
Cis-Acting Factors Causing Secondary Epimutations: Impact on the Risk for Cancer and Other Diseases

 , , ,
, , ,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Origin and Consequences of Epimutations
3. Aberrant Methylation as the Initial Step of Carcinogenesis in Hereditary Cancers
4. Primary Epimutations and Environmental Factors
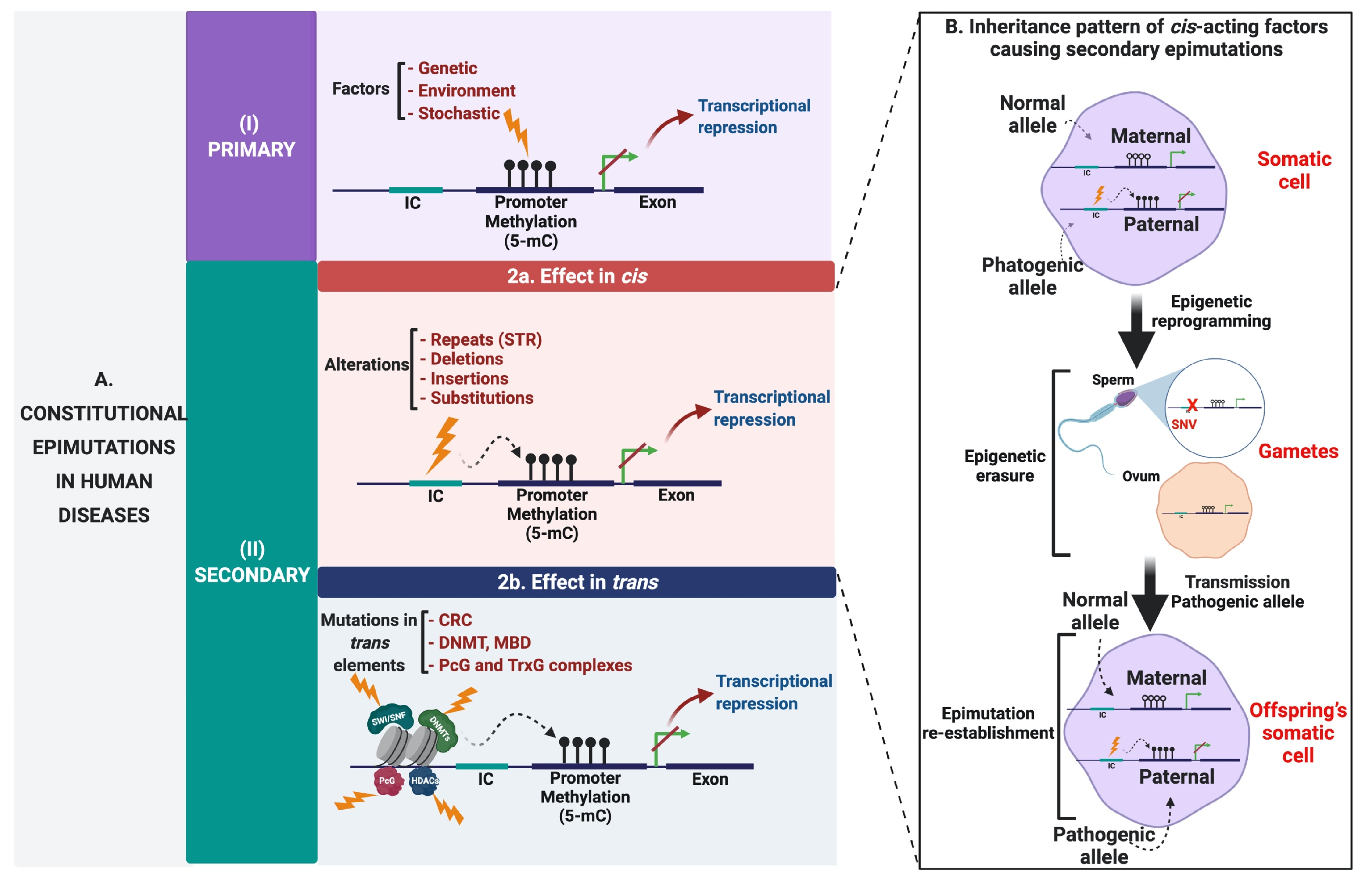
5. Secondary Epimutations
6. Cis-Acting Factors Causing Secondary Epimutations: Historical Evidence

7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2017, 18, 5–18. [Google Scholar] [CrossRef]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef]
- Ding, H.X.; Lv, Z.; Yuan, Y.; Xu, Q. MiRNA Polymorphisms and Cancer Prognosis: A Systematic Review and Meta-Analysis. Front. Oncol. 2018, 8, 596. [Google Scholar] [CrossRef]
- Carlevaro-Fita, J.; Lanzos, A.; Feuerbach, L.; Hong, C.; Mas-Ponte, D.; Pedersen, J.S.; Drivers, P.; Functional Interpretation, G.; Johnson, R.; Consortium, P. Cancer LncRNA Census reveals evidence for deep functional conservation of long noncoding RNAs in tumorigenesis. Commun. Biol. 2020, 3, 56. [Google Scholar] [CrossRef]
- Verduci, L.; Tarcitano, E.; Strano, S.; Yarden, Y.; Blandino, G. CircRNAs: Role in human diseases and potential use as biomarkers. Cell Death. Dis. 2021, 12, 468. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Hansen, T.B.; Veno, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Mir, M.; Bickmore, W.; Furlong, E.E.M.; Narlikar, G. Chromatin topology, condensates and gene regulation: Shifting paradigms or just a phase? Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Schuster-Böckler, B.; Lehner, B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 2012, 488, 504–507. [Google Scholar] [CrossRef]
- Smith, K.S.; Liu, L.L.; Ganesan, S.; Michor, F.; De, S. Nuclear topology modulates the mutational landscapes of cancer genomes. Nat. Struct. Mol. Biol. 2017, 24, 1000–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Entrevan, M.; Schuettengruber, B.; Cavalli, G. Regulation of Genome Architecture and Function by Polycomb Proteins. Trends Cell Biol. 2016, 26, 511–525. [Google Scholar] [CrossRef]
- Ito, T.; Teo, Y.V.; Evans, S.A.; Neretti, N.; Sedivy, J.M. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep. 2018, 22, 3480–3492. [Google Scholar] [CrossRef] [Green Version]
- Mills, A.A. Throwing the cancer switch: Reciprocal roles of polycomb and trithorax proteins. Nat. Rev. Cancer 2010, 10, 669–682. [Google Scholar] [CrossRef]
- Soares, J.; Pinto, A.E.; Cunha, C.V.; Andre, S.; Barao, I.; Sousa, J.M.; Cravo, M. Global DNA hypomethylation in breast carcinoma: Correlation with prognostic factors and tumor progression. Cancer 1999, 85, 112–118. [Google Scholar] [CrossRef]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Statham, A.; Stirzaker, C.; Molloy, P.L.; Frommer, M. DNA methylation: Bisulphite modification and analysis. Nat. Protoc. 2006, 1, 2353–2364. [Google Scholar] [CrossRef] [PubMed]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, E32. [Google Scholar] [CrossRef] [Green Version]
- Khulan, B.; Thompson, R.F.; Ye, K.; Fazzari, M.J.; Suzuki, M.; Stasiek, E.; Figueroa, M.E.; Glass, J.L.; Chen, Q.; Montagna, C.; et al. Comparative isoschizomer profiling of cytosine methylation: The HELP assay. Genome Res. 2006, 16, 1046–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatada, I.; Fukasawa, M.; Kimura, M.; Morita, S.; Yamada, K.; Yoshikawa, T.; Yamanaka, S.; Endo, C.; Sakurada, A.; Sato, M.; et al. Genome-wide profiling of promoter methylation in human. Oncogene 2006, 25, 3059–3064. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, J.; Heyn, H.; Moran, S.; Serra-Musach, J.; Pujana, M.A.; Bibikova, M.; Esteller, M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011, 6, 692–702. [Google Scholar] [CrossRef]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Yuan, Z.; Tan, T.; Wang, J.; Zhang, J.; Luo, H.J.; Xia, Y.; Ji, W.; Gao, F. Improved tagmentation-based whole-genome bisulfite sequencing for input DNA from less than 100 mammalian cells. Epigenomics 2015, 7, 47–56. [Google Scholar] [CrossRef]
- Laird, P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef]
- Silverman, B.R.; Shi, J. Alterations of epigenetic regulators in pancreatic cancer and their clinical implications. Int. J. Mol. Sci. 2016, 17, 2138. [Google Scholar] [CrossRef] [Green Version]
- Ji, P.; Wang, X.; Xie, N.; Li, Y. N6-Methyladenosine in RNA and DNA: An Epitranscriptomic and Epigenetic Player Implicated in Determination of Stem Cell Fate. Stem Cells Int. 2018, 2018, 3256524. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Yokochi, T.; Robertson, K.D. Preferential methylation of unmethylated DNA by mammalian de novo DNA methyltransferase Dnmt3a. J. Biol. Chem. 2002, 277, 11735–11745. [Google Scholar] [CrossRef] [Green Version]
- Laird, P.W. The power and the promise of DNA methylation markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef]
- Tang, W.W.C.; Dietmann, S.; Irie, N.; Leitch, H.G.; Floros, V.I.; Bradshaw, C.R.; Hackett, J.A.; Chinnery, P.F.; Surani, M.A. A unique gene regulatory network resets the human germline epigenome for development. Cell 2015, 161, 1453–1467. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.P.; Chaillet, J.R.; Bestor, T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998, 20, 116–117. [Google Scholar] [CrossRef]
- Hark, A.T.; Schoenherr, C.J.; Katz, D.J.; Ingram, R.S.; Levorse, J.M.; Tilghman, S.M. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 2000, 405, 486–489. [Google Scholar] [CrossRef]
- Cotton, A.M.; Price, E.M.; Jones, M.J.; Balaton, B.P.; Kobor, M.S.; Brown, C.J. Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum. Mol. Genet. 2015, 24, 1528–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerman, K.; Sebastiani, P.; Jacques, P.; Liu, S.; Demeo, D.; Ordovás, J.M. DNA methylation modules associate with incident cardiovascular disease and cumulative risk factor exposure. Clin. Epigenetics 2019, 11, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Agha, G.; Baccarelli, A.A. The Role of DNA Methylation in Cardiovascular Risk and Disease. Methodol. Asp. Study Des. Data Anal. Epidemiol. Stud. 2016, 118, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Choong, M.K.; Tsafnat, G. Genetic and epigenetic biomarkers of colorectal cancer. Clin. Gastroenterol. Hepatol. 2012, 10, 9–15. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357. [Google Scholar] [CrossRef] [Green Version]
- Perry, A.S.; Watson, R.W.G.; Lawler, M.; Hollywood, D. The epigenome as a therapeutic target in prostate cancer. Nat. Rev. Urol. 2010, 7, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Oey, H.; Whitelaw, E. On the meaning of the word ‘epimutation’. Trends Genet. 2014, 30, 519–520. [Google Scholar] [CrossRef] [PubMed]
- Hesson, L.B.; Hitchins, M.P.; Ward, R.L. Epimutations and cancer predisposition: Importance and mechanisms. Curr. Opin. Genet. Dev. 2010, 20, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. The inheritance of epigenetic defects. Science 1987, 238, 163–170. [Google Scholar] [CrossRef]
- Haertle, L.; Maierhofer, A.; Bock, J.; Lehnen, H.; Bottcher, Y.; Bluher, M.; Schorsch, M.; Potabattula, R.; El Hajj, N.; Appenzeller, S.; et al. Hypermethylation of the non-imprinted maternal MEG3 and paternal MEST alleles is highly variable among normal individuals. PLoS ONE 2017, 12, e0184030. [Google Scholar] [CrossRef] [Green Version]
- Horsthemke, B. Epimutations in human disease. Curr. Top. Microbiol. Immunol. 2006, 310, 45–59. [Google Scholar] [CrossRef]
- Watson, I.R.; Takahashi, K.; Futreal, P.A.; Chin, L. Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 2013, 14, 703–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohrenweiser, H.W.; Jones, I.M. Review of the molecular characteristics of gene mutations of the germline and somatic cells of the human. Mutat. Res. 1990, 231, 87–108. [Google Scholar] [CrossRef]
- Hitchins, M.P.; Rapkins, R.W.; Kwok, C.T.; Srivastava, S.; Wong, J.J.; Khachigian, L.M.; Polly, P.; Goldblatt, J.; Ward, R.L. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5’UTR. Cancer Cell 2011, 20, 200–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloane, M.A.; Ward, R.L.; Hesson, L.B. Defining the criteria for identifying constitutional epimutations. Clin. Epigenetics 2016, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Santos, F.; Hendrich, B.; Reik, W.; Dean, W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev. Biol. 2002, 241, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitchins, M.P. Constitutional epimutation as a mechanism for cancer causality and heritability? Nat. Rev. Cancer 2015, 15, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Hitchins, M.P.; Ward, R.L. Erasure of MLH1 methylation in spermatozoa-implications for epigenetic inheritance. Nat. Genet. 2007, 39, 1289. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, S.A.; Tomizawa, S.; Krueger, F.; Ruf, N.; Carli, N.; Segonds-Pichon, A.; Sato, S.; Hata, K.; Andrews, S.R.; Kelsey, G. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat. Genet. 2011, 43, 811–814. [Google Scholar] [CrossRef] [Green Version]
- Lonning, P.E.; Eikesdal, H.P.; Loes, I.M.; Knappskog, S. Constitutional Mosaic Epimutations-a hidden cause of cancer? Cell Stress 2019, 3, 118–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cropley, J.E.; Martin, D.I.; Suter, C.M. Germline epimutation in humans. Pharmacogenomics 2008, 9, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Toguchida, J.; Ohtani, N.; Yandell, D.W.; Rapaport, J.M.; Dryja, T.P. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am. J. Hum. Genet. 1991, 48, 880–888. [Google Scholar] [PubMed]
- Herman, J.G.; Latif, F.; Weng, Y.; Lerman, M.I.; Zbar, B.; Liu, S.; Samid, D.; Duan, D.S.; Gnarra, J.R.; Linehan, W.M.; et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 9700–9704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, M.F.; Loda, M.; Gaida, G.M.; Lipman, J.; Mishra, R.; Goldman, H.; Jessup, J.M.; Kolodner, R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997, 57, 808–811. [Google Scholar] [PubMed]
- Dobrovic, A.; Simpfendorfer, D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res. 1997, 57, 3347–3350. [Google Scholar]
- Banno, K.; Kisu, I.; Yanokura, M.; Tsuji, K.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Yamagami, W.; Nomura, H.; Tominaga, E.; et al. Epimutation and cancer: A new carcinogenic mechanism of Lynch syndrome (Review). Int. J. Oncol. 2012, 41, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Petronis, A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 2010, 465, 721–727. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Tucker, T.; Friedman, J.M. Pathogenesis of hereditary tumors: Beyond the "two-hit" hypothesis. Clin. Genet. 2002, 62, 345–357. [Google Scholar] [CrossRef]
- Mossman, D.; Scott, R.J. Epimutations, inheritance and causes of aberrant DNA methylation in cancer. Hered. Cancer Clin. Pr. 2006, 4, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Hitchins, M.P. The role of epigenetics in Lynch syndrome. Fam. Cancer 2013, 12, 189–205. [Google Scholar] [CrossRef]
- Gelli, E.; Pinto, A.M.; Somma, S.; Imperatore, V.; Cannone, M.G.; Hadjistilianou, T.; De Francesco, S.; Galimberti, D.; Currò, A.; Bruttini, M.; et al. Evidence of predisposing epimutation in retinoblastoma. Hum. Mutat. 2019, 40, 201–206. [Google Scholar] [CrossRef]
- Esteller, M.; Fraga, M.F.; Guo, M.; Garcia-Foncillas, J.; Hedenfalk, I.; Godwin, A.K.; Trojan, J.; Vaurs-Barrière, C.; Bignon, Y.J.; Ramus, S.; et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum. Mol. Genet. 2001, 10, 3001–3007. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.Y.; Asiedu, C.K.; Supakar, P.C.; Khan, R.; Ehrlich, K.C.; Ehrlich, M. Binding sites in mammalian genes and viral gene regulatory regions recognized by methylated DNA-binding protein. Nucleic Acids Res. 1990, 18, 6253–6260. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.; Tanner, S.M.; Byrd, J.C.; Angerman, E.B.; Perko, J.D.; Chen, S.S.; Hackanson, B.; Grever, M.R.; Lucas, D.M.; Matkovic, J.J.; et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 2007, 129, 879–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazzoli, I.; Loda, M.; Garber, J.; Syngal, S.; Kolodner, R.D. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res. 2002, 62, 3925–3928. [Google Scholar] [PubMed]
- Suter, C.M.; Martin, D.I.; Ward, R.L. Germline epimutation of MLH1 in individuals with multiple cancers. Nat. Genet. 2004, 36, 497–501. [Google Scholar] [CrossRef]
- Tiffon, C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. Int. J. Mol. Sci. 2018, 19, 3425. [Google Scholar] [CrossRef] [Green Version]
- Moulton, T.; Crenshaw, T.; Hao, Y.; Moosikasuwan, J.; Lin, N.; Dembitzer, F.; Hensle, T.; Weiss, L.; McMorrow, L.; Loew, T.; et al. Epigenetic lesions at the H19 locus in Wilms’ tumour patients. Nat. Genet. 1994, 7, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Argos, M. Arsenic Exposure and Epigenetic Alterations: Recent Findings Based on the Illumina 450K DNA Methylation Array. Curr. Environ. Health Rep. 2015, 2, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philibert, R.A.; Beach, S.R.; Brody, G.H. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics 2012, 7, 1331–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeilinger, S.; Kuhnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.P.; Ottaviano, R.; Unterberger, E.B.; Lempiainen, H.; Muller, A.; Terranova, R.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; Lyall, M.J.; et al. Loss of Tet1-Associated 5-Hydroxymethylcytosine Is Concomitant with Aberrant Promoter Hypermethylation in Liver Cancer. Cancer Res. 2016, 76, 3097–3108. [Google Scholar] [CrossRef] [Green Version]
- Meehan, R.R.; Thomson, J.P.; Lentini, A.; Nestor, C.E.; Pennings, S. DNA methylation as a genomic marker of exposure to chemical and environmental agents. Curr. Opin. Chem. Biol. 2018, 45, 48–56. [Google Scholar] [CrossRef]
- Liu, L.; Wylie, R.C.; Andrews, L.G.; Tollefsbol, T.O. Aging, cancer and nutrition: The DNA methylation connection. Mech. Ageing Dev. 2003, 124, 989–998. [Google Scholar] [CrossRef]
- McCabe, D.C.; Caudill, M.A. DNA methylation, genomic silencing, and links to nutrition and cancer. Nutr. Rev. 2005, 63, 183–195. [Google Scholar] [CrossRef]
- Zeisel, S. Choline, Other Methyl-Donors and Epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef]
- Brunaud, L.; Alberto, J.M.; Ayav, A.; Gerard, P.; Namour, F.; Antunes, L.; Braun, M.; Bronowicki, J.P.; Bresler, L.; Gueant, J.L. Effects of vitamin B12 and folate deficiencies on DNA methylation and carcinogenesis in rat liver. Clin. Chem. Lab. Med. 2003, 41, 1012–1019. [Google Scholar] [CrossRef]
- Wu, J.C.; Santi, D.V. On the mechanism and inhibition of DNA cytosine methyltransferases. Prog. Clin. Biol. Res. 1985, 198, 119–129. [Google Scholar]
- Ross, S.A. Diet and DNA methylation interactions in cancer prevention. Ann. N. Y. Acad. Sci. 2003, 983, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Garfinkel, M.D.; Ruden, D.M. Chromatin effects in nutrition, cancer, and obesity. Nutrition 2004, 20, 56–62. [Google Scholar] [CrossRef]
- Kim, Y.I. Folate and DNA methylation: A mechanistic link between folate deficiency and colorectal cancer? Cancer Epidemiol. Prev. Biomark. 2004, 13, 511–519. [Google Scholar]
- Morgan, A.E.; Davies, T.J.; Mc Auley, M.T. The role of DNA methylation in ageing and cancer. Proc. Nutr. Soc. 2018, 77, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. Epigenetics provides a new generation of oncogenes and tumour-suppressor genes. Br. J. Cancer 2006, 94, 179–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitchins, M.P.; Wong, J.J.; Suthers, G.; Suter, C.M.; Martin, D.I.; Hawkins, N.J.; Ward, R.L. Inheritance of a cancer-associated MLH1 germ-line epimutation. N. Engl. J. Med. 2007, 356, 697–705. [Google Scholar] [CrossRef] [Green Version]
- Pineda, M.; Mur, P.; Iniesta, M.D.; Borras, E.; Campos, O.; Vargas, G.; Iglesias, S.; Fernandez, A.; Gruber, S.B.; Lazaro, C.; et al. MLH1 methylation screening is effective in identifying epimutation carriers. Eur. J. Hum. Genet. 2012, 20, 1256–1264. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, R.J.; McDowell, T.L.; Raman, S.; O’Rourke, D.M.; Garrick, D.; Ayyub, H.; Higgs, D.R. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat. Genet. 2000, 24, 368–371. [Google Scholar] [CrossRef] [Green Version]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkerk, A.J.; Holden, J.J.; Fenwick, R.G., Jr.; Warren, S.T.; et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Stoger, R.; Kajimura, T.M.; Brown, W.T.; Laird, C.D. Epigenetic variation illustrated by DNA methylation patterns of the fragile-X gene FMR1. Hum. Mol. Genet. 1997, 6, 1791–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielinska, B.; Blaydes, S.M.; Buiting, K.; Yang, T.; Krajewska-Walasek, M.; Horsthemke, B.; Brannan, C.I. De novo deletions of SNRPN exon 1 in early human and mouse embryos result in a paternal to maternal imprint switch. Nat. Genet. 2000, 25, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Perk, J.; Makedonski, K.; Lande, L.; Cedar, H.; Razin, A.; Shemer, R. The imprinting mechanism of the Prader-Willi/Angelman regional control center. EMBO J. 2002, 21, 5807–5814. [Google Scholar] [CrossRef] [Green Version]
- Dittrich, B.; Buiting, K.; Korn, B.; Rickard, S.; Buxton, J.; Saitoh, S.; Nicholls, R.D.; Poustka, A.; Winterpacht, A.; Zabel, B.; et al. Imprint switching on human chromosome 15 may involve alternative transcripts of the SNRPN gene. Nat. Genet. 1996, 14, 163–170. [Google Scholar] [CrossRef]
- Glenn, C.C.; Saitoh, S.; Jong, M.T.; Filbrandt, M.M.; Surti, U.; Driscoll, D.J.; Nicholls, R.D. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am. J. Hum. Genet. 1996, 58, 335–346. [Google Scholar]
- Sutcliffe, J.S.; Nakao, M.; Christian, S.; Orstavik, K.H.; Tommerup, N.; Ledbetter, D.H.; Beaudet, A.L. Deletions of a differentially methylated CpG island at the SNRPN gene define a putative imprinting control region. Nat. Genet. 1994, 8, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K.; Saitoh, S.; Gross, S.; Dittrich, B.; Schwartz, S.; Nicholls, R.D.; Horsthemke, B. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat. Genet. 1995, 9, 395–400. [Google Scholar] [CrossRef]
- Dittrich, B.; Buiting, K.; Gross, S.; Horsthemke, B. Characterization of a methylation imprint in the Prader-Willi syndrome chromosome region. Hum. Mol. Genet. 1993, 2, 1995–1999. [Google Scholar] [CrossRef]
- Buiting, K.; Dittrich, B.; Robinson, W.P.; Guitart, M.; Abeliovich, D.; Lerer, I.; Horsthemke, B. Detection of aberrant DNA methylation in unique Prader-Willi syndrome patients and its diagnostic implications. Hum. Mol. Genet. 1994, 3, 893–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glenn, C.C.; Nicholls, R.D.; Robinson, W.P.; Saitoh, S.; Niikawa, N.; Schinzel, A.; Horsthemke, B.; Driscoll, D.J. Modification of 15q11-q13 DNA methylation imprints in unique Angelman and Prader-Willi patients. Hum. Mol. Genet. 1993, 2, 1377–1382. [Google Scholar] [CrossRef] [Green Version]
- Glenn, C.C.; Porter, K.A.; Jong, M.T.; Nicholls, R.D.; Driscoll, D.J. Functional imprinting and epigenetic modification of the human SNRPN gene. Hum. Mol. Genet. 1993, 2, 2001–2005. [Google Scholar] [CrossRef]
- Buiting, K.; Gross, S.; Lich, C.; Gillessen-Kaesbach, G.; el-Maarri, O.; Horsthemke, B. Epimutations in Prader-Willi and Angelman syndromes: A molecular study of 136 patients with an imprinting defect. Am. J. Hum. Genet. 2003, 72, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Maarri, O.; Buiting, K.; Peery, E.G.; Kroisel, P.M.; Balaban, B.; Wagner, K.; Urman, B.; Heyd, J.; Lich, C.; Brannan, C.I.; et al. Maternal methylation imprints on human chromosome 15 are established during or after fertilization. Nat. Genet. 2001, 27, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Barbour, V.M.; Tufarelli, C.; Sharpe, J.A.; Smith, Z.E.; Ayyub, H.; Heinlein, C.A.; Sloane-Stanley, J.; Indrak, K.; Wood, W.G.; Higgs, D.R. alpha-thalassemia resulting from a negative chromosomal position effect. Blood 2000, 96, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Hitchins, M.P. Inheritance of epigenetic aberrations (constitutional epimutations) in cancer susceptibility. Adv. Genet. 2010, 70, 201–243. [Google Scholar] [CrossRef]
- Tufarelli, C.; Stanley, J.A.; Garrick, D.; Sharpe, J.A.; Ayyub, H.; Wood, W.G.; Higgs, D.R. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat. Genet. 2003, 34, 157–165. [Google Scholar] [CrossRef]
- Sparago, A.; Cerrato, F.; Vernucci, M.; Ferrero, G.B.; Silengo, M.C.; Riccio, A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat. Genet. 2004, 36, 958–960. [Google Scholar] [CrossRef] [PubMed]
- Niemitz, E.L.; DeBaun, M.R.; Fallon, J.; Murakami, K.; Kugoh, H.; Oshimura, M.; Feinberg, A.P. Microdeletion of LIT1 in familial Beckwith-Wiedemann syndrome. Am. J. Hum. Genet. 2004, 75, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Ligtenberg, M.J.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.; et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat. Genet. 2009, 41, 112–117. [Google Scholar] [CrossRef]
- Pinheiro, H.; Bordeira-Carrico, R.; Seixas, S.; Carvalho, J.; Senz, J.; Oliveira, P.; Inacio, P.; Gusmao, L.; Rocha, J.; Huntsman, D.; et al. Allele-specific CDH1 downregulation and hereditary diffuse gastric cancer. Hum. Mol. Genet. 2010, 19, 943–952. [Google Scholar] [CrossRef] [Green Version]
- Rapkins, R.W.; Wang, F.; Nguyen, H.N.; Cloughesy, T.F.; Lai, A.; Ha, W.; Nowak, A.K.; Hitchins, M.P.; McDonald, K.L. The MGMT promoter SNP rs16906252 is a risk factor for MGMT methylation in glioblastoma and is predictive of response to temozolomide. Neuro Oncol. 2015, 17, 1589–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gueant, J.L.; Chery, C.; Oussalah, A.; Nadaf, J.; Coelho, D.; Josse, T.; Flayac, J.; Robert, A.; Koscinski, I.; Gastin, I.; et al. A PRDX1 mutant allele causes a MMACHC secondary epimutation in cblC patients. Nat. Commun. 2018, 9, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.G.R.; van Veen, E.M.; Byers, H.J.; Wallace, A.J.; Ellingford, J.M.; Beaman, G.; Santoyo-Lopez, J.; Aitman, T.J.; Eccles, D.M.; Lalloo, F.I.; et al. A Dominantly Inherited 5’ UTR Variant Causing Methylation-Associated Silencing of BRCA1 as a Cause of Breast and Ovarian Cancer. Am. J. Hum. Genet. 2018, 103, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, W.N.; Luharia, A.; Evans, G.A.; Raza, H.; Haire, A.C.; Grundy, R.; Bowdin, S.C.; Riccio, A.; Sebastio, G.; Bliek, J.; et al. Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2005, 13, 1025–1032. [Google Scholar] [CrossRef]
- Elliott, M.; Bayly, R.; Cole, T.; Temple, I.K.; Maher, E.R. Clinical features and natural history of Beckwith-Wiedemann syndrome: Presentation of 74 new cases. Clin. Genet. 1994, 46, 168–174. [Google Scholar] [CrossRef]
- Elliott, M.; Maher, E.R. Beckwith-Wiedemann syndrome. J. Med. Genet. 1994, 31, 560–564. [Google Scholar] [CrossRef] [Green Version]
- Beckwith, J.B. National Wilms Tumor Study: An update for pathologists. Pediatr. Dev. Pathol. 1998, 1, 79–84. [Google Scholar] [CrossRef]
- Everman, D.B.; Shuman, C.; Dzolganovski, B.; O’Riordan M, A.; Weksberg, R.; Robin, N.H. Serum alpha-fetoprotein levels in Beckwith-Wiedemann syndrome. J. Pediatr. 2000, 137, 123–127. [Google Scholar] [CrossRef]
- Frevel, M.A.; Sowerby, S.J.; Petersen, G.B.; Reeve, A.E. Methylation sequencing analysis refines the region of H19 epimutation in Wilms tumor. J. Biol. Chem. 1999, 274, 29331–29340. [Google Scholar] [CrossRef] [Green Version]
- DeBaun, M.R.; Niemitz, E.L.; Feinberg, A.P. Association of In Vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am. J. Hum. Genet. 2003, 72, 156–160. [Google Scholar] [CrossRef] [Green Version]
- DeBaun, M.R.; Niemitz, E.L.; McNeil, D.E.; Brandenburg, S.A.; Lee, M.P.; Feinberg, A.P. Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. Am. J. Hum. Genet. 2002, 70, 604–611. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.P.; DeBaun, M.R.; Mitsuya, K.; Galonek, H.L.; Brandenburg, S.; Oshimura, M.; Feinberg, A.P. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc. Natl. Acad. Sci. USA 1999, 96, 5203–5208. [Google Scholar] [CrossRef] [Green Version]
- Slager, S.L.; Kay, N.E. Familial chronic lymphocytic leukemia: What does it mean to me? Clin. Lymphoma Myeloma 2009, 9 (Suppl. 3), S194–S197. [Google Scholar] [CrossRef] [Green Version]
- Hitchins, M.P. Finding the needle in a haystack: Identification of cases of Lynch syndrome with MLH1 epimutation. Fam. Cancer 2016, 15, 413–422. [Google Scholar] [CrossRef]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef]
- Marmol, I.; Sanchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [Green Version]
- Peltomaki, P.; Vasen, H. Mutations associated with HNPCC predisposition–Update of ICG-HNPCC/INSiGHT mutation database. Dis. Markers 2004, 20, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Cunningham, J.M.; Christensen, E.R.; Tester, D.J.; Kim, C.Y.; Roche, P.C.; Burgart, L.J.; Thibodeau, S.N. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998, 58, 3455–3460. [Google Scholar]
- Hitchins, M.P.; Owens, S.E.; Kwok, C.T.; Godsmark, G.; Algar, U.F.; Ramesar, R.S. Identification of new cases of early-onset colorectal cancer with an MLH1 epimutation in an ethnically diverse South African cohort. Clin. Genet. 2011, 80, 428–434. [Google Scholar] [CrossRef]
- Wong, J.J.; Hawkins, N.J.; Ward, R.L.; Hitchins, M.P. Methylation of the 3p22 region encompassing MLH1 is representative of the CpG island methylator phenotype in colorectal cancer. Mod. Pathol. 2011, 24, 396–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitchins, M.; Williams, R.; Cheong, K.; Halani, N.; Lin, V.A.; Packham, D.; Ku, S.; Buckle, A.; Hawkins, N.; Burn, J.; et al. MLH1 germline epimutations as a factor in hereditary nonpolyposis colorectal cancer. Gastroenterology 2005, 129, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.L.; Yuen, S.T.; Kong, C.K.; Chan, Y.W.; Chan, A.S.; Ng, W.F.; Tsui, W.Y.; Lo, M.W.; Tam, W.Y.; Li, V.S.; et al. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat. Genet. 2006, 38, 1178–1183. [Google Scholar] [CrossRef]
- Chan, T.L.; Chan, Y.W.; Ho, J.W.; Chan, C.; Chan, A.S.; Chan, E.; Lam, P.W.; Tse, C.W.; Lee, K.C.; Lau, C.W.; et al. MSH2 c.1452-1455delAATG is a founder mutation and an important cause of hereditary nonpolyposis colorectal cancer in the southern Chinese population. Am. J. Hum. Genet. 2004, 74, 1035–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niessen, R.C.; Hofstra, R.M.; Westers, H.; Ligtenberg, M.J.; Kooi, K.; Jager, P.O.; de Groote, M.L.; Dijkhuizen, T.; Olderode-Berends, M.J.; Hollema, H.; et al. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes. Chromosomes Cancer 2009, 48, 737–744. [Google Scholar] [CrossRef]
- Lynch, H.T.; Kaurah, P.; Wirtzfeld, D.; Rubinstein, W.S.; Weissman, S.; Lynch, J.F.; Grady, W.; Wiyrick, S.; Senz, J.; Huntsman, D.G. Hereditary diffuse gastric cancer: Diagnosis, genetic counseling, and prophylactic total gastrectomy. Cancer 2008, 112, 2655–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pharoah, P.D.; Guilford, P.; Caldas, C.; International Gastric Cancer Linkage, C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001, 121, 1348–1353. [Google Scholar] [CrossRef]
- Pegg, A.E. Mammalian O6-alkylguanine-DNA alkyltransferase: Regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990, 50, 6119–6129. [Google Scholar]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Toyota, M.; Sanchez-Cespedes, M.; Capella, G.; Peinado, M.A.; Watkins, D.N.; Issa, J.P.; Sidransky, D.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res. 2000, 60, 2368–2371. [Google Scholar] [PubMed]
- Christians, A.; Hartmann, C.; Benner, A.; Meyer, J.; von Deimling, A.; Weller, M.; Wick, W.; Weiler, M. Prognostic value of three different methods of MGMT promoter methylation analysis in a prospective trial on newly diagnosed glioblastoma. PLoS ONE 2012, 7, e33449. [Google Scholar] [CrossRef]
- Costello, J.F.; Futscher, B.W.; Kroes, R.A.; Pieper, R.O. Methylation-related chromatin structure is associated with exclusion of transcription factors from and suppressed expression of the O-6-methylguanine DNA methyltransferase gene in human glioma cell lines. Mol. Cell Biol. 1994, 14, 6515–6521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, J.F.; Futscher, B.W.; Tano, K.; Graunke, D.M.; Pieper, R.O. Graded methylation in the promoter and body of the O6-methylguanine DNA methyltransferase (MGMT) gene correlates with MGMT expression in human glioma cells. J. Biol. Chem. 1994, 269, 17228–17237. [Google Scholar] [CrossRef]
- Qian, X.C.; Brent, T.P. Methylation hot spots in the 5’ flanking region denote silencing of the O6-methylguanine-DNA methyltransferase gene. Cancer Res. 1997, 57, 3672–3677. [Google Scholar] [PubMed]
- Watkins, D.; Rosenblatt, D.S. Inborn errors of cobalamin absorption and metabolism. Am. J. Med. Genet. C Semin. Med. Genet. 2011, 157C, 33–44. [Google Scholar] [CrossRef]
- Jönsson, G.; Staaf, J.; Vallon-Christersson, J.; Ringnér, M.; Gruvberger-Saal, S.K.; Saal, L.H.; Holm, K.; Hegardt, C.; Arason, A.; Fagerholm, R.; et al. The retinoblastoma gene undergoes rearrangements in BRCA1-deficient basal-like breast cancer. Cancer Res. 2012, 72, 4028–4036. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, R.L.; Nemeth, E.; Tran, H.; Shvartsman, H.; Cass, I.; Narod, S.; Karlan, B.Y. BRCA1 promoter region hypermethylation in ovarian carcinoma: A population-based study. Cancer Res. 2000, 60, 5329–5333. [Google Scholar]
- Laner, A.; Benet-Pages, A.; Neitzel, B.; Holinski-Feder, E. Analysis of 3297 individuals suggests that the pathogenic germline 5’-UTR variant BRCA1 c.-107A > T is not common in south-east Germany. Fam. Cancer 2020, 19, 211–213. [Google Scholar] [CrossRef]
- Garg, P.; Jadhav, B.; Rodriguez, O.L.; Patel, N.; Martin-Trujillo, A.; Jain, M.; Metsu, S.; Olsen, H.; Paten, B.; Ritz, B.; et al. A Survey of Rare Epigenetic Variation in 23,116 Human Genomes Identifies Disease-Relevant Epivariations and CGG Expansions. Am. J. Hum. Genet. 2020, 107, 654–669. [Google Scholar] [CrossRef]
- Heyn, H.; Moran, S.; Hernando-Herraez, I.; Sayols, S.; Gomez, A.; Sandoval, J.; Monk, D.; Hata, K.; Marques-Bonet, T.; Wang, L.; et al. DNA methylation contributes to natural human variation. Genome Res. 2013, 23, 1363–1372. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz de la Cruz, M.; de la Cruz Montoya, A.H.; Rojas Jiménez, E.A.; Martínez Gregorio, H.; Díaz Velásquez, C.E.; Paredes de la Vega, J.; de la Cruz Hernández-Hernández, F.; Vaca Paniagua, F. Cis-Acting Factors Causing Secondary Epimutations: Impact on the Risk for Cancer and Other Diseases. Cancers 2021, 13, 4807. https://doi.org/10.3390/cancers13194807
Ruiz de la Cruz M, de la Cruz Montoya AH, Rojas Jiménez EA, Martínez Gregorio H, Díaz Velásquez CE, Paredes de la Vega J, de la Cruz Hernández-Hernández F, Vaca Paniagua F. Cis-Acting Factors Causing Secondary Epimutations: Impact on the Risk for Cancer and Other Diseases. Cancers. 2021; 13(19):4807. https://doi.org/10.3390/cancers13194807
Chicago/Turabian StyleRuiz de la Cruz, Miguel, Aldo Hugo de la Cruz Montoya, Ernesto Arturo Rojas Jiménez, Héctor Martínez Gregorio, Clara Estela Díaz Velásquez, Jimena Paredes de la Vega, Fidel de la Cruz Hernández-Hernández, and Felipe Vaca Paniagua. 2021. "Cis-Acting Factors Causing Secondary Epimutations: Impact on the Risk for Cancer and Other Diseases" Cancers 13, no. 19: 4807. https://doi.org/10.3390/cancers13194807
APA StyleRuiz de la Cruz, M., de la Cruz Montoya, A. H., Rojas Jiménez, E. A., Martínez Gregorio, H., Díaz Velásquez, C. E., Paredes de la Vega, J., de la Cruz Hernández-Hernández, F., & Vaca Paniagua, F. (2021). Cis-Acting Factors Causing Secondary Epimutations: Impact on the Risk for Cancer and Other Diseases. Cancers, 13(19), 4807. https://doi.org/10.3390/cancers13194807