
Targeting the p53 Pathway in CLL: State of the Art and Future Perspectives


Abstract
:Simple Summary
Abstract
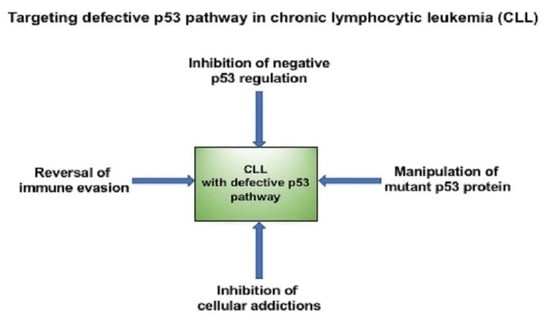
1. Introduction
2. p53 Pathway Defects as Adverse Prognostic Biomarkers in CLL
3. Inhibiting the Inhibitor: Targeting MDM2 to Upregulate Wild-Type p53
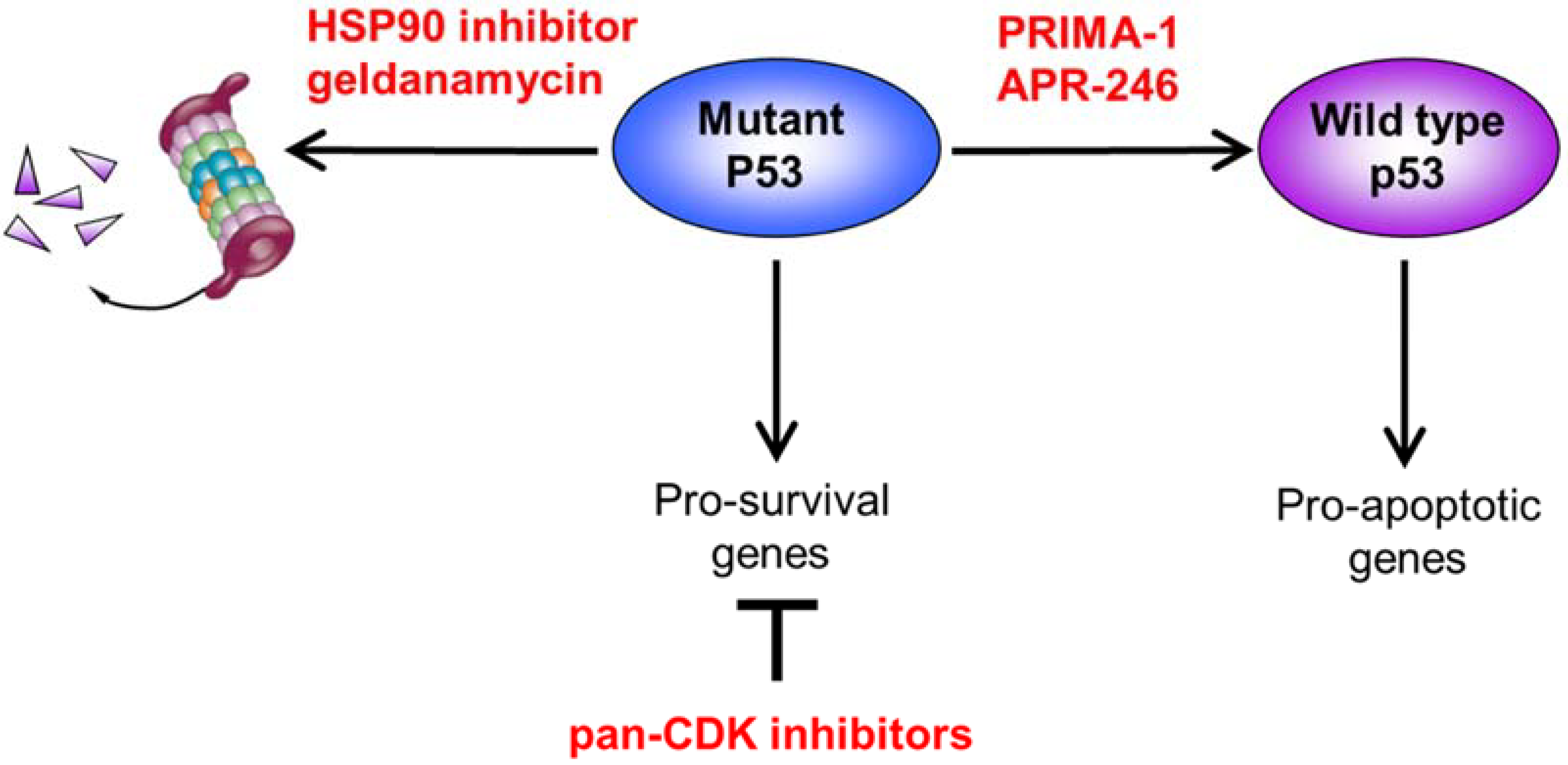
4. Confronting Mutant p53: Strategies to Restore the Tumor-Suppressive Function
4.1. Inhibiting CDK to Restore Transcriptional Control of Apoptosis and Prosurvival Responses
4.2. Altering Mutant p53 Conformation to Restore Wild-Type Function
4.3. Destabilizing Mutant p53 through HSP90 Inhibition
5. Novel Synthetically Lethal Strategies Exploiting p53 Pathway Defects
5.1. Exacerbating Oxidative-Stress-Induced Cytotoxicity
5.2. Targeting Replication Stress through ATR/Chk1 Inhibition
5.3. Targeting PARP, DNA-PK, and USP7 Addiction
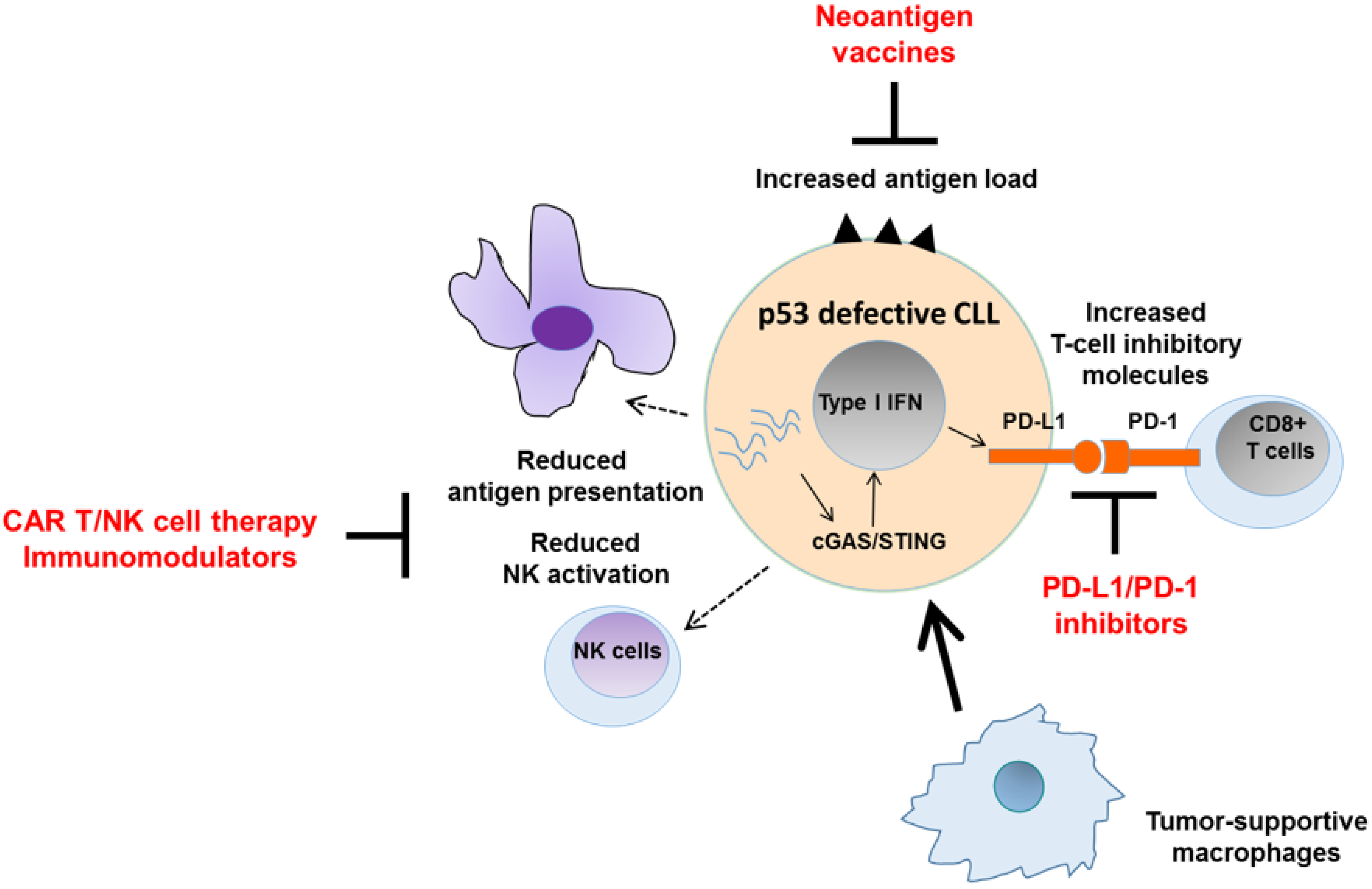
6. Harnessing the Immunogenicity of p53 Pathway Defects
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Kwok, M.; Oldreive, C.; Rawstron, A.C.; Goel, A.; Papatzikas, G.; Jones, R.E.; Drennan, S.; Agathanggelou, A.; Sharma-Oates, A.; Evans, P.; et al. Integrative analysis of spontaneous CLL regression highlights genetic and microenvironmental interdependency in CLL. Blood 2020, 135, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, I.; Chiaretti, S.; Tavolaro, S.; De Propris, M.S.; Maggio, R.; Mancini, F.; Peragine, N.; Santangelo, S.; Marinelli, M.; Mauro, F.R.; et al. Spontaneous regression of chronic lymphocytic leukemia: Clinical and biologic features of 9 cases. Blood 2009, 114, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.; Nadeu, F.; Colomer, D.; Campo, E. Chronic lymphocytic leukemia: From molecular pathogenesis to novel therapeutic strategies. Haematologica 2020, 105, 2205–2217. [Google Scholar] [CrossRef]
- Guièze, R.; Wu, C.J. Genomic and epigenomic heterogeneity in chronic lymphocytic leukemia. Blood 2015, 126, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purroy, N.; Wu, C.J. Coevolution of Leukemia and Host Immune Cells in Chronic Lymphocytic Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7, a026740. [Google Scholar] [CrossRef] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Cremona, C.; Behrens, A. ATM signalling and cancer. Oncogene 2013, 33, 3351–3360. [Google Scholar] [CrossRef] [Green Version]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Pettitt, A.R.; Sherrington, P.D.; Stewart, G.; Cawley, J.C.; Taylor, A.M.R.; Stankovic, T. p53 dysfunction in B-cell chronic lymphocytic leukemia: Inactivation of ATM as an alternative toTP53 mutation. Blood 2001, 98, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosravi, R.; Maya, R.; Gottlieb, T.; Oren, M.; Shiloh, Y.; Shkedy, D. Rapid ATM-dependent phosphoryla-tion of MDM2 precedes p53 accumulation in response to DNA damage. Proc. Natl. Acad. Sci. USA 1999, 26, 14973–14977. [Google Scholar] [CrossRef] [Green Version]
- Stankovic, T.; Stewart, G.S.; Fegan, C.; Biggs, P.; Last, J.; Byrd, P.J.; Keenan, R.D.; Moss, P.A.H.; Taylor, A.M.R. Ataxia telangiectasia mutated-deficient B-cell chronic lymphocytic leukemia occurs in pregerminal center cells and results in defective damage response and unrepaired chromosome damage. Blood 2002, 99, 300–309. [Google Scholar] [CrossRef]
- Austen, B.; Powell, J.E.; Alvi, A.; Edwards, I.; Hooper, L.; Starczynski, J.; Taylor, A.M.R.; Fegan, C.; Moss, P.; Stankovic, T. Mutations in the ATM gene lead to impaired overall and treatment-free survival that is independent of IGVH mutation status in patients with B-CLL. Blood 2005, 106, 3175–3182. [Google Scholar] [CrossRef] [Green Version]
- Malcikova, J.; Smardova, J.; Rocnova, L.; Tichy, B.; Kuglik, P.; Vranova, V.; Cejkova, S.; Svitakova, M.; Francova, H.S.; Brychtova, Y.; et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: Selection, impact on survival, and response to DNA damage. Blood 2009, 114, 5307–5314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenz, T.; Eichhorst, B.; Busch, R.; Denzel, T.; Häbe, S.; Winkler, D.; Bühler, A.; Edelmann, J.; Bergmann, M.; Hopfinger, G.; et al. TP53 mutation and survival in chronic lymphocytic leukemia. J. Clin. Oncol. 2010, 28, 4473–4479. [Google Scholar] [CrossRef]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic Aberrations and Survival in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austen, B.; Skowronska, A.; Baker, C.; Powell, J.E.; Gardiner, A.; Oscier, D.; Majid, A.; Dyer, M.; Siebert, R.; Taylor, A.M.; et al. Mutation Status of the Residual ATM Allele Is an Important Determinant of the Cellular Response to Chemotherapy and Survival in Patients with Chronic Lymphocytic Leukemia Containing an 11q Deletion. J. Clin. Oncol. 2007, 25, 5448–5457. [Google Scholar] [CrossRef] [PubMed]
- Skowronska, A.; Parker, A.; Ahmed, G.; Oldreive, C.; Davis, Z.; Richards, S.; Dyer, M.; Matutes, E.; González, D.; Taylor, A.M.R.; et al. Biallelic ATM Inactivation Significantly Reduces Survival in Patients Treated on the United Kingdom Leukemia Research Fund Chronic Lymphocytic Leukemia 4 Trial. J. Clin. Oncol. 2012, 30, 4524–4532. [Google Scholar] [CrossRef]
- Steele, A.J.; Prentice, A.G.; Hoffbrand, A.V.; Yogashangary, B.C.; Hart, S.M.; Nacheva, E.P.; Howard-Reeves, J.D.; Duke, V.M.; Kottaridis, P.D.; Cwynarski, K.; et al. p53-mediated apoptosis of CLL cells: Evidence for a transcription-independent mechanism. Blood 2008, 112, 3827–3834. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2017, 25, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Oren, M.; Rotter, V. Mutant p53 Gain-of-Function in Cancer. Cold Spring Harb. Perspect. Biol. 2009, 2, a001107. [Google Scholar] [CrossRef]
- Malcikova, J.; Stanokozubik, K.; Tichy, B.; Kantorova, B.; Pavlova, S.; Tom, N.; Radova, L.; Smardova, J.; Pardy, F.; Doubek, M.; et al. Detailed analysis of therapy-driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia 2014, 29, 877–885. [Google Scholar] [CrossRef]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.S.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, D.; Martinez, P.; Wade, R.; Hockley, S.; Oscier, D.; Matutes, E.; Dearden, C.E.; Richards, S.M.; Catovsky, D.; Morgan, G.J. Mutational Status of the TP53 Gene as a Predictor of Response and Survival in Patients with Chronic Lymphocytic Leukemia: Results From the LRF CLL4 Trial. J. Clin. Oncol. 2011, 29, 2223–2229. [Google Scholar] [CrossRef]
- Rossi, D.; Khiabanian, H.; Spina, V.; Ciardullo, C.; Bruscaggin, A.; Famà, R.; Rasi, S.; Monti, S.; Deambrogi, C.; De Paoli, L.; et al. Clinical impact of small TP53 mutat-ed subclones in chronic lymphocytic leukemia. Blood 2014, 14, 2139–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stilgenbauer, S.; Schnaiter, A.; Paschka, P.; Zenz, T.; Rossi, M.; Döhner, K.; Bühler, A.; Böttcher, S.; Ritgen, M.; Kneba, M.; et al. Gene mutations and treatment outcome in chronic lymphocytic leu-kemia: Results from the CLL8 trial. Blood 2014, 123, 3247–3254. [Google Scholar] [CrossRef] [Green Version]
- Schuh, A.; Becq, J.; Humphray, S.; Alexa, A.; Burns, A.; Clifford, R.; Feller, S.M.; Grocock, R.; Henderson, S.; Khrebtukova, I.; et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood 2012, 120, 4191–4196. [Google Scholar] [CrossRef] [Green Version]
- Gruber, M.; Bozic, I.; Leshchiner, I.; Livitz, D.; Stevenson, K.; Rassenti, L.; Rosebrock, D.; Taylor-Weiner, A.; Olive, O.; Goyetche, R.; et al. Growth dynamics in naturally progressing chronic lymphocytic leukaemia. Nature 2019, 570, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.J.; Kwok, M.; Gould, C.; Oldreive, C.E.; Mao, J.; Parry, H.; Smith, E.; Agathanggelou, A.; Pratt, G.; Taylor, A.M.R.; et al. Dynamic changes in clonal cytogenetic architecture during progression of chronic lymphocytic leukemia in patients and patient-derived murine xenografts. Oncotarget 2017, 8, 44749–44760. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Bennett, F.L.; O’Connor, S.J.M.; Kwok, M.; Fenton, J.A.L.; Plummer, M.; De Tute, R.; Owen, R.G.; Richards, S.; Jack, A.S.; et al. Monoclonal B-Cell Lymphocytosis and Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2008, 359, 575–583. [Google Scholar] [CrossRef]
- Barrio, S.; Shanafelt, T.D.; Ojha, J.; Chaffee, K.G.; Secreto, C.; Kortüm, K.M.; Pathangey, S.; Van-Dyke, D.L.; Slager, S.L.; Fonseca, R.; et al. Genomic characterization of high-count MBL cases indicates that early detection of driver mutations and subclonal expansion are predictors of adverse clinical outcome. Leukemia 2016, 31, 170–176. [Google Scholar] [CrossRef]
- Kwok, M.; Rawstron, A.C.; Varghese, A.; Evans, P.A.S.; O’Connor, S.J.M.; Doughty, C.; Newton, D.J.; Moreton, P.; Hillmen, P. Minimal residual disease is an independent predictor for 10-year survival in CLL. Blood 2016, 128, 2770–2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttcher, S.; Ritgen, M.; Fischer, K.; Stilgenbauer, S.; Busch, R.M.; Fingerle-Rowson, G.; Fink, A.M.; Bühler, A.; Zenz, T.; Wenger, M.K.; et al. Minimal Residual Disease Quantification Is an Independent Predictor of Progression-Free and Overall Survival in Chronic Lymphocytic Leukemia: A Multivariate Analysis from the Randomized GCLLSG CLL8 Trial. J. Clin. Oncol. 2012, 30, 980–988. [Google Scholar] [CrossRef]
- O’Brien, S.; Furman, R.R.; Coutre, S.; Flinn, I.W.; Burger, J.A.; Blum, K.; Sharman, J.; Wierda, W.; Jones, J.; Zhao, W.; et al. Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: A 5-year experience. Blood 2018, 131, 1910–1919. [Google Scholar] [CrossRef]
- Byrd, J.C.; Hillmen, P.; O’Brien, S.; Barrientos, J.C.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; Barr, P.M.; et al. Long-term follow-up of the RESONATE phase 3 trial of ibrutinib vs ofatumumab. Blood 2019, 133, 2031–2042. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.R.; Hillmen, P.; O’Brien, S.; Barrientos, J.C.; Reddy, N.M.; Coutre, S.E.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; Barr, P.M.; et al. Extended follow-up and impact of high-risk prognostic factors from the phase 3 RESONATE study in patients with previously treated CLL/SLL. Leukemia 2018, 32, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kater, A.P.; Wu, J.Q.; Kipps, T.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Robak, T.; de la Serna, J.; et al. Venetoclax Plus Rituximab in Relapsed Chronic Lymphocytic Leukemia: 4-Year Results and Evaluation of Impact of Genomic Complexity and Gene Mutations from the MURANO Phase III Study. J. Clin. Oncol. 2020, 38, 4042–4054. [Google Scholar] [CrossRef] [PubMed]
- Tausch, E.; Schneider, C.; Robrecht, S.; Zhang, C.; Dolnik, A.; Bloehdorn, J.; Bahlo, J.; Al-Sawaf, O.; Ritgen, M.; Fink, A.-M.; et al. Prognostic and predictive impact of genetic markers in patients with CLL treated with obinutuzumab and venetoclax. Blood 2020, 135, 2402–2412. [Google Scholar] [CrossRef]
- Dicker, F.; Herholz, H.; Schnittger, S.; Nakao, A.; Patten, N.; Wu, L.; Kern, W.; Haferlach, T. The detection of TP53 mutations in chronic lymphocytic leukemia independently predicts rapid disease progression and is highly correlated with a complex aberrant karyotype. Leukemia 2009, 23, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Leeksma, A.C.; Taylor, J.; Wu, B.; Gardner, J.R.; He, J.; Nahas, M.; Gonen, M.; Alemayehu, W.G.; Te Raa, D.; Walther, T.; et al. Clonal diversity predicts adverse outcome in chronic lymphocytic leukemia. Leukemia 2019, 33, 390–402. [Google Scholar] [CrossRef]
- Baliakas, P.; Jeromin, S.; Iskas, M.; Puiggros, A.; Plevova, K.; Nguyen-Khac, F.; Davis, Z.; Rigolin, G.M.; Visentin, A.; Xochelli, A.; et al. Cytogenetic complexity in chronic lymphocytic leukemia: Definitions, associations, and clinical impact. Blood 2019, 133, 1205–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Cole-Man, J.; Jones, D.; et al. BTK(C481S)-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2017, 35, 1437–1443. [Google Scholar] [CrossRef] [Green Version]
- Maddocks, K.J.; Ruppert, A.S.; Lozanski, G.; Heerema, N.A.; Zhao, W.; Abruzzo, L.V.; Lozanski, A.; Davis, M.; Gordon, A.L.; Smith, L.L.; et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients with Chronic Lymphocytic Leukemia. JAMA Oncol. 2015, 1, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Anderson, M.A.; Gong, J.-N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2018, 9, 342–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guièze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernández-Sánchez, M.; Zurita, A.M.; Sun, J.; Hacken, E.T.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384.e13. [Google Scholar] [CrossRef] [PubMed]
- Guarini, A.; Peragine, N.; Messina, M.; Marinelli, M.; Ilari, C.; Cafforio, L.; Raponi, S.; Bonina, S.; Mariglia, P.; Mauro, F.R.; et al. Unravelling the suboptimal response ofTP53-mutated chronic lymphocytic leukaemia to ibrutinib. Br. J. Haematol. 2018, 184, 392–396. [Google Scholar] [CrossRef] [Green Version]
- Chigrinova, E.; Rinaldi, A.; Kwee, I.; Rossi, D.; Rancoita, P.; Strefford, P.J.C.; Oscier, D.; Stamatopoulos, K.; Papadaki, T.; Berger, F.; et al. Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood 2013, 122, 2673–2682. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, G.; Khiabanian, H.; Holmes, A.B.; Wang, J.; Messina, M.; Mullighan, C.G.; Pasqualucci, L.; Rabadan, R.; Dalla-Favera, R. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J. Exp. Med. 2013, 210, 2273–2288. [Google Scholar] [CrossRef] [Green Version]
- Klintman, J.; Appleby, N.; Stamatopoulos, B.; Ridout, K.; Eyre, T.A.; Robbe, P.; Pascua, L.L.; Knight, S.J.L.; Dreau, H.; Cabes, M.; et al. Genomic and transcriptomic correlates of Richter transformation in chronic lymphocytic leukemia. Blood 2021, 137, 2800–2816. [Google Scholar] [CrossRef]
- Kojima, K.; Konopleva, M.; McQueen, T.; O’Brien, S.; Plunkett, W.; Andreeff, M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood 2006, 108, 993–1000. [Google Scholar] [CrossRef]
- Coll-Mulet, L.; Iglesias-Serret, D.; Santidrián, A.F.; Cosialls, A.M.; de Frias, M.; Castaño, E.; Campàs, C.; Barragán, M.; de Sevilla, A.F.; Domingo, A.; et al. MDM2 antagonists activate p53 and synergize with genotoxic drugs in B-cell chronic lymphocytic leukemia cells. Blood 2006, 107, 4109–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saddler, C.; Ouillette, P.; Kujawski, L.; Shangary, S.; Talpaz, M.; Kaminski, M.; Erba, H.; Shedden, K.; Wang, S.; Malek, S.N. Comprehensive biomarker and genomic analysis identifies p53 status as the major determinant of response to MDM2 inhibitors in chronic lymphocytic leukemia. Blood 2008, 111, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Ciardullo, C.; Aptullahoglu, E.; Woodhouse, L.; Lin, W.-Y.; Wallis, J.P.; Marr, H.; Marshall, S.; Bown, N.; Willmore, E.; Lunec, J. Non-genotoxic MDM2 inhibition selectively induces a pro-apoptotic p53 gene signature in chronic lymphocytic leukemia cells. Haematologica 2019, 104, 2429–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvi, A.J.; Austen, B.; Weston, V.J.; Fegan, C.; MacCallum, D.; Gianella-Borradori, A.; Lane, D.P.; Hubank, M.; Powell, J.E.; Wei, W.; et al. A novel CDK inhibitor, CYC202 (R-roscovitine), overcomes the defect in p53-dependent apoptosis in B-CLL by down-regulation of genes involved in transcription regulation and survival. Blood 2005, 105, 4484–4491. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Keating, M.J.; Gandhi, V.; Plunkett, W. Transcription inhibition by flavopiridol: Mechanism of chronic lymphocytic leukemia cell death. Blood 2005, 106, 2513–2519. [Google Scholar] [CrossRef]
- Chen, R.; Wierda, W.G.; Chubb, S.; Hawtin, R.E.; Fox, J.A.; Keating, M.J.; Gandhi, V.; Plunkett, W. Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood 2009, 113, 4637–4645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Germano, S.; Clements, C.; Samuel, J.; Shelmani, G.; Jayne, S.; Dyer, M.; Macip, S. Pro-survival signal inhibition by CDK inhibitor dinaciclib in Chronic Lymphocytic Leukaemia. Br. J. Haematol. 2016, 175, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Jaskova, Z.; Pavlova, S.; Malcikova, J.; Brychtova, Y.; Trbusek, M. PRIMA-1(MET) cytotoxic effect correlates with p53 protein reduction in TP53-mutated chronic lymphocytic leukemia cells. Leuk. Res. 2020, 89, 106288. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.; Rockliffe, N.; Johnson, G.G.; Sherrington, P.D.; Pettitt, A.R. Hsp90 inhibition has opposing effects on wild-type and mutant p53 and induces p21 expression and cytotoxicity irrespective of p53/ATM status in chronic lymphocytic leukaemia cells. Oncogene 2007, 27, 2445–2455. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, G.; Pelicano, H.; Liao, J.; Huang, J.; Feng, L.; Keating, M.J.; Huang, P. Targeting p53-deficient chronic lymphocytic leukemia cells in vitro and in vivo by ROS-mediated mechanism. Oncotarget 2016, 7, 71378–71389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trachootham, D.; Zhang, H.; Zhang, W.; Feng, L.; Du, M.; Zhou, Y.; Chen, Z.; Pelicano, H.; Plunkett, W.; Wierda, W.G.; et al. Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism. Blood 2008, 112, 1912–1922. [Google Scholar] [CrossRef]
- Agathanggelou, A.; Weston, V.J.; Perry, T.; Davies, N.J.; Skowronska, A.; Payne, D.T.; Fossey, J.S.; Oldreive, C.E.; Wei, W.; Pratt, G.; et al. Targeting the Ataxia Telangiectasia Mutated-null phenotype in chronic lymphocytic leukemia with pro-oxidants. Haematologica 2015, 100, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [Green Version]
- Boudny, M.; Zemanova, J.; Khirsariya, P.; Borsky, M.; Verner, J.; Cerna, J.; Oltova, A.; Seda, V.; Mraz, M.; Jaros, J.; et al. Novel CHK1 inhibitor MU380 exhibits significant single-agent activity in TP53-mutated chronic lymphocytic leukemia cells. Haematologica 2019, 104, 2443–2455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemanova, J.; Hylse, O.; Collakova, J.; Vesely, P.; Oltova, A.; Borsky, M.; Zaprazna, K.; Kasparkova, M.; Janovska, P.; Verner, J.; et al. Chk1 inhibition significantly potentiates activity of nucleoside analogs in TP53-mutated B-lymphoid cells. Oncotarget 2016, 7, 62091–62106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, V.J.; Oldreive, C.E.; Skowronska, A.; Oscier, D.G.; Pratt, G.; Dyer, M.J.S.; Smith, G.; Powell, J.E.; Rudzki, Z.; Kearns, P.; et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 2010, 116, 4578–4587. [Google Scholar] [CrossRef] [Green Version]
- Riabinska, A.; Daheim, M.; Herter-Sprie, G.S.; Winkler, J.; Fritz, C.; Hallek, M.; Thomas, R.K.; Kreuzer, K.-A.; Frenzel, L.P.; Monfared, P.; et al. Therapeutic Targeting of a Robust Non-Oncogene Addiction to PRKDC in ATM-Defective Tumors. Sci. Transl. Med. 2013, 5, 189ra78. [Google Scholar] [CrossRef]
- Willmore, E.; Elliott, S.L.; Mainou-Fowler, T.; Summerfield, G.P.; Jackson, G.H.; O’Neill, F.; Lowe, C.; Carter, A.; Harris, R.; Pettitt, A.R.; et al. DNA-Dependent Protein Kinase Is a Therapeutic Target and an Indicator of Poor Prognosis in B-Cell Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2008, 14, 3984–3992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agathanggelou, A.; Smith, E.; Davies, N.J.; Kwok, M.; Zlatanou, A.; Oldreive, C.E.; Mao, J.; Da Costa, D.; Yadollahi, S.; Perry, T.; et al. USP7 inhibition alters homologous recombination repair and targets CLL cells independently of ATM/p53 functional status. Blood 2017, 130, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Shangary, S.; Wang, S. Targeting the MDM2-p53 Interaction for Cancer Therapy. Clin. Cancer Res. 2008, 14, 5318–5324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gryshchenko, I.; Hofbauer, S.; Stoecher, M.; Daniel, P.T.; Steurer, M.; Gaiger, A.; Eigenberger, K.; Greil, R.; Tinhofer, I. MDM2 SNP309 Is Associated with Poor Outcome in B-Cell Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2008, 26, 2252–2257. [Google Scholar] [CrossRef]
- Asslaber, D.; Piñón, J.D.; Seyfried, I.; Desch, P.; Stöcher, M.; Tinhofer, I.; Egle, A.; Merkel, O.; Greil, R. microRNA-34a expression correlates with MDM2 SNP309 polymorphism and treatment-free survival in chronic lymphocytic leukemia. Blood 2010, 115, 4191–4197. [Google Scholar] [CrossRef] [Green Version]
- Ljungström, V.; Cortese, D.; Young, E.; Pandzic, T.; Mansouri, L.; Plevova, K.; Ntoufa, S.; Baliakas, P.; Clifford, R.; Sutton, L.-A.; et al. Whole-exome sequencing in relapsing chronic lymphocytic leukemia: Clinical impact of recurrent RPS15 mutations. Blood 2016, 127, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Brummelkamp, T.R.; Fabius, A.W.M.; Mullenders, J.; Madiredjo, M.; Velds, A.; Kerkhoven, R.M.; Bernards, R.; Beijersbergen, R. An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat. Chem. Biol. 2006, 2, 202–206. [Google Scholar] [CrossRef] [Green Version]
- Andreeff, M.; Kelly, K.R.; Yee, K.W.; Assouline, S.; Strair, R.K.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2015, 22, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Ringshausen, I.; O’Shea, C.C.; Finch, A.J.; Swigart, L.B.; Evan, G.I. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell 2005, 10, 501–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, M.H.; Shen, H.; Maki, C.G. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene 2011, 30, 4678–4686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Suh, Y.-A.; Fuller, M.Y.; Jackson, J.; Xiong, S.; Terzian, T.; Quintás-Cardama, A.; Bankson, J.; El-Naggar, A.K.; Lozano, G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J. Clin. Investig. 2011, 121, 893–904. [Google Scholar] [CrossRef]
- Chapeau, E.A.; Gembarska, A.; Durand, E.Y.; Mandon, E.; Estadieu, C.; Romanet, V.; Wiesmann, M.; Tiedt, R.; Lehar, J.; de Weck, A.; et al. Resistance mechanisms to TP53-MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an Arf mouse model. Proc. Natl. Acad. Sci. USA 2017, 114, 3151–3156. [Google Scholar] [CrossRef] [Green Version]
- Byrd, J.C.; Lin, T.S.; Dalton, J.T.; Wu, D.; Phelps, M.A.; Fischer, B.; Moran, M.; Blum, K.A.; Rovin, B.; Brooker-McEldowney, M.; et al. Flavopiridol administered using a pharmacologically derived schedule is associated with marked clinical efficacy in refractory, genetically high-risk chronic lymphocytic leukemia. Blood 2006, 109, 399–404. [Google Scholar] [CrossRef]
- Tong, W.-G.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.D.; Badros, A.Z.; Popplewell, L.; Coutre, S.; Fox, J.A.; et al. Phase I and Pharmacologic Study of SNS-032, a Potent and Selective Cdk2, 7, and 9 Inhibitor, in Patients with Advanced Chronic Lymphocytic Leukemia and Multiple Myeloma. J. Clin. Oncol. 2010, 28, 3015–3022. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Jones, J.; Johnson, A.J.; Andritsos, L.A.; Maddocks, K.; Jaglowski, S.; Hessler, J.; Grever, M.R.; Im, E.; Zhou, H.; et al. Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia 2015, 29, 1524–1529. [Google Scholar] [CrossRef] [Green Version]
- Ghia, P.; Scarfò, L.; Perez, S.; Pathiraja, K.; DeRosier, M.; Small, K.; Sisk, C.M.; Patton, N. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 2017, 129, 1876–1878. [Google Scholar] [CrossRef] [PubMed]
- Deneberg, S.; Cherif, H.; Lazarevic, V.; Andersson, P.-O.; Von Euler, M.; Juliusson, G.; Lehmann, S. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016, 6, e447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, G.; Yap, C.; Oldreive, C.; Slade, D.; Bishop, R.; Griffiths, M.; Dyer, M.; Fegan, C.; Oscier, D.; Pettitt, A.; et al. A multi-centre phase I trial of the PARP inhibitor olaparib in patients with relapsed chronic lymphocytic leukaemia, T-prolymphocytic leukaemia or mantle cell lymphoma. Br. J. Haematol. 2017, 182, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.; Mita, M.; Mahipal, A.; Nemunaitis, J.; Massard, C.; Mikkelsen, T.; Cruz, C.; Paz-Ares, L.; Hidalgo, M.; Rathkopf, D.; et al. First-In-Human Phase I Study of a Dual mTOR Kinase And DNA-PK Inhibitor (CC-115) In Advanced Malignancy. Cancer Manag. Res. 2019, 11, 10463–10476. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Burkhardt, U.E.; Hainz, U.; Stevenson, K.; Goldstein, N.R.; Pasek, M.; Naito, M.; Wu, D.; Ho, V.T.; Alonso, A.; Hammond, N.N.; et al. Autologous CLL cell vaccination early after transplant induces leukemia-specific T cells. J. Clin. Investig. 2013, 123, 3756–3765. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, T.; Hubank, M.; Cronin, D.; Stewart, G.S.; Fletcher, D.; Bignell, C.R.; Alvi, A.J.; Austen, B.; Weston, V.J.; Fegan, C.; et al. Microarray analysis reveals that TP53- and ATM-mutant B-CLLs share a defect in activating proapoptotic responses after DNA damage but are distinguished by major differences in activating prosurvival responses. Blood 2004, 103, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.J.; Yeh, Y.-Y.; Smith, L.L.; Wagner, A.J.; Hessler, J.; Gupta, S.; Flynn, J.M.; Jones, J.; Zhang, X.; Bannerji, R.; et al. The novel cyclin-dependent kinase inhibitor dinaciclib (SCH727965) promotes apoptosis and abrogates microenvironmental cytokine protection in chronic lymphocytic leukemia cells. Leukemia 2012, 26, 2554–2557. [Google Scholar] [CrossRef] [Green Version]
- Kitada, S.; Zapata, J.M.; Andreeff, M.; Reed, J.C. Protein kinase inhibitors flavopiridol and 7-hydroxy-staurosporine down-regulate antiapoptosis proteins in B-cell chronic lymphocytic leukemia. Blood 2000, 96, 393–397. [Google Scholar] [CrossRef]
- Nahi, H.; Lehmann, S.; Mollgard, L.; Bengtzen, S.; Selivanova, G.; Wiman, K.; Paul, C.; Merup, M. Effects of PRIMA-1 on chronic lymphocytic leukaemia cells with and without hemizygous p53 deletion. Br. J. Haematol. 2004, 127, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system x(C)(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, S.; Bykov, V.N.; Ali, D.; Andrén, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in Vivo: A First-in-Human Study With p53-Targeting Compound APR-246 in Refractory Hematologic Malignancies and Prostate Cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, F.H.S.M.M.B.J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Diedrich, D.; Frieg, B.; Ahlert, H.; Stein, S.; Bopp, B.; Lang, F.; Zhang, T.; Kröger, T.; Ernst, T.; et al. Targeting HSP90 dimerization via the C terminus is effective in imatinib-resistant CML and lacks the heat shock response. Blood 2018, 132, 307–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingallina, E.; Sorrentino, G.; Bertolio, R.; Lisek, K.; Zannini, A.; Azzolin, L.; Severino, L.U.; Scaini, D.; Mano, M.; Mantovani, F.; et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell Biol. 2018, 20, 28–35. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neil, N.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2019, 19, 23–38. [Google Scholar] [CrossRef]
- Samuel, J.; Jayne, S.; Chen, Y.; Majid, A.; Wignall, A.; Wormull, T.; Najeeb, H.; Luo, J.-L.; Jones, G.D.; Macip, S.; et al. Posttranscriptional Upregulation of p53 by Reactive Oxygen Species in Chronic Lymphocytic Leukemia. Cancer Res. 2016, 76, 6311–6319. [Google Scholar] [CrossRef] [Green Version]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Li, S.; Swaroop, M.; Guan, K.; Oberley, L.W.; Sun, Y. Transcriptional Activation of the Human Glutathione Peroxidase Promoter by p53. J. Biol. Chem. 1999, 274, 12061–12066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, K.-A.; Nakamura, Y.; Arakawa, H. Identification of ALDH4 as a p53-inducible gene and its protective role in cellular stresses. J. Hum. Genet. 2004, 49, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Sablina, A.A.; Feinstein, E.; Koonin, E.V.; Chumakov, P. Regeneration of Peroxiredoxins by p53-Regulated Sestrins, Homologs of Bacterial AhpD. Science 2004, 304, 596–600. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z.H. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Kozlov, S.; Lavin, M.; Person, M.D.; Paull, T.T. ATM Activation by Oxidative Stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tripathi, D.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.-H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nature 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lee, J.-H.; Paull, T.T.; Gehrke, S.; D’Alessandro, A.; Dou, Q.; Gladyshev, V.N.; Schroeder, E.A.; Steyl, S.K.; Christian, B.E.; et al. Mitochondrial redox sensing by the kinase ATM maintains cellular antioxidant capacity. Sci. Signal. 2018, 11, eaaq0702. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2010, 30, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013, 210, 1529–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.-Y.; Graves, P.R.; Thoma, R.S.; Wu, Z.; Shaw, A.S.; Piwnica-Worms, H. Mitotic and G2 Checkpoint Control: Regulation of 14-3-3 Protein Binding by Phosphorylation of Cdc25C on Serine-216. Science 1997, 277, 1501–1505. [Google Scholar] [CrossRef]
- Sorensen, C.; Syljuåsen, R.G.; Falck, J.; Schroeder, T.; Rönnstrand, L.; Khanna, K.K.; Zhou, B.-B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Dungrawala, H.; Rose, K.L.; Bhat, K.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 2015, 59, 998–1010. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.Q.; Blow, J.J. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J. Cell Biol. 2010, 191, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-H.; Jones, M.J.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J.; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-Mediated Phosphorylation of FANCI Regulates Dormant Origin Firing in Response to Replication Stress. Mol. Cell 2015, 58, 323–338. [Google Scholar] [CrossRef] [Green Version]
- Kwok, M.; Stankovic, T. Targeting the Ataxia Telangiectasia and Rad3 Signaling Pathway to Overcome Chemoresistance in Cancer. In Targeting Cell Survival Pathways to Enhance Response to Chemotherapy; Johnson, D.E., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 203–230. [Google Scholar] [CrossRef]
- Kwok, M.; Stankovic, T. Targeting replication stress in sporadic tumors. In DNA Replication and Double Strand Break Repair: Mechanisms and Clinical Significance; Grand, R.J., Ed.; Garland Science: New York, NY, USA, 2019; pp. 329–337. [Google Scholar]
- Toledo, L.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Castedo, M.; Perfettini, J.L.; Roumie, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [Green Version]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.G.; Reaper, P.M.; Pettitt, A.R.; Sherrington, P.D. The ATR-p53 pathway is suppressed in noncycling normal and malignant lymphocytes. Oncogene 2004, 23, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; El-Khouiery, A.; Soria, J.-C.; Lopez, J.; Berges, A.; Cheung, S.A.; Irurzun-Arana, I.; Goldwin, A.; et al. Ceralasertib (AZD6738), an Oral ATR Kinase Inhibitor, in Combination with Carboplatin in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Kim, S.T.; Smith, S.A.; Mortimer, P.; Loembé, A.-B.; Cho, H.; Kim, K.-M.; Smith, C.; Willis, S.; Irurzun-Arana, I.; Berges, A.; et al. Phase I Study of Ceralasertib (AZD6738), a Novel DNA Damage Repair Agent, in Combination with Weekly Paclitaxel in Refractory Cancer. Clin. Cancer Res. 2021, 27, 4700–4709. [Google Scholar] [CrossRef]
- Gilad, O.; Nabet, B.; Ragland, R.L.; Schoppy, D.W.; Smith, K.D.; Durham, A.C.; Brown, E.J. Combining ATR Suppression with Oncogenic Ras Synergistically Increases Genomic Instability, Causing Synthetic Lethality or Tumorigenesis in a Dosage-Dependent Manner. Cancer Res. 2010, 70, 9693–9702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.Y.; Tan, K.V.; Cornelissen, B. PARP Inhibitors in Cancer Diagnosis and Therapy. Clin. Cancer Res. 2020, 27, 1585–1594. [Google Scholar] [CrossRef]
- Knittel, G.; Rehkämper, T.; Korovkina, D.; Liedgens, P.; Fritz, C.; Torgovnick, A.; Al-Baldawi, Y.; Al-Maarri, M.; Cun, Y.; Fedorchenko, O.; et al. Two mouse models reveal an actionable PARP1 dependence in aggressive chronic lymphocytic leukemia. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Herriott, A.; Tudhope, S.J.; Junge, G.; Rodrigues, N.; Patterson, M.J.; Woodhouse, L.; Lunec, J.; Hunter, J.E.; Mulligan, E.A.; Cole, M.; et al. PARP1 expression, activity and ex vivo sensitivity to the PARP inhibitor, talazoparib (BMN 673), in chronic lymphocytic leukaemia. Oncotarget 2015, 6, 43978–43991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, M.; Murina, O.; Reijns, M.; Agathanggelou, A.; Challis, R.; Tarnauskaitė, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef]
- Klein, U.; Lia, M.; Crespo, M.; Siegel, R.; Shen, Q.; Mo, T.; Ambesi-Impiombato, A.; Califano, A.; Migliazza, A.; Bhagat, G.; et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 2010, 17, 28–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerritelli, S.M.; Crouch, R.J. The Balancing Act of Ribonucleotides in DNA. Trends Biochem. Sci. 2016, 41, 434–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; O’Connor, M.J.; De Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- Deriano, L.; Guipaud, O.; Merle-Béral, H.; Binet, J.L.; Ricoul, M.; Potocki-Veronese, G.; Favaudon, V.; Mac-iorowski, Z.; Muller, C.; Salles, B.; et al. Human chronic lymphocytic leukemia B cells can escape DNA damage-induced apoptosis through the nonhomologous end-joining DNA repair pathway. Blood 2005, 105, 4776–4783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijssen, R.; Ter Burg, J.; Garrick, B.; van Bochove, G.G.; Brown, J.R.; Fernandes, S.M.; Rodríguez, M.S.; Michot, J.-M.; Hallek, M.; Eichhorst, B.; et al. Dual TORK/DNA-PK inhibition blocks critical signaling pathways in chronic lymphocytic leukemia. Blood 2016, 128, 574–583. [Google Scholar] [CrossRef]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A Dynamic Role of HAUSP in the p53-Mdm2 Pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar] [CrossRef]
- Turnbull, A.P.; Ioannidis, S.; Krajewski, W.W.; Pinto-Fernandez, A.; Heride, C.; Martin, A.C.L.; Tonkin, L.M.; Townsend, E.C.; Buker, S.M.; Lancia, J.D.; et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 2017, 550, 481–486. [Google Scholar] [CrossRef]
- Kategaya, L.; Di Lello, P.; Rougé, L.; Pastor, R.; Clark, K.R.; Drummond, J.; Kleinheinz, T.; Lin, E.; Upton, J.-P.; Prakash, S.; et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 2017, 550, 534–538. [Google Scholar] [CrossRef] [Green Version]
- Carrá, G.; Panuzzo, C.; Torti, D.; Parvis, G.; Crivellaro, S.; Familiari, U.; Volante, M.; Morena, D.; Lingua, M.F.; Brancaccio, M.; et al. Therapeutic inhibition of USP7-PTEN network in chronic lymphocytic leukemia: A strategy to overcome TP53 mutated/deleted clones. Oncotarget 2017, 8, 35508–35522. [Google Scholar] [CrossRef] [Green Version]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef]
- Agathangelidis, A.; Ljungström, V.; Scarfo’, L.; Fazi, C.; Gounari, M.; Pandzic, T.; Sutton, L.-A.; Stamatopoulos, K.; Tonon, G.; Rosenquist, R.; et al. Highly similar genomic landscapes in monoclonal B-cell lymphocytosis and ultra-stable chronic lymphocytic leukemia with low frequency of driver mutations. Haematologica 2018, 103, 865–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsay, A.G.; Johnson, A.J.; Lee, A.M.; Görgün, G.; Le Dieu, R.; Blum, W.; Byrd, J.C.; Gribben, J.G. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J. Clin. Investig. 2008, 118, 2427–2437. [Google Scholar] [CrossRef]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef]
- Pascutti, M.F.; Jak, M.; Tromp, J.M.; Derks, I.A.M.; Remmerswaal, E.B.M.; Thijssen, R.; Van Attekum, M.H.A.; Van Bochove, G.G.; Luijks, D.M.; Pals, S.T.; et al. IL-21 and CD40L signals from autologous T cells can induce antigen-independent proliferation of CLL cells. Blood 2013, 122, 3010–3019. [Google Scholar] [CrossRef] [Green Version]
- Rajasagi, M.; Shukla, S.A.; Fritsch, E.F.; Keskin, D.B.; DeLuca, D.; Carmona, E.; Zhang, W.; Sougnez, C.; Cibulskis, K.; Sidney, J.; et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 2014, 124, 453–462. [Google Scholar] [CrossRef]
- Kowalewski, D.J.; Schuster, H.; Backert, L.; Berlin, C.; Kahn, S.; Kanz, L.; Salih, H.R.; Rammensee, H.G.; Steva-novic, S.; Stickel, J.S. HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc. Natl. Acad. Sci. USA 2015, 112, E6258–E6260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, B.S.; Roessner, P.M.; Yazdanparast, H.; Colomer, D.; Campo, E.; Kugler, S.; Yosifov, D.; Stilgenbauer, S.; Schmidt, M.; Gabriel, R.; et al. Control of chronic lymphocytic leukemia development by clonally-expanded CD8+ T-cells that undergo functional exhaustion in secondary lymphoid tissues. Leukemia 2018, 33, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-cell Responses to TP53 “Hotspot” Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin. Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malekzadeh, P.; Yossef, R.; Cafri, G.; Paria, B.C.; Lowery, F.J.; Jafferji, M.; Good, M.L.; Sachs, A.; Copeland, A.; Kim, S.P.; et al. Antigen Experienced T Cells from Peripheral Blood Recognize p53 Neoantigens. Clin. Cancer Res. 2020, 26, 1267–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.K.; Anker, J.F.; Oh, M.S.; Bais, P.; Namburi, S.; Agte, S.; Giles, F.J.; Chuang, J.H. Mutations in DNA repair genes are associated with increased neoantigen burden and a distinct immunophenotype in lung squamous cell carcinoma. Sci. Rep. 2019, 9, 7949–7960. [Google Scholar] [CrossRef]
- Zhu, K.; Wang, J.; Zhu, J.; Jiang, J.; Shou, J.; Chen, X. p53 induces TAP1 and enhances the transport of MHC class I peptides. Oncogene 1999, 18, 7740–7747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Niu, D.D.; Lai, L.Y.; Ren, E.C. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat. Commun. 2013, 4, 2359. [Google Scholar] [CrossRef]
- Textor, S.; Fiegler, N.; Arnold, A.; Porgador, A.; Hofmann, T.G.; Cerwenka, A. Human NK Cells Are Alerted to Induction of p53 in Cancer Cells by Upregulation of the NKG2D Ligands ULBP1 and ULBP2. Cancer Res. 2011, 71, 5998–6009. [Google Scholar] [CrossRef] [Green Version]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.H.; Peltier, H.J.; Ye, Y.P.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar] [CrossRef] [Green Version]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.; Saha, S.; Bettke, J.; Nagar, R.; Parrales, A.; Iwakuma, T.; van der Velden, A.W.; Martinez, L.A. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 2021, 39, 494–508.e5. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Hon-da, K.; et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003, 424, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Marcus, A.; Mao, A.J.; Lensink-Vasan, M.; Wang, L.; Vance, R.E.; Raulet, D.H. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 2018, 49, 754–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schadt, L.; Sparano, C.; Schweiger, N.A.; Silina, K.; Cecconi, V.; Lucchiari, G.; Yagita, H.; Guggisberg, E.; Saba, S.; Nascakova, Z.; et al. Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 2019, 29, 1236–1248.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jonsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappa B Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Ding, W.; LaPlant, B.R.; Call, T.G.; Parikh, S.A.; Leis, J.F.; He, R.; Shanafelt, T.D.; Sinha, S.; Le-Rademacher, J.; Feldman, A.L.; et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood 2017, 129, 3419–3427. [Google Scholar] [CrossRef] [PubMed]
- Parry, H.M.; Mirajkar, N.; Cutmore, N.; Zuo, J.; Long, H.; Kwok, M.; Oldrieve, C.; Hudson, C.; Stankovic, T.; Paneesha, S.; et al. Long-Term Ibrutinib Therapy Reverses CD8+ T Cell Exhaustion in B Cell Chronic Lymphocytic Leukaemia. Front. Immunol. 2019, 10, 2832. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Goyama, S.; Liu, X.X.; Tamura, M.; Asada, S.; Tanaka, Y.; Tomofusa Fukuyama, T.; Wunderlich, M.; O’Brien, E.; Mizukawa, B.; et al. Antitumor immunity augments the therapeutic effects of p53 activation on acute myeloid leukemia. Nat. Commun. 2019, 10, 4869. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Q.; Mulford, I.J.; Sharp, F.; Liang, J.; Kurtulus, S.; Trabucco, G.; Quinn, D.S.; Longmire, T.A.; Patel, N.; Patil, R.; et al. Inhibition of MDM2 Promotes Antitumor Responses in p53 Wild-Type Cancer Cells through Their Interaction with the Immune and Stromal Microenvironment. Cancer Res. 2021, 81, 3079–3091. [Google Scholar] [CrossRef]
- Zhou, J.; Kryczek, I.; Li, S.; Li, X.; Aguilar, A.; Wei, S.; Grove, S.; Vatan, L.; Yu, J.; Yan, Y.; et al. The ubiquitin ligase MDM2 sustains STAT5 stability to control T cell-mediated antitumor immunity. Nat. Immunol. 2021, 22, 460–470. [Google Scholar] [CrossRef]
- Vendetti, F.P.; Karukonda, P.; Clump, D.A.; Teo, T.; LaLonde, R.; Nugent, K.; Ballew, M.; Kiesel, B.F.; Beumer, J.H.; Sarkar, S.N.; et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell–dependent antitumor activity following radiation. J. Clin. Investig. 2018, 128, 3926–3940. [Google Scholar] [CrossRef]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.; Paget, J.T.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Harding, S.M.; Natesan, R.; Tian, L.; Benci, J.L.; Li, W.; Minn, A.J.; Asangani, I.A.; Greenberg, R.A. Cell Cycle Checkpoints Cooperate to Suppress DNA- and RNA-Associated Molecular Pattern Recognition and Anti-Tumor Immune Responses. Cell Rep. 2020, 32, 108080. [Google Scholar] [CrossRef] [PubMed]
- Farkkila, A.; Gulhan, D.C.; Casado, J.; Jacobson, C.A.; Nguyen, H.; Kochupurakkal, B.; Zoltan Maliga, Z.; Yapp, C.; Chen, Y.-A.; Shapiro, D.; et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat. Commun. 2020, 11, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.-A.; Park, Y.H.; Delord, J.-P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Gángó, A.; Alpar, D.; Galik, B.; Marosvári, D.; Kiss, R.; Fésüs, V.; Aczél, D.; Eyüpoglu, E.; Nagy, N.; Nagy, Á.; et al. Dissection of subclonal evolution by temporal mutation profiling in chronic lymphocytic leukemia patients treated with ibrutinib. Int. J. Cancer 2020, 146, 85–93. [Google Scholar] [CrossRef]
- Herling, C.D.; Abedpour, N.; Weiss, J.; Schmitt, A.; Jachimowicz, R.D.; Merkel, O.; Cartolano, M.; Oberbeck, S.; Mayer, P.; Berg, V.; et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat. Commun. 2018, 9, 727. [Google Scholar] [CrossRef] [PubMed]
- Voltan, R.; Rimondi, E.; Melloni, E.; Rigolin, G.M.; Casciano, F.; Arcidiacono, M.V.; Celeghini, C.; Cuneo, A.; Zauli, G.; Secchiero, P. Ibrutinib synergizes with MDM-2 inhibitors in promoting cytotoxicity in B chronic lymphocytic leukemia. Oncotarget 2016, 7, 70623–70638. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Tsai, J.; Thompson, P.A.; Chen, Y.; Xiong, P.; Liu, C.; Burrows, F.; Sivina, M.; Burger, J.A.; Keating, M.J.; et al. The multi-kinase inhibitor TG02 induces apoptosis and blocks B-cell receptor signaling in chronic lymphocytic leukemia through dual mechanisms of action. Blood Cancer J. 2021, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Quijada-Álamo, M.; Hernández-Sánchez, M.; Alonso-Pérez, V.; Rodríguez-Vicente, A.E.; García-Tuñón, I.; Martín-Izquierdo, M.; Hernández-Sánchez, J.M.; Herrero, A.B.; Bastida, J.M.; Segundo, L.S.; et al. CRISPR/Cas9-generated models uncover therapeutic vulnerabilities of del(11q) CLL cells to dual BCR and PARP inhibition. Leukemia 2020, 34, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.; Konopleva, M.; Andreeff, M. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 2017, 32, 748–760.e746. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutation Frequency | Deletion Frequency | Number of Patients Analyzed | Reference |
|---|---|---|---|---|
| ND | 18% | 325 | Döhner et al., 2000 [17] | |
| 32% | 4% | 50 | Stankovic et al., 2002 [13] | |
| 12% | 3% | 155 | Austen et al., 2005 [14] | |
| ATM | ND | 22% | 330 | Malcikova et al., 2009 [15] |
| 14.7% | 30% | 224 | Skowronska et al., 2012 [19] | |
| 8% | 15% | 160 | Landau et al., 2013 [25] | |
| 15% | 22% | 538 | Landau et al., 2015 [26] | |
| ND | 7% | 325 | Döhner et al., 2000 [17] | |
| 12% | 6% | 50 | Stankovic et al., 2002 [13] | |
| 4% | ND | 155 | Austen et al., 2005 [14] | |
| 5% | 11% | 400 | Malcikova et al., 2009 [15] | |
| TP53 | 8.5% | 5% | 328 | Zenz et al., 2010 [16] |
| 7.6% | 6% | 529 | Gonzalez et al., 2011 [27] | |
| 15% | ND | 309 | Rossi et al., 2014 [28] | |
| 11.5% | 7% | 635 | Stilgenbauer et al., 2014 [29] | |
| 13% | 13% | 160 | Landau et al., 2013 [25] | |
| 7% | 6.3% | 538 | Landau et al., 2015 [26] |
| Compound | Therapeutic Strategy | Cellular Effect | Reference |
|---|---|---|---|
| Targeting MDM2/p53 Axis | |||
| Nutlin (RG7388) | MDM2 inhibition | Stabilization of wild-type p53, induction of p53 target genes and p53-mediated apoptosis in ATM deficient tumors | Kojima et al., 2006 [53] Coll-Mulet et al., 2006 [54] Saddler et al., 2008 [55] Ciardullo et al., 2019 [56] |
| Restoration of p53 tumor suppressor function | |||
| Roscovitine (CYC202) Flavopiridol SNS-032 Dinaciclib | Pan-CDK inhibition | Suppression of p53-dependent pro-survival transcription in ATM- and p53-deficient tumors | Alvi et al., 2005 [57] Chen et al., 2005 [58] Chen et al., 2009 [59] Chen et al., 2016 [60] |
| PRIMA 1 APR-246 | Refolding of mutant p53 | Restoration of p53 wild- type properties and induction of cytotoxicity | Nahi et al., 2004 [61] Jaskova et al., 2020 [62] |
| Geldanamycin | HSP90 inhibition | Destabilization and degradation of mutant p53 and induction of cytotoxicity | Alexandrova et al., 2015 [63] Lin et al., 2008 [64] |
| Synthetic lethality | |||
| PEITC Parthenolide | Depletion of cellular glutathione Induction of ROS | Exacerbation of oxidative stress to intolerable levels in p53- and ATM-deficient tumors | Liu et al., 2016 [65] Trachootham et al., 2008 [66] Agathanggelou et al., 2015 [67] |
| ATR inhibitor AZD6738 (ceralasertib) | Exploiting synthetically lethal interaction between ATR and ATM or p53 | Exacerbation of replication stress in ATM- and p53-deficient tumors and induction of cellular death | Kwok et al., 2016 [68] |
| Chk1 inhibitor MU380 | Exploiting synthetically lethal interaction between Chk1 and p53 | Significant chemosensitization of TP53-mutant CLL cells and potentiation of nucleoside analog activity | Boudny et al., 2019 [69] Zemanova et al., 2016 [70] |
| PARP inhibitor olaparib | Exploiting synthetically lethal interaction between PARP1 and ATM | Exacerbation of unrepaired DNA damage and induction of cellular death | Weston et al., 2010 [71] |
| DNAPK inhibitors KU-0060648 and NU7441 | Exploiting synthetically lethal interaction between DNAPK and ATM | Exacerbation of unrepaired DNA damage and selective killing of ATM-defective CLL cells | Riabinska et al., 2013 [72] Willmore et al., 2008 [73] |
| USP7 inhibitor HBX19818 | Inhibition of HRR in ATM- and p53-deficient cells | Accumulation of DNA damage that leads to DNA fragmentation and necrotic cell death via unrestrained PARylation | Agathanggelou et al., 2017 [74] |
| Compound | Clinical Trial | Observation | Reference |
|---|---|---|---|
| Nutlin RG7112 | Phase I | Of 20 patients enrolled on the trial, one achieved partial response, whereas the majority maintained stable disease | Andreeff et al., 2016 [82] |
| Flavopiridol | Phase I | Successful induction of partial remission was observed in 45% of patients, with tumor lysis syndrome being the main dose-limiting toxicity | Byrd et al., 2007 [87] |
| SNS-032 | Phase I | A single CLL patient responded out of 19 patients enrolled in the trial | Tong et al., 2010 [88] |
| Dinaciclib | Phase I/II Phase III | Partial response was observed in 28 of 52 patients with relapsed CLL in the first study [89], and in 8 of 20 patients in the second study [90] | Flynn et al., 2015 [89] Ghia et al., 2017 [90] |
| APR-246 (PRIMA-1) | Phase I | This study involved refractory AML and CLL patients. Clinical response was observed in a single CLL patient. APR-246 was well tolerated, with the most common adverse effects being of neurological nature | Deneberg et al., 2016 [91] |
| APR-246 + venetoclax | Phase I | Ongoing clinical trial | NCT04419389 |
| ATR inhibitor ceralasertib + ibrutinib | Phase I | Ongoing clinical trial | NCT03328273 |
| PARP inhibitor olaparib | Phase I | Nine CLL patients were enrolled in this trial. While on twice-daily olaparib, patients with ATM pathway alterations displayed a longer median PFS of 83 days compared to 38 days among those with an intact ATM pathway | Pratt et al., 2017 [92] |
| CC-115, a dual TORK/DNA-PK inhibitor | Phase I | Among 8 patients with ATM defective relapsed/refractory CLL or small lymphocytic lymphoma (SLL), a partial response was observed in 3 patients | Munster et al., 2019 [93] |
| CAR T/NK therapy | Pilot Phase I/II | Substantial elimination of CLL tumor cells | Porter et al., 2011 [94] Porter et al., 2015 [95] Liu et al., 2020 [96] |
| Neoantigen vaccines | Phase I | CD8+ T cells from vaccinated patients react against autologous CLL tumor | Burkhardt et al., 2013 [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwok, M.; Agathanggelou, A.; Davies, N.; Stankovic, T. Targeting the p53 Pathway in CLL: State of the Art and Future Perspectives. Cancers 2021, 13, 4681. https://doi.org/10.3390/cancers13184681
Kwok M, Agathanggelou A, Davies N, Stankovic T. Targeting the p53 Pathway in CLL: State of the Art and Future Perspectives. Cancers. 2021; 13(18):4681. https://doi.org/10.3390/cancers13184681
Chicago/Turabian StyleKwok, Marwan, Angelo Agathanggelou, Nicholas Davies, and Tatjana Stankovic. 2021. "Targeting the p53 Pathway in CLL: State of the Art and Future Perspectives" Cancers 13, no. 18: 4681. https://doi.org/10.3390/cancers13184681
APA StyleKwok, M., Agathanggelou, A., Davies, N., & Stankovic, T. (2021). Targeting the p53 Pathway in CLL: State of the Art and Future Perspectives. Cancers, 13(18), 4681. https://doi.org/10.3390/cancers13184681