
Impact of KMT2A Rearrangement and CSPG4 Expression in Pediatric Acute Myeloid Leukemia

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Genetic Data
2.3. Flow Cytometric Data
2.4. Statistical Analysis
3. Results
3.1. Patient Characteristics
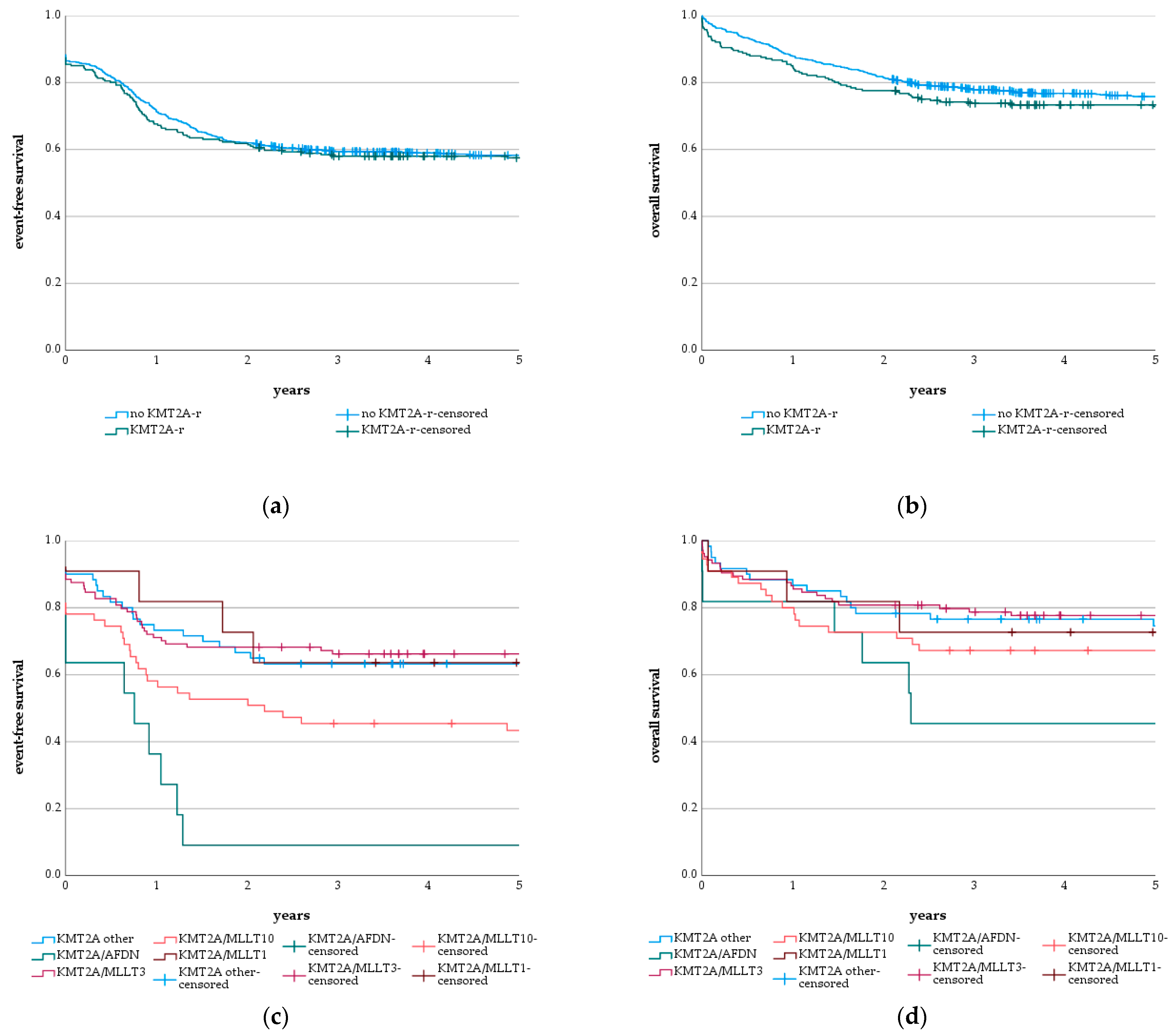
3.2. KMT2A-r and Subgroups
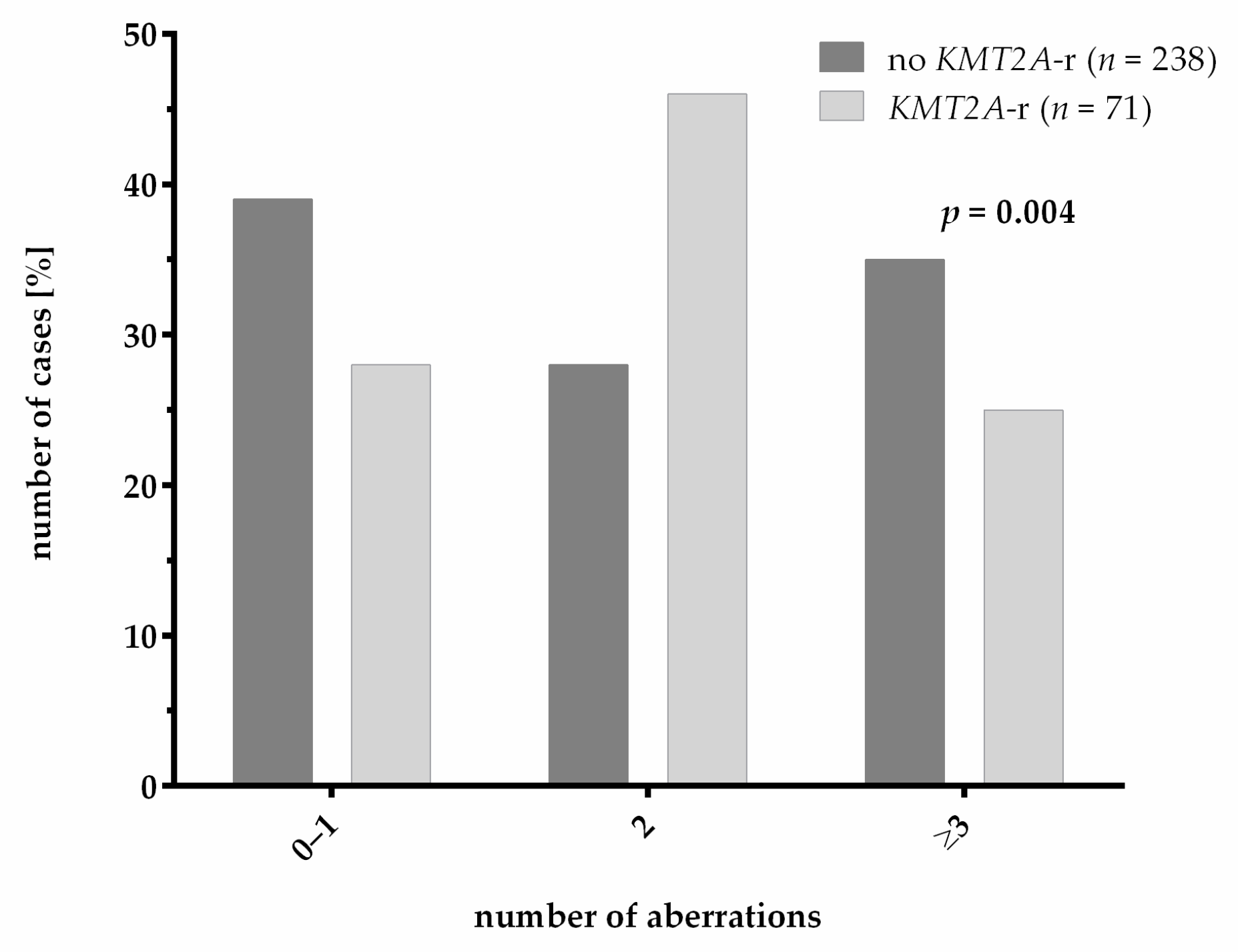
3.3. Genetics
3.4. CSPG4 Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zwaan, C.M.; Kolb, E.A.; Reinhardt, D.; Abrahamsson, J.; Adachi, S.; Aplenc, R.; De Bont, E.S.J.M.; de Moerloose, B.; Dworzak, M.; Gibson, B.E.S.; et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 2949–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balgobind, B.V.; Hollink, I.H.I.M.; Arentsen-Peters, S.T.C.J.M.; Zimmermann, M.; Harbott, J.; Beverloo, H.B.; von Bergh, A.R.M.; Cloos, J.; Kaspers, G.J.L.; de Haas, V.; et al. Integrative analysis of type-I and type-II aberrations underscores the genetic heterogeneity of pediatric acute myeloid leukemia. Haematologica 2011, 96, 1478–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdmann, F.; Kaatsch, P.; Grabow, D.; Spix, C. German Childhood Cancer Registry—Annual Report 2019 (1980–2018). Available online: https://www.kinderkrebsregister.de/dkkr/ergebnisse/jahresberichte/jahresbericht-2019.html (accessed on 12 July 2021).
- Rasche, M.; Zimmermann, M.; Borschel, L.; Bourquin, J.-P.; Dworzak, M.; Klingebiel, T.; Lehrnbecher, T.; Creutzig, U.; Klusmann, J.-H.; Reinhardt, D. Successes and challenges in the treatment of pediatric acute myeloid leukemia: A retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 2018, 32, 2167–2177. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef]
- Erfurth, F.; Hemenway, C.S.; de Erkenez, A.C.; Domer, P.H. MLL fusion partners AF4 and AF9 interact at subnuclear foci. Leukemia 2004, 18, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL Targets SET Domain Methyltransferase Activity to Hox Gene Promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Hoshii, T.; Armstrong, S.A. Mixed-Lineage Leukemia Fusions and Chromatin in Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7, a026658. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; den Boer, M.L.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Balgobind, B.V.; Raimondi, S.C.; Harbott, J.; Zimmermann, M.; Alonzo, T.A.; Auvrignon, A.; Beverloo, H.B.; Chang, M.; Creutzig, U.; Dworzak, M.N.; et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: Results of an international retrospective study. Blood 2009, 114, 2489–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bill, M.; Mrózek, K.; Kohlschmidt, J.; Eisfeld, A.-K.; Walker, C.J.; Nicolet, D.; Papaioannou, D.; Blachly, J.S.; Orwick, S.; Carroll, A.J.; et al. Mutational landscape and clinical outcome of patients with de novo acute myeloid leukemia and rearrangements involving 11q23/KMT2A. Proc. Natl. Acad. Sci. USA 2020, 117, 26340–26346. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Schnittger, S.; Poetzinger, F.; Kohlmann, A.; Stiel, A.; Eder, C.; Fasan, A.; Kern, W.; Haferlach, T.; Haferlach, C. High incidence of RAS signalling pathway mutations in MLL-rearranged acute myeloid leukemia. Leukemia 2013, 27, 1933–1936. [Google Scholar] [CrossRef] [Green Version]
- Smith, F.O.; Rauch, C.; Williams, D.E.; March, C.J.; Arthur, D.; Hilden, J.; Lampkin, B.C.; Buckley, J.D.; Buckley, C.V.; Woods, W.G.; et al. The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood 1996, 87, 1123–1133. [Google Scholar] [PubMed]
- Hilden, J.M.; Smith, F.O.; Frestedt, J.L.; McGlennen, R.; Howells, W.B.; Sorensen, P.H.; Arthur, D.C.; Woods, W.G.; Buckley, J.; Bernstein, I.D.; et al. MLL gene rearrangement, cytogenetic 11q23 abnormalities, and expression of the NG2 molecule in infant acute myeloid leukemia. Blood 1997, 89, 3801–3805. [Google Scholar] [CrossRef]
- Behm, F.G.; Smith, F.O.; Raimondi, S.C.; Pui, C.H.; Bernstein, I.D. Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood 1996, 87, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Wuchter, C.; Harbott, J.; Schoch, C.; Schnittger, S.; Borkhardt, A.; Karawajew, L.; Ratei, R.; Ruppert, V.; Haferlach, T.; Creutzig, U.; et al. Detection of acute leukemia cells with mixed lineage leukemia (MLL) gene rearrangements by flow cytometry using monoclonal antibody 7.1. Leukemia 2000, 14, 1232–1238. [Google Scholar] [CrossRef] [Green Version]
- Mauvieux, L.; Delabesse, E.; Bourquelot, P.; Radford-Weiss, I.; Bennaceur, A.; Flandrin, G.; Valensi, F.; MacIntyre, E.A. NG2 expression in MLL rearranged acute myeloid leukaemia is restricted to monoblastic cases. Br. J. Haematol. 1999, 107, 674–676. [Google Scholar] [CrossRef]
- Cimino, G.; Rapanotti, M.C.; Elia, L.; Biondi, A.; Fizzotti, M.; Testi, A.M.; Tosti, S.; Croce, C.M.; Canaani, E.; Mandelli, F. ALL-1 gene rearrangements in acute myeloid leukemia: Association with M4-M5 French-American-British classification subtypes and young age. Cancer Res. 1995, 55, 1625–1628. [Google Scholar]
- Emerenciano, M.; Meyer, C.; Mansur, M.B.; Marschalek, R.; Pombo-de-Oliveira, M.S. The distribution of MLL breakpoints correlates with outcome in infant acute leukaemia. Br. J. Haematol. 2013, 161, 224–236. [Google Scholar] [CrossRef]
- Creutzig, U.; van den Heuvel-Eibrink, M.M.; Gibson, B.; Dworzak, M.N.; Adachi, S.; de Bont, E.; Harbott, J.; Hasle, H.; Johnston, D.; Kinoshita, A.; et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: Recommendations from an international expert panel. Blood 2012, 120, 3187–3205. [Google Scholar] [CrossRef] [PubMed]
- Dworzak, M.N.; Buldini, B.; Gaipa, G.; Ratei, R.; Hrusak, O.; Luria, D.; Rosenthal, E.; Bourquin, J.-P.; Sartor, M.; Schumich, A.; et al. AIEOP-BFM consensus guidelines 2016 for flow cytometric immunophenotyping of Pediatric acute lymphoblastic leukemia. Cytometry B Clin. Cytom. 2018, 94, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, Y.; Shiba, N.; Yamato, G.; Ohki, K.; Tabuchi, K.; Sotomatsu, M.; Tomizawa, D.; Kinoshita, A.; Arakawa, H.; Saito, A.M.; et al. Patients aged less than 3 years with acute myeloid leukaemia characterize a molecularly and clinically distinct subgroup. Br. J. Haematol. 2020, 188, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Rasche, M.; Zimmermann, M.; Steidel, E.; Alonzo, T.; Aplenc, R.; Bourquin, J.-P.; Boztug, H.; Cooper, T.; Gamis, A.S.; Gerbing, R.B.; et al. Survival Following Relapse in Children with Acute Myeloid Leukemia: A Report from AML-BFM and COG. Cancers 2021, 13, 2336. [Google Scholar] [CrossRef]
- Rasche, M.; Steidel, E.; Kondryn, D.; von Neuhoff, N.; Sramkova, L.; Creutzig, U.; Dworzak, M.; Reinhardt, D. Impact of a Risk-Adapted Treatment Approach in Pediatric AML: A Report of the AML-BFM Registry 2012. Blood 2019, 134, 293. [Google Scholar] [CrossRef]
- Klusmann, J.-H.; Reinhardt, D.; Zimmermann, M.; Kremens, B.; Vormoor, J.; Dworzak, M.; Creutzig, U.; Klingebiel, T. The role of matched sibling donor allogeneic stem cell transplantation in pediatric high-risk acute myeloid leukemia: Results from the AML-BFM 98 study. Haematologica 2012, 97, 21–29. [Google Scholar] [CrossRef]
- Creutzig, U.; Rössig, C.; Dworzak, M.; Stary, J.; von Stackelberg, A.; Wössmann, W.; Zimmermann, M.; Reinhardt, D. Exchange Transfusion and Leukapheresis in Pediatric Patients with AML With High Risk of Early Death by Bleeding and Leukostasis. Pediatr. Blood Cancer 2016, 63, 640–645. [Google Scholar] [CrossRef]
- Matsuo, H.; Yoshida, K.; Nakatani, K.; Harata, Y.; Higashitani, M.; Ito, Y.; Kamikubo, Y.; Shiozawa, Y.; Shiraishi, Y.; Chiba, K.; et al. Fusion partner-specific mutation profiles and KRAS mutations as adverse prognostic factors in MLL-rearranged AML. Blood Adv. 2020, 4, 4623–4631. [Google Scholar] [CrossRef]
- Coenen, E.A.; Raimondi, S.C.; Harbott, J.; Zimmermann, M.; Alonzo, T.A.; Auvrignon, A.; Beverloo, H.B.; Chang, M.; Creutzig, U.; Dworzak, M.N.; et al. Prognostic significance of additional cytogenetic aberrations in 733 de novo pediatric 11q23/MLL-rearranged AML patients: Results of an international study. Blood 2011, 117, 7102–7111. [Google Scholar] [CrossRef]
- Camós, M.; Esteve, J.; Jares, P.; Colomer, D.; Rozman, M.; Villamor, N.; Costa, D.; Carrió, A.; Nomdedéu, J.; Montserrat, E.; et al. Gene expression profiling of acute myeloid leukemia with translocation t(8;16)(p11;p13) and MYST3-CREBBP rearrangement reveals a distinctive signature with a specific pattern of HOX gene expression. Cancer Res. 2006, 66, 6947–6954. [Google Scholar] [CrossRef] [Green Version]
- de Rooij, J.D.E.; Hollink, I.H.I.M.; Arentsen-Peters, S.T.C.J.M.; van Galen, J.F.; Berna Beverloo, H.; Baruchel, A.; Trka, J.; Reinhardt, D.; Sonneveld, E.; Zimmermann, M.; et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 2013, 27, 2280–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paggetti, J.; Largeot, A.; Aucagne, R.; Jacquel, A.; Lagrange, B.; Yang, X.-J.; Solary, E.; Bastie, J.-N.; Delva, L. Crosstalk between leukemia-associated proteins MOZ and MLL regulates HOX gene expression in human cord blood CD34+ cells. Oncogene 2010, 29, 5019–5031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreeff, M.; Ruvolo, V.; Gadgil, S.; Zeng, C.; Coombes, K.; Chen, W.; Kornblau, S.; Barón, A.E.; Drabkin, H.A. HOX expression patterns identify a common signature for favorable AML. Leukemia 2008, 22, 2041–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.; Iwasaki, M.; Somervaille, T.C.P.; So, C.W.E.; So, C.W.E.; Cleary, M.L. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007, 21, 2762–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | All Patients | Patients with KMT2A-r | Patients without KMT2A-r | p-Value | |
|---|---|---|---|---|---|
| Number | 967 (100%) | 241 (25%) | 726 (75%) | - | |
| Age | Median | 8.3 | 3.3 | 10.0 | <0.001 T (***) |
| Range | 0.0–18.0 | 0.0–17.9 | 0.0–18.0 | ||
| Gender | Male | 491 (100%) | 134 (27%) | 357 (73%) | 0.084 C (n.s.) |
| Female | 476 (100%) | 107 (22%) | 369 (78%) | ||
| WBC count [×103/µL] | Median | 16.1 | 19.3 | 15.8 | 0.001 T (**) |
| Range | 0.0–817.1 | 0.2–585.0 | 0.0–817.1 | ||
| Hemoglobin [g/dL] | Median | 8.3 | 8.3 | 8.3 | 0.156 T (n.s.) |
| Range | 2.1–18.3 | 2.1–17.1 | 3.1–18.3 | ||
| Platelet count [×103/µL] | Median | 66.0 | 89.0 | 60.0 | <0.001 T (***) |
| Range | 2.0–1370.0 | 5.0–468.0 | 2.0–1370.0 | ||
| Risk groups | Standard risk | 244 (27%) | 4 (2%) | 240 (35%) | <0.001 C (***) |
| Intermediate risk | 552 (61%) | 185 (81%) | 367 (54%) | ||
| High risk | 114 (13%) | 40 (17%) | 74 (11%) | ||
| No data | 57 | 12 | 45 | - | |
| Morphologic subtype (FAB classification) | M0 | 19 (2%) | 2 (1%) | 17 (%) | <0.001 C (***) |
| M1 | 112 (14%) | 6 (3%) | 106 (18%) | ||
| M2 | 175 (22%) | 4 (2%) | 171 (28%) | ||
| M4 | 134 (17%) | 36 (18%) | 98 (16%) | ||
| M4 eo | 67 (8%) | 0 (0%) | 67 (11%) | ||
| M5 | 198 (25%) | 147 (73%) | 51 (8%) | ||
| M6 | 12 (1%) | 1 (0%) | 11 (2%) | ||
| M7 | 88 (11%) | 6 (3%) | 82 (14%) | ||
| No data | 162 | 39 | 123 | - | |
| CSPG4 expression | Strong positive | 31 (9%) | 23 (27%) | 8 (3%) | <0.001 C (***) |
| Weak positive | 55 (16%) | 36 (42%) | 19 (7%) | ||
| Negative | 260 (75%) | 26 (31%) | 234 (90%) | ||
| No data | 621 | 156 | 465 | - | |
| Features | Total | Alive | Relapsed | Dead (Early Death) | Non- Response | Secondary Malignancy |
|---|---|---|---|---|---|---|
| no KMT2A-r | 726 | 547 | 206 | 179 (20) | 97 | 6 |
| KMT2A/MLLT3 | 104 | 81 | 20 | 23 (7) | 12 | 1 |
| KMT2A/MLLT10 | 55 | 37 | 16 | 18 (5) | 12 | 2 |
| KMT2A/AFDN | 11 | 5 | 7 | 6 (2) | 4 | 0 |
| KMT2A/MLLT1 | 11 | 8 | 3 | 3 (1) | 1 | 0 |
| KMT2A/MLLT11 | 8 | 7 | 1 | 1 (1) | 1 | 0 |
| KMT2A/AFF1 | 3 | 2 | 1 | 1 (0) | 0 | 0 |
| KMT2A/SEPTIN9 | 3 | 3 | 0 | 0 (0) | 0 | 0 |
| KMT2A/ELL | 3 | 2 | 1 | 1 (0) | 0 | 0 |
| KMT2A/MLLT6 | 2 | 2 | 0 | 0 (0) | 0 | 0 |
| KMT2A/PRPF19 | 2 | 1 | 0 | 1 (0) | 0 | 0 |
| KMT2A/ABI2 | 1 | 1 | 0 | 0 (0) | 0 | 0 |
| KMT2A/KNL1 | 1 | 1 | 1 | 0 (0) | 0 | 0 |
| KMT2A/USP2 | 1 | 1 | 0 | 0 (0) | 0 | 0 |
| KMT2A-r not further specified | 36 | 24 | 10 | 12 (2) | 4 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmeister, L.M.; Orhan, E.; Walter, C.; Niktoreh, N.; Hanenberg, H.; von Neuhoff, N.; Reinhardt, D.; Schneider, M. Impact of KMT2A Rearrangement and CSPG4 Expression in Pediatric Acute Myeloid Leukemia. Cancers 2021, 13, 4817. https://doi.org/10.3390/cancers13194817
Hoffmeister LM, Orhan E, Walter C, Niktoreh N, Hanenberg H, von Neuhoff N, Reinhardt D, Schneider M. Impact of KMT2A Rearrangement and CSPG4 Expression in Pediatric Acute Myeloid Leukemia. Cancers. 2021; 13(19):4817. https://doi.org/10.3390/cancers13194817
Chicago/Turabian StyleHoffmeister, Lina Marie, Eser Orhan, Christiane Walter, Naghmeh Niktoreh, Helmut Hanenberg, Nils von Neuhoff, Dirk Reinhardt, and Markus Schneider. 2021. "Impact of KMT2A Rearrangement and CSPG4 Expression in Pediatric Acute Myeloid Leukemia" Cancers 13, no. 19: 4817. https://doi.org/10.3390/cancers13194817
APA StyleHoffmeister, L. M., Orhan, E., Walter, C., Niktoreh, N., Hanenberg, H., von Neuhoff, N., Reinhardt, D., & Schneider, M. (2021). Impact of KMT2A Rearrangement and CSPG4 Expression in Pediatric Acute Myeloid Leukemia. Cancers, 13(19), 4817. https://doi.org/10.3390/cancers13194817