protein (YAP) and transcriptional coactivator with a PDZ-binding motif (TAZ): A nexus between hypoxia and cancer. Acta Pharm. Sin. B 2020, 10, 947–960. [Google Scholar] [CrossRef]
- Ma, B.; Chen, Y.; Chen, L.; Cheng, H.; Mu, C.; Li, J.; Gao, R.; Zhou, C.; Cao, L.; Liu, J.; et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat. Cell Biol. 2015, 17, 95–103. [Google Scholar] [CrossRef]
- Hooglugt, A.; van der Stoel, M.M.; Boon, R.A.; Huveneers, S. Endothelial YAP/TAZ Signaling in Angiogenesis and Tumor Vasculature. Front. Oncol. 2020, 10, 612802. [Google Scholar] [CrossRef]
- Neto, F.; Klaus-Bergmann, A.; Ong, Y.T.; Alt, S.; Vion, A.C.; Szymborska, A.; Carvalho, J.R.; Hollfinger, I.; Bartels-Klein, E.; Franco, C.A.; et al. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. eLife 2018, 7, e31037. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.H.; Kim, J.; Park, D.Y.; Bae, H.; Lee, D.H.; Kim, K.H.; Hong, S.P.; Jang, S.P.; Kubota, Y.; et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J. Clin. Investig. 2017, 127, 3441–3461. [Google Scholar] [CrossRef]
- Wang, X.; Freire Valls, A.; Schermann, G.; Shen, Y.; Moya, I.M.; Castro, L.; Urban, S.; Solecki, G.M.; Winkler, F.; Riedemann, L.; et al. YAP/TAZ Orchestrate VEGF Signaling during Developmental Angiogenesis. Dev. Cell 2017, 42, 462–478.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelson, H.; Fredlund, E.; Ovenberger, M.; Landberg, G.; Pahlman, S. Hypoxia-induced dedifferentiation of tumor cells--a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin. Cell Dev. Biol. 2005, 16, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Jogi, A.; Ora, I.; Nilsson, H.; Lindeheim, A.; Makino, Y.; Poellinger, L.; Axelson, H.; Pahlman, S. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc. Natl. Acad. Sci. USA 2002, 99, 7021–7026. [Google Scholar] [CrossRef] [Green Version]
- Cangelosi, D.; Morini, M.; Zanardi, N.; Sementa, A.R.; Muselli, M.; Conte, M.; Garaventa, A.; Pfeffer, U.; Bosco, M.C.; Varesio, L.; et al. Hypoxia Predicts Poor Prognosis in Neuroblastoma Patients and Associates with Biological Mechanisms Involved in Telomerase Activation and Tumor Microenvironment Reprogramming. Cancers 2020, 12, 2343. [Google Scholar] [CrossRef]
- Pahlman, S.; Mohlin, S. Hypoxia and hypoxia-inducible factors in neuroblastoma. Cell Tissue Res. 2018, 372, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Meitar, D.; Crawford, S.E.; Rademaker, A.W.; Cohn, S.L. Tumor angiogenesis correlates with metastatic disease, N-myc amplification, and poor outcome in human neuroblastoma. J. Clin. Oncol. 1996, 14, 405–414. [Google Scholar] [CrossRef]
- Giussani, M.; Triulzi, T.; Sozzi, G.; Tagliabue, E. Tumor Extracellular Matrix Remodeling: New Perspectives as a Circulating Tool in the Diagnosis and Prognosis of Solid Tumors. Cells 2019, 8, 81. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Eble, J.A.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef]
- Jang, M.; An, J.; Oh, S.W.; Lim, J.Y.; Kim, J.; Choi, J.K.; Cheong, J.H.; Kim, P. Matrix stiffness epigenetically regulates the oncogenic activation of the Yes-associated protein in gastric cancer. Nat. Biomed. Eng. 2021, 5, 114–123. [Google Scholar] [CrossRef]
- Lam, W.A.; Cao, L.; Umesh, V.; Keung, A.J.; Sen, S.; Kumar, S. Extracellular matrix rigidity modulates neuroblastoma cell differentiation and N-myc expression. Mol. Cancer 2010, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Abyaneh, H.S.; Regenold, M.; McKee, T.D.; Allen, C.; Gauthier, M.A. Towards extracellular matrix normalization for improved treatment of solid tumors. Theranostics 2020, 10, 1960–1980. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- White, S.M.; Murakami, S.; Yi, C. The complex entanglement of Hippo-Yap/Taz signaling in tumor immunity. Oncogene 2019, 38, 2899–2909. [Google Scholar] [CrossRef]
- Guo, X.; Zhao, Y.; Yan, H.; Yang, Y.; Shen, S.; Dai, X.; Ji, X.; Ji, F.; Gong, X.G.; Li, L.; et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev. 2017, 31, 247–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Li, W.; Wang, S.; Zhang, P.; Wang, Q.; Xiao, J.; Zhang, C.; Zheng, X.; Xu, X.; Xue, S.; et al. YAP Aggravates Inflammatory Bowel Disease by Regulating M1/M2 Macrophage Polarization and Gut Microbial Homeostasis. Cell Rep. 2019, 27, 1176–1189.e5. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Khan, S.K.; Liu, Y.; Xu, R.; Park, O.; He, Y.; Cha, B.; Gao, B.; Yang, Y. Hepatic Hippo signaling inhibits protumoural microenvironment to suppress hepatocellular carcinoma. Gut 2018, 67, 1692–1703. [Google Scholar] [CrossRef]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [Green Version]
- Stampouloglou, E.; Cheng, N.; Federico, A.; Slaby, E.; Monti, S.; Szeto, G.L.; Varelas, X. Yap suppresses T-cell function and infiltration in the tumor microenvironment. PLoS Biol. 2020, 18, e3000591. [Google Scholar] [CrossRef]
- Lebid, A.; Chung, L.; Pardoll, D.M.; Pan, F. YAP Attenuates CD8 T Cell-Mediated Anti-tumor Response. Front. Immunol. 2020, 11, 580. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kim, C.G.; Kim, S.K.; Shin, S.J.; Choe, E.A.; Park, S.H.; Shin, E.C.; Kim, J. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol. Res. 2018, 6, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Miao, J.; Hsu, P.C.; Yang, Y.L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef] [Green Version]
- Zeine, R.; Salwen, H.R.; Peddinti, R.; Tian, Y.; Guerrero, L.; Yang, Q.; Chlenski, A.; Cohn, S.L. Presence of cancer-associated fibroblasts inversely correlates with Schwannian stroma in neuroblastoma tumors. Mod. Pathol. 2009, 22, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Wienke, J.; Dierselhuis, M.P.; Tytgat, G.A.M.; Kunkele, A.; Nierkens, S.; Molenaar, J.J. The immune landscape of neuroblastoma: Challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur. J. Cancer 2021, 144, 123–150. [Google Scholar] [CrossRef]
- Asgharzadeh, S.; Salo, J.A.; Ji, L.; Oberthuer, A.; Fischer, M.; Berthold, F.; Hadjidaniel, M.; Liu, C.W.; Metelitsa, L.S.; Pique-Regi, R.; et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J. Clin. Oncol. 2012, 30, 3525–3532. [Google Scholar] [CrossRef] [Green Version]
- Nolan, J.C.; Frawley, T.; Tighe, J.; Soh, H.; Curtin, C.; Piskareva, O. Preclinical models for neuroblastoma: Advances and challenges. Cancer Lett. 2020, 474, 53–62. [Google Scholar] [CrossRef]
- Corallo, D.; Frabetti, S.; Candini, O.; Gregianin, E.; Dominici, M.; Fischer, H.; Aveic, S. Emerging Neuroblastoma 3D In Vitro Models for Pre-Clinical Assessments. Front. Immunol. 2020, 11, 584214. [Google Scholar] [CrossRef]
- Ornell, K.J.; Coburn, J.M. Developing preclinical models of neuroblastoma: Driving therapeutic testing. BMC Biomed. Eng. 2019, 1, 33. [Google Scholar] [CrossRef]
- Monferrer, E.; Martin-Vano, S.; Carretero, A.; Garcia-Lizarribar, A.; Burgos-Panadero, R.; Navarro, S.; Samitier, J.; Noguera, R. A three-dimensional bioprinted model to evaluate the effect of stiffness on neuroblastoma cell cluster dynamics and behavior. Sci. Rep. 2020, 10, 6370. [Google Scholar] [CrossRef] [Green Version]
- Quinn, C.H.; Beierle, A.M.; Beierle, E.A. Artificial Tumor Microenvironments in Neuroblastoma. Cancers 2021, 13, 1629. [Google Scholar] [CrossRef]
- Braekeveldt, N.; Wigerup, C.; Tadeo, I.; Beckman, S.; Sanden, C.; Jonsson, J.; Erjefalt, J.S.; Berbegall, A.P.; Borjesson, A.; Backman, T.; et al. Neuroblastoma patient-derived orthotopic xenografts reflect the microenvironmental hallmarks of aggressive patient tumours. Cancer Lett. 2016, 375, 384–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef]
- Zhu, S.; Thomas Look, A. Neuroblastoma and Its Zebrafish Model. Adv. Exp. Med. Biol. 2016, 916, 451–478. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Hong, W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics 2020, 10, 3622–3635. [Google Scholar] [CrossRef] [PubMed]
- Brodowska, K.; Al-Moujahed, A.; Marmalidou, A.; Meyer Zu Horste, M.; Cichy, J.; Miller, J.W.; Gragoudas, E.; Vavvas, D.G. The clinically used photosensitizer Verteporfin (VP) inhibits YAP-TEAD and human retinoblastoma cell growth in vitro without light activation. Exp. Eye Res. 2014, 124, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Wang, F.; Wang, Y.; Li, T.; Xiu, P.; Zhong, J.; Sun, X.; Li, J. Verteporfin suppresses cell survival, angiogenesis and vasculogenic mimicry of pancreatic ductal adenocarcinoma via disrupting the YAP-TEAD complex. Cancer Sci. 2017, 108, 478–487. [Google Scholar] [CrossRef]
- Dasari, V.R.; Mazack, V.; Feng, W.; Nash, J.; Carey, D.J.; Gogoi, R. Verteporfin exhibits YAP-independent anti-proliferative and cytotoxic effects in endometrial cancer cells. Oncotarget 2017, 8, 28628–28640. [Google Scholar] [CrossRef] [Green Version]
- Pobbati, A.V.; Rubin, B.P. Protein-Protein Interaction Disruptors of the YAP/TAZ-TEAD Transcriptional Complex. Molecules 2020, 25, 6001. [Google Scholar] [CrossRef]
- Zhou, Z.; Hu, T.; Xu, Z.; Lin, Z.; Zhang, Z.; Feng, T.; Zhu, L.; Rong, Y.; Shen, H.; Luk, J.M.; et al. Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. FASEB J. 2015, 29, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.T.; Konradi, A.W.; Feng, Y.; Peng, X.; Qiao, S.; Post, L. Targeting the Hippo-YAP pathway with novel small-molecule inhibitors of the YAP-TEAD transcription activity [abstract]. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Laraba, L.; de Guibert, J.G.; Tang, T.T.; Post, L.; Parkinson, D.B. TEAD inhibition reduces tumor proliferation in Merlin null meningioma and schwannoma [abstract]. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Tang, T.T.; Konradi, A.W.; Feng, Y.; Peng, X.; Ma, M.; Li, J.; Yu, F.X.; Guan, K.L.; Post, L. Small Molecule Inhibitors of TEAD Auto-palmitoylation Selectively Inhibit Proliferation and Tumor Growth of NF2-deficient Mesothelioma. Mol. Cancer 2021, 20, 986–998. [Google Scholar] [CrossRef]
- Higuchi, T.; Nakamura, M.; Shimada, K.; Ishida, E.; Hirao, K.; Konishi, N. HRK inactivation associated with promoter methylation and LOH in prostate cancer. Prostate 2008, 68, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Obata, T.; Toyota, M.; Satoh, A.; Sasaki, Y.; Ogi, K.; Akino, K.; Suzuki, H.; Murai, M.; Kikuchi, T.; Mita, H.; et al. Identification of HRK as a target of epigenetic inactivation in colorectal and gastric cancer. Clin. Cancer Res. 2003, 9, 6410–6418. [Google Scholar] [PubMed]
- Li, H.; Cai, Q.; Wu, H.; Vathipadiekal, V.; Dobbin, Z.C.; Li, T.; Hua, X.; Landen, C.N.; Birrer, M.J.; Sanchez-Beato, M.; et al. SUZ12 promotes human epithelial ovarian cancer by suppressing apoptosis via silencing HRK. Mol. Cancer Res. 2012, 10, 1462–1472. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Chen, X.; Chen, N.; Nie, L.; Li, X.; Li, Q.; Zeng, H.; Zhou, Q. Synergistic silencing by promoter methylation and reduced AP-2alpha transactivation of the proapoptotic HRK gene confers apoptosis resistance and enhanced tumor growth. Am. J. Pathol. 2013, 182, 84–95. [Google Scholar] [CrossRef]
- Hillmer, R.E.; Link, B.A. The Roles of Hippo Signaling Transducers Yap and Taz in Chromatin Remodeling. Cells 2019, 8, 502. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Mao, B.; Cheng, C.; Zou, Z.; Gao, J.; Yang, Y.; Lei, T.; Qi, X.; Yuan, Z.; Xu, W.; et al. YAP promotes breast cancer metastasis by repressing growth differentiation factor-15. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, J.; Sun, S.; Chen, X.; Zhang, H.; Li, B.; Sun, S. YAP/TAZ-mediated activation of serine metabolism and methylation regulation is critical for LKB1-deficient breast cancer progression. Biosci. Rep. 2017, 37, BSR20171072. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.S. Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phimmachanh, M.; Han, J.Z.R.; O’Donnell, Y.E.I.; Latham, S.L.; Croucher, D.R. Histone Deacetylases and Histone Deacetylase Inhibitors in Neuroblastoma. Front. Cell Dev. Biol. 2020, 8, 578770. [Google Scholar] [CrossRef] [PubMed]
- Jubierre, L.; Jimenez, C.; Rovira, E.; Soriano, A.; Sabado, C.; Gros, L.; Llort, A.; Hladun, R.; Roma, J.; Toledo, J.S.; et al. Targeting of epigenetic regulators in neuroblastoma. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Faiao-Flores, F.; Emmons, M.F.; Durante, M.A.; Kinose, F.; Saha, B.; Fang, B.; Koomen, J.M.; Chellappan, S.P.; Maria-Engler, S.S.; Rix, U.; et al. HDAC Inhibition Enhances the In Vivo Efficacy of MEK Inhibitor Therapy in Uveal Melanoma. Clin. Cancer Res. 2019, 25, 5686–5701. [Google Scholar] [CrossRef]
- Yamada, T.; Amann, J.M.; Tanimoto, A.; Taniguchi, H.; Shukuya, T.; Timmers, C.; Yano, S.; Shilo, K.; Carbone, D.P. Histone Deacetylase Inhibition Enhances the Antitumor Activity of a MEK Inhibitor in Lung Cancer Cells Harboring RAS Mutations. Mol. Cancer 2018, 17, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Holden, J.K.; Crawford, J.J.; Noland, C.L.; Schmidt, S.; Zbieg, J.R.; Lacap, J.A.; Zang, R.; Miller, G.M.; Zhang, Y.; Beroza, P.; et al. Small Molecule Dysregulation of TEAD Lipidation Induces a Dominant-Negative Inhibition of Hippo Pathway Signaling. Cell Rep. 2020, 31, 107809. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Focus | Comments | References |
|---|---|---|
| Tumorigenesis |
| [51,53,54,55] |
| Metastasis |
| [51,55,56] |
| Therapy Resistance |
| [51,52,53] |
| Mesenchymal Properties |
| [51,57,58,59] |
| Therapy | Mechanism and Effects | References |
|---|---|---|
| Verteporfin |
| [159,160,161] |
| Cyclic Peptides (i.e., Peptide 17) |
| [162,163] |
| TEAD auto-palmitoylation inhibitors |
| [166,179] |
| Combination therapies |
| [113,135,175,176,177,178] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, J.; Goldsmith, K.C. A New Player in Neuroblastoma: YAP and Its Role in the Neuroblastoma Microenvironment. Cancers 2021, 13, 4650. https://doi.org/10.3390/cancers13184650
Shim J, Goldsmith KC. A New Player in Neuroblastoma: YAP and Its Role in the Neuroblastoma Microenvironment. Cancers. 2021; 13(18):4650. https://doi.org/10.3390/cancers13184650
Chicago/Turabian StyleShim, Jenny, and Kelly C. Goldsmith. 2021. "A New Player in Neuroblastoma: YAP and Its Role in the Neuroblastoma Microenvironment" Cancers 13, no. 18: 4650. https://doi.org/10.3390/cancers13184650
APA StyleShim, J., & Goldsmith, K. C. (2021). A New Player in Neuroblastoma: YAP and Its Role in the Neuroblastoma Microenvironment. Cancers, 13(18), 4650. https://doi.org/10.3390/cancers13184650