
Carfilzomib Enhances the Suppressive Effect of Ruxolitinib in Myelofibrosis

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Cell Line Preparation and Measurement of Sensitivity to Ruxolitinib
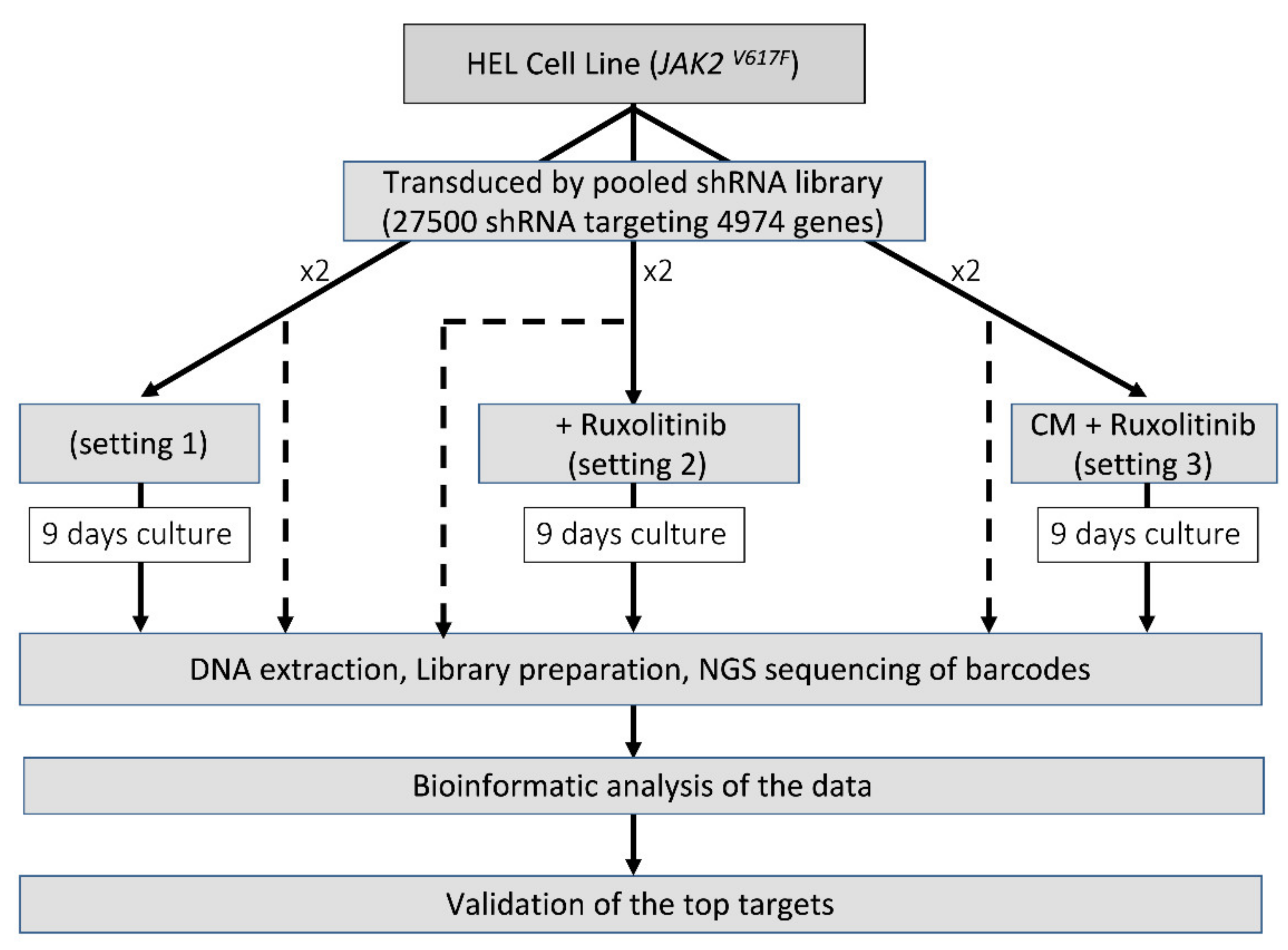
2.3. Pooled-shRNA Library Screen
2.4. DNA Extraction, shRNA, Bioinformatics and Statistical Analyses
2.5. Chemical Validation of Targets
2.6. Colony Formation Assay
2.7. RNA Sequencing and Analysis
3. Results
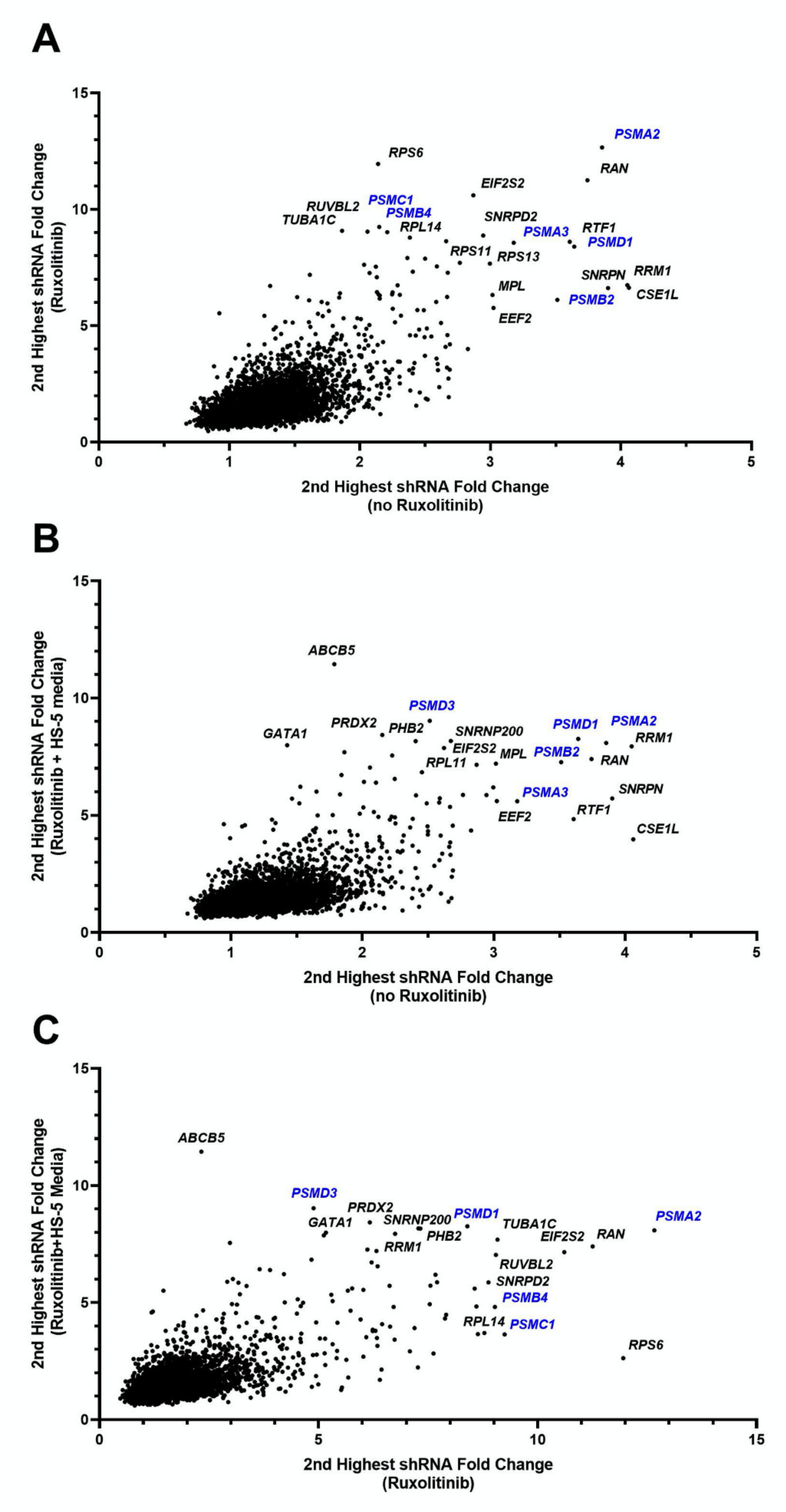
3.1. Pooled-shRNA Library Screen Identified Proteasome Family Activity Essential for the Viability of the HEL Cells in the Presence or Absence of Ruxolitinib
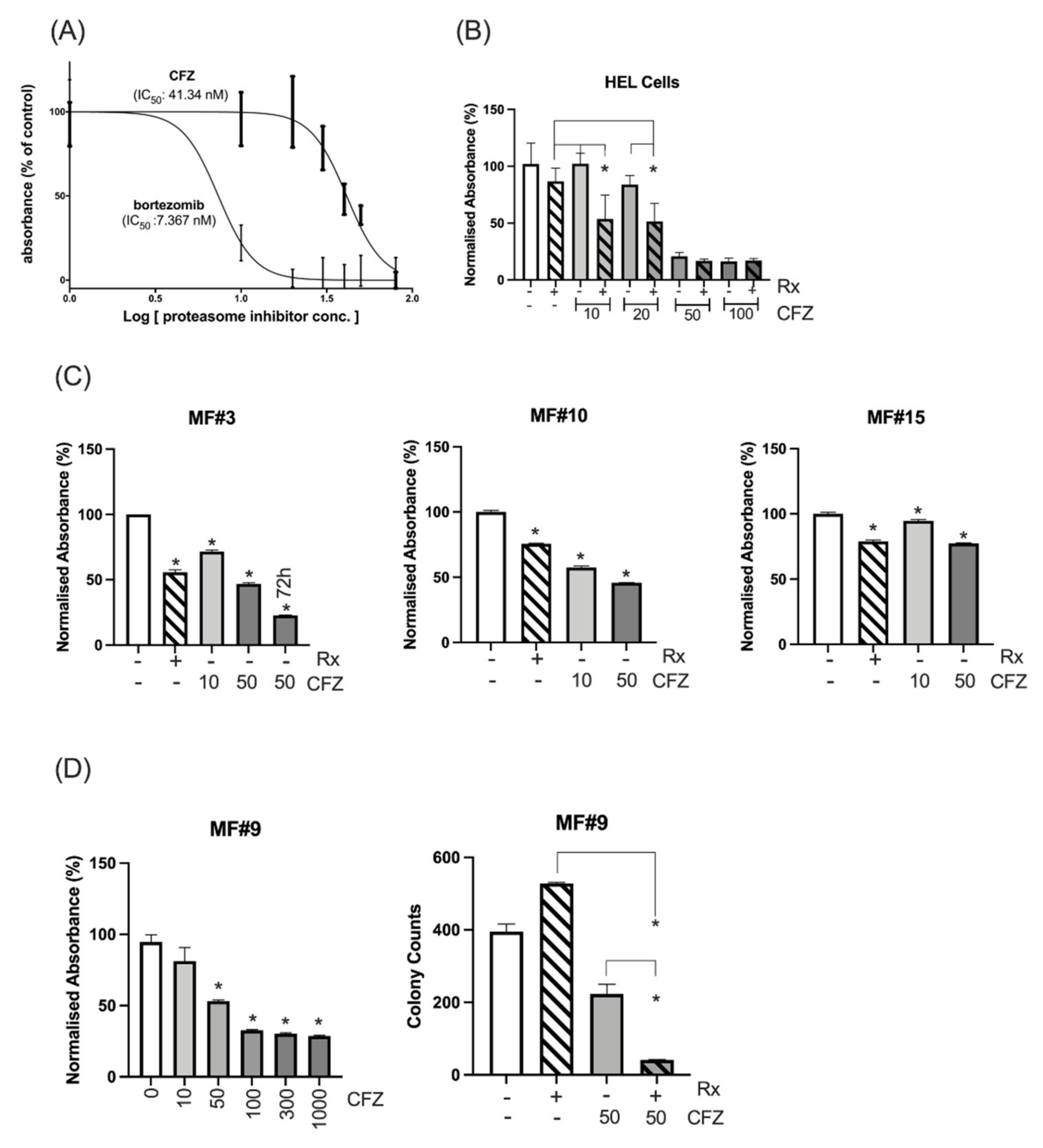
3.2. HEL Cells and Primary MF Cells Demonstrated High Sensitivity to Combined Inhibition of Proteasomes and JAK2/STAT5
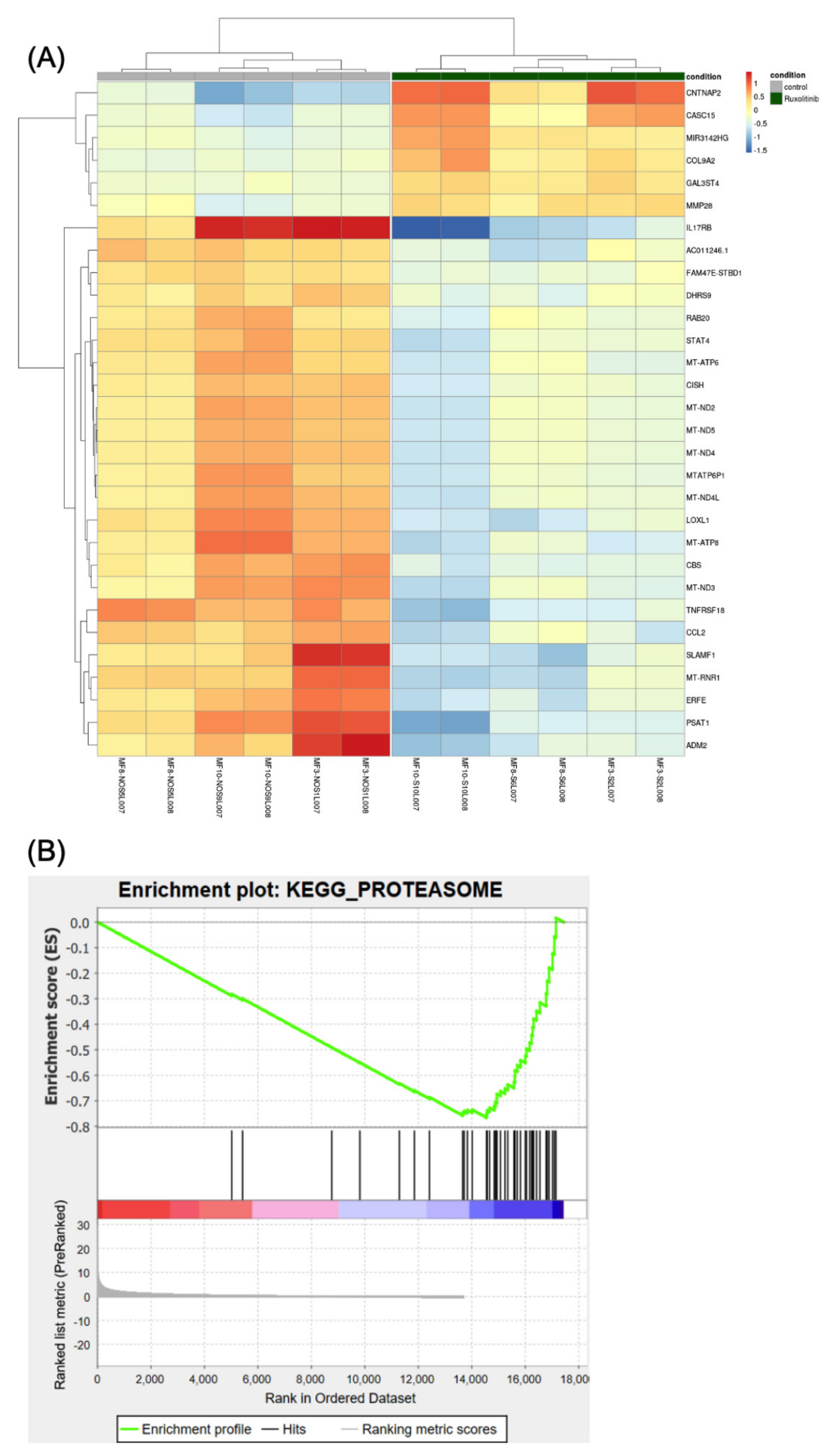
3.3. Gene Expression Analysis Shows Downregulation of Proteasome Genes by Ruxolitinib
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vallapureddy, R.R.; Mudireddy, M.; Penna, D.; Lasho, T.L.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Begna, K.H.; Gangat, N.; Pardanani, A.; et al. Leukemic transformation among 1306 patients with primary myelofibrosis: Risk factors and development of a predictive model. Blood Cancer J. 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Crispino, J.D.; Stein, B. Myelofibrosis in 2019: Moving beyond JAK2 inhibition. Blood Cancer J. 2019, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A. Primary myelofibrosis: 2014 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2014, 89, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Hoffman, R. Ruxolitinib: The first FDA approved therapy for the treatment of myelofibrosis. Clin. Cancer Res. 2012, 18, 3008–3014. [Google Scholar] [CrossRef] [Green Version]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.R.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.; Deininger, M.; Miller, C.B.; Silver, R.T.; et al. Results of COMFORT-I, a randomized double-blind phase III trial of JAK 1/2 inhibitor INCB18424 (424) versus placebo (PB) for patients with myelofibrosis (MF). J. Clin. Oncol. 2011, 29, 6500. [Google Scholar] [CrossRef]
- Passamonti, F.; Maffioli, M.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Barbui, T.; Caramazza, D.; Pieri, L.; Rumi, E.; Gisslinger, H.; et al. Impact of ruxolitinib on the natural history of primary myelofibrosis: A comparison of the DIPSS and the COMFORT-2 cohorts. Blood 2014, 123, 1833–1835. [Google Scholar] [CrossRef] [Green Version]
- Verstovsek, S.; Gotlib, J.; Mesa, R.A.; Vannucchi, A.M.; Kiladjian, J.J.; Cervantes, F.; Harrison, C.N.; Paquette, R.; Sun, W.; Naim, A.; et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J. Hematol. Oncol. 2017, 10, 156. [Google Scholar] [CrossRef] [Green Version]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar] [CrossRef] [Green Version]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Jayavelu, A.K.; Schnoder, T.M.; Perner, F.; Herzog, C.; Meiler, A.; Krishnamoorthy, G.; Huber, N.; Mohr, J.; Edelmann-Stephan, B.; Austin, R.; et al. Splicing factor YBX1 mediates persistence of JAK2-mutated neoplasms. Nature 2020, 588, 157–163. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Li, Z.W.; Dalton, W.S. Tumor microenvironment and drug resistance in hematologic malignancies. Blood Rev. 2006, 20, 333–342. [Google Scholar] [CrossRef]
- Wang, Y.; Fiskus, W.; Chong, D.G.; Buckley, K.M.; Natarajan, K.; Rao, R.; Joshi, A.; Balusu, R.; Koul, S.; Chen, J.; et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 2009, 114, 5024–5033. [Google Scholar] [CrossRef]
- Fiedler, W.; Henke, R.P.; Ergun, S.; Schumacher, U.; Gehling, U.M.; Vohwinkel, G.; Kilic, N.; Hossfeld, D.K. Derivation of a new hematopoietic cell line with endothelial features from a patient with transformed myeloproliferative syndrome: A case report. Cancer 2000, 88, 344–351. [Google Scholar] [CrossRef]
- Fiskus, W.; Verstovsek, S.; Manshouri, T.; Rao, R.; Balusu, R.; Venkannagari, S.; Rao, N.N.; Ha, K.; Smith, J.E.; Hembruff, S.L.; et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 7347–7358. [Google Scholar] [CrossRef] [Green Version]
- Traer, E.; MacKenzie, R.; Snead, J.; Agarwal, A.; Eiring, A.M.; O’Hare, T.; Druker, B.J.; Deininger, M.W. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia 2012, 26, 1140–1143. [Google Scholar] [CrossRef] [Green Version]
- Khorashad, J.S.; Eiring, A.M.; Mason, C.C.; Gantz, K.C.; Bowler, A.D.; Redwine, H.M.; Yu, F.; Kraft, I.L.; Pomicter, A.D.; Reynolds, K.R.; et al. shRNA library screening identifies nucleocytoplasmic transport as a mediator of BCR-ABL1 kinase-independent resistance. Blood 2015, 125, 1772–1781. [Google Scholar] [CrossRef] [Green Version]
- Quintas-Cardama, A.; Vaddi, K.; Liu, P.; Manshouri, T.; Li, J.; Scherle, P.A.; Caulder, E.; Wen, X.; Li, Y.; Waeltz, P.; et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: Therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010, 115, 3109–3117. [Google Scholar] [CrossRef]
- Chase, A.; Bryant, C.; Score, J.; Haferlach, C.; Grossmann, V.; Schwaab, J.; Hofmann, W.K.; Reiter, A.; Cross, N.C. Ruxolitinib as potential targeted therapy for patients with JAK2 rearrangements. Haematologica 2013, 98, 404–408. [Google Scholar] [CrossRef]
- Mason, C.C. Four study design principles for genetic investigations using next generation sequencing. BMJ 2017, 359, j4069. [Google Scholar] [CrossRef] [Green Version]
- Mason, C.C.; Fiol, C.R.; Baker, M.J.; Nadal-Melsio, E.; Yebra-Fernandez, E.; Bicalho, L.; Chowdhury, A.; Albert, M.; Reid, A.G.; Claudiani, S.; et al. Identification of genetic targets in acute myeloid leukaemia for designing targeted therapy. Br. J. Haematol. 2020. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Di Rosa, M.; Giallongo, C.; Romano, A.; Tibullo, D.; Li Volti, G.; Musumeci, G.; Barbagallo, I.; Imbesi, R.; Castrogiovanni, P.; Palumbo, G.A. Immunoproteasome Genes Are Modulated in CD34(+) JAK2(V617F) Mutated Cells from Primary Myelofibrosis Patients. Int. J. Mol. Sci. 2020, 21, 2926. [Google Scholar] [CrossRef]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Increased Expression of Proteasome-Related Genes In Patients with Primary Myelofibrosis. Blood 2010, 116, 4117. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hajek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Spicka, I.; Oriol, A.; Hajek, R.; Rosinol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef] [Green Version]
- Verstovsek, S.; Yeleswaram, S.; Hou, K.; Chen, X.; Erickson-Viitanen, S. Sustained-release ruxolitinib: Findings from a phase 1 study in healthy subjects and a phase 2 study in patients with myelofibrosis. Hematol. Oncol. 2018, 36, 701–708. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, K.P.; Siegel, D.S.; Vesole, D.H.; Lee, P.; Rosen, S.T.; Zojwalla, N.; Holahan, J.R.; Lee, S.; Wang, Z.; Badros, A. Phase I study of 30-minute infusion of carfilzomib as single agent or in combination with low-dose dexamethasone in patients with relapsed and/or refractory multiple myeloma. J. Clin. Oncol. 2015, 33, 732–739. [Google Scholar] [CrossRef]
- Suzuki, K.; Ri, M.; Chou, T.; Sugiura, I.; Takezako, N.; Sunami, K.; Ishida, T.; Izumi, T.; Ozaki, S.; Shumiya, Y.; et al. Carfilzomib, lenalidomide and dexamethasone in patients with heavily pretreated multiple myeloma: A phase 1 study in Japan. Cancer Sci. 2017, 108, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Faure, A.; Hayes, M.; Sugden, B. How Kaposi’s sarcoma-associated herpesvirus stably transforms peripheral B cells towards lymphomagenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 16519–16528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon-Gabriel, C.P.; Foerster, K.; Saleem, S.; Bleckmann, D.; Benkisser-Petersen, M.; Thornton, N.; Umezawa, K.; Decker, S.; Burger, M.; Veelken, H.; et al. Microenvironmental stromal cells abrogate NF-kappaB inhibitor-induced apoptosis in chronic lymphocytic leukemia. Haematologica 2018, 103, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Tabe, Y.; Konopleva, M. Role of Microenvironment in Resistance to Therapy in AML. Curr. Hematol. Malig. Rep. 2015, 10, 96–103. [Google Scholar] [CrossRef]
- Loscocco, F.; Visani, G.; Galimberti, S.; Curti, A.; Isidori, A. BCR-ABL Independent Mechanisms of Resistance in Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 939. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Pomicter, A.D.; Tantravahi, S.; Mason, C.C.; Senina, A.V.; Ahmann, J.M.; Wang, Q.; Than, H.; Patel, A.B.; Heaton, W.L.; et al. Nuclear-Cytoplasmic Transport Is a Therapeutic Target in Myelofibrosis. Clin. Cancer Res. 2019, 25, 2323–2335. [Google Scholar] [CrossRef]
- Besse, A.; Besse, L.; Kraus, M.; Mendez-Lopez, M.; Bader, J.; Xin, B.T.; de Bruin, G.; Maurits, E.; Overkleeft, H.S.; Driessen, C. Proteasome Inhibition in Multiple Myeloma: Head-to-Head Comparison of Currently Available Proteasome Inhibitors. Cell Chem. Biol. 2019, 26, 340–351. [Google Scholar] [CrossRef]
- Vangala, J.R.; Dudem, S.; Jain, N.; Kalivendi, S.V. Regulation of PSMB5 protein and beta subunits of mammalian proteasome by constitutively activated signal transducer and activator of transcription 3 (STAT3): Potential role in bortezomib-mediated anticancer therapy. J. Biol. Chem. 2014, 289, 12612–12622. [Google Scholar] [CrossRef] [Green Version]
- Kusoglu, A.; Bagca, B.G.; Ozates Ay, N.P.; Saydam, G.; Avci, C.B. Ruxolitinib Regulates the Autophagy Machinery in Multiple Myeloma Cells. Anticancer Agents Med. Chem. 2020. [Google Scholar] [CrossRef]
- Caocci, G.; La Nasa, G. Could ruxolitinib be effective in patients with COVID-19 infection at risk of acute respiratory distress syndrome (ARDS)? Ann. Hematol. 2020, 99, 1675–1676. [Google Scholar] [CrossRef]
- Pickering, A.M.; Koop, A.L.; Teoh, C.Y.; Ermak, G.; Grune, T.; Davies, K.J. The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem. J. 2010, 432, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Reinacker, K.; Dimayuga, E.; Nukala, V.; Drake, J.; Butterfield, D.A.; Dunn, J.C.; Martin, S.; Bruce-Keller, A.J.; Keller, J.N. Role of the proteasome in protein oxidation and neural viability following low-level oxidative stress. FEBS Lett. 2003, 546, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Aiken, C.T.; Kaake, R.M.; Wang, X.; Huang, L. Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell. Proteom. 2011, 10, R110.006924. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A. Regulation of Cytokine Production by the Unfolded Protein Response; Implications for Infection and Autoimmunity. Front. Immunol. 2018, 9, 422. [Google Scholar] [CrossRef]
- Khateb, A.; Ronai, Z.A. Unfolded Protein Response in Leukemia: From Basic Understanding to Therapeutic Opportunities. Trends Cancer 2020, 6, 960–973. [Google Scholar] [CrossRef]
- Salati, S.; Genovese, E.; Carretta, C.; Zini, R.; Bartalucci, N.; Prudente, Z.; Pennucci, V.; Ruberti, S.; Rossi, C.; Rontauroli, S.; et al. Calreticulin Ins5 and Del52 mutations impair unfolded protein and oxidative stress responses in K562 cells expressing CALR mutants. Sci. Rep. 2019, 9, 10558. [Google Scholar] [CrossRef] [Green Version]
- Nam, A.S.; Kim, K.T.; Chaligne, R.; Izzo, F.; Ang, C.; Taylor, J.; Myers, R.M.; Abu-Zeinah, G.; Brand, R.; Omans, N.D.; et al. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature 2019, 571, 355–360. [Google Scholar] [CrossRef]
- Tillmann, S.; Olschok, K.; Schroder, S.K.; Butow, M.; Baumeister, J.; Kalmer, M.; Preussger, V.; Weinbergerova, B.; Kricheldorf, K.; Mayer, J.; et al. The Unfolded Protein Response Is a Major Driver of LCN2 Expression in BCR-ABL- and JAK2V617F-Positive MPN. Cancers 2021, 13, 4210. [Google Scholar] [CrossRef]
- Digaleh, H.; Kiaei, M.; Khodagholi, F. Nrf2 and Nrf1 signaling and ER stress crosstalk: Implication for proteasomal degradation and autophagy. Cell. Mol. Life Sci. 2013, 70, 4681–4694. [Google Scholar] [CrossRef]
- Zhao, G.L.; Yu, L.M.; Gao, W.L.; Duan, W.X.; Jiang, B.; Liu, X.D.; Zhang, B.; Liu, Z.H.; Zhai, M.E.; Jin, Z.X.; et al. Berberine protects rat heart from ischemia/reperfusion injury via activating JAK2/STAT3 signaling and attenuating endoplasmic reticulum stress. Acta Pharmacol. Sin. 2016, 37, 354–367. [Google Scholar] [CrossRef]
- Goda, A.E.; Erikson, R.L.; Sakai, T.; Ahn, J.S.; Kim, B.Y. Preclinical evaluation of bortezomib/dipyridamole novel combination as a potential therapeutic modality for hematologic malignancies. Mol. Oncol. 2015, 9, 309–322. [Google Scholar] [CrossRef]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. The ubiquitin-proteasome system and activation of NF-kappaB: Involvement of the ubiquitin ligase KPC1 in p105 processing and tumor suppression. Mol. Cell. Oncol. 2015, 2, e1054552. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.J. Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell Biol. 2005, 7, 758–765. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.A.C.; Malkova, O.; Engle, E.K.; Miner, C.A.; Fulbright, M.C.; Behbehani, G.K.; Collins, T.B.; Bandyopadhyay, S.; Zhou, A.; Nolan, G.P.; et al. Mass cytometry analysis reveals hyperactive NF Kappa B signaling in myelofibrosis and secondary acute myeloid leukemia. Leukemia 2017, 31, 1962–1974. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.B.; Meng, F.R.; Fang, Y.X.; Wu, X.; Zhang, C.W.; Liu, Y.; Liu, D.; Li, G.Q.; Feng, F.B.; Qiu, H.Y. Inhibition of NF-kappaB signaling pathway induces apoptosis and suppresses proliferation and angiogenesis of human fibroblast-like synovial cells in rheumatoid arthritis. Medicine 2018, 97, e10920. [Google Scholar] [CrossRef]
- Wang, C.Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S., Jr. NF-kappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef]
- Bours, V.; Bentires-Alj, M.; Hellin, A.C.; Viatour, P.; Robe, P.; Delhalle, S.; Benoit, V.; Merville, M.P. Nuclear factor-kappa B, cancer, and apoptosis. Biochem. Pharmacol. 2000, 60, 1085–1089. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Guang, M.H.; Zhuo, H.Q.; Min, X.H.; Yao, Q.; Gu, A.Q.; Wu, S.H.; Zhang, D.B.; Lu, J.Y.; Chen, Y.; et al. Carfilzomib Inhibits Constitutive NF-kappaB Activation in Mantle Cell Lymphoma B Cells and Leads to the Induction of Apoptosis. Acta Haematol. 2017, 137, 106–112. [Google Scholar] [CrossRef]
- Vrabel, D.; Pour, L.; Sevcikova, S. The impact of NF-kappaB signaling on pathogenesis and current treatment strategies in multiple myeloma. Blood Rev. 2019, 34, 56–66. [Google Scholar] [CrossRef]
- Ghermezi, M.; Spektor, T.M.; Berenson, J.R. The role of JAK inhibitors in multiple myeloma. Clin. Adv. Hematol. Oncol. 2019, 17, 500–505. [Google Scholar] [PubMed]
- Wagner-Ballon, O.; Pisani, D.F.; Gastinne, T.; Tulliez, M.; Chaligne, R.; Lacout, C.; Aurade, F.; Villeval, J.L.; Gonin, P.; Vainchenker, W.; et al. Proteasome inhibitor bortezomib impairs both myelofibrosis and osteosclerosis induced by high thrombopoietin levels in mice. Blood 2007, 110, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesa, R.A.; Verstovsek, S.; Rivera, C.; Pardanani, A.; Hussein, K.; Lasho, T.; Wu, W.; Tefferi, A. Bortezomib therapy in myelofibrosis: A phase II clinical trial. Leukemia 2008, 22, 1636–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barosi, G.; Gattoni, E.; Guglielmelli, P.; Campanelli, R.; Facchetti, F.; Fisogni, S.; Goldberg, J.; Marchioli, R.; Hoffman, R.; Vannucchi, A.M.; et al. Phase I/II study of single-agent bortezomib for the treatment of patients with myelofibrosis. Clinical and biological effects of proteasome inhibition. Am. J. Hematol. 2010, 85, 616–619. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Chng, W.J.; Feng, S. Carfilozomib versus bortezomib for relapsed or refractory myeloma—Authors’ reply. Lancet Oncol. 2016, 17, e126. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Levitsky, K.; Parlati, F.; Bennett, M.K.; Arastu-Kapur, S.; Kellerman, L.; Woo, T.F.; Wong, A.F.; Papadopoulos, K.P.; Niesvizky, R.; et al. Clinical activity of carfilzomib correlates with inhibition of multiple proteasome subunits: Application of a novel pharmacodynamic assay. Br. J. Haematol. 2016, 173, 884–895. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Roussou, M.; Gavriatopoulou, M.; Psimenou, E.; Ziogas, D.; Eleutherakis-Papaiakovou, E.; Fotiou, D.; Migkou, M.; Kanellias, N.; Panagiotidis, I.; et al. Cardiac and renal complications of carfilzomib in patients with multiple myeloma. Blood Adv. 2017, 1, 449–454. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Y.; Hsieh, F.S.; Chu, P.Y.; Tsai, W.C.; Huang, C.T.; Yu, Y.B.; Huang, T.T.; Ko, P.S.; Hung, M.H.; Wang, W.L.; et al. Carfilzomib induces leukaemia cell apoptosis via inhibiting ELK1/KIAA1524 (Elk-1/CIP2A) and activating PP2A not related to proteasome inhibition. Br. J. Haematol. 2017, 177, 726–740. [Google Scholar] [CrossRef] [Green Version]
- Ponder, K.G.; Matulis, S.M.; Hitosugi, S.; Gupta, V.A.; Sharp, C.; Burrows, F.; Nooka, A.K.; Kaufman, J.L.; Lonial, S.; Boise, L.H. Dual inhibition of Mcl-1 by the combination of carfilzomib and TG02 in multiple myeloma. Cancer Biol. Ther. 2016, 17, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Roberts, L.; Chen, Z.; Merta, P.J.; Glaser, K.B.; Shah, O.J. JAK2V617F drives Mcl-1 expression and sensitizes hematologic cell lines to dual inhibition of JAK2 and Bcl-xL. PLoS ONE 2015, 10, e0114363. [Google Scholar] [CrossRef] [Green Version]
- Bartalucci, N.; Calabresi, L.; Balliu, M.; Martinelli, S.; Rossi, M.C.; Villeval, J.L.; Annunziato, F.; Guglielmelli, P.; Vannucchi, A.M. Inhibitors of the PI3K/mTOR pathway prevent STAT5 phosphorylation in JAK2V617F mutated cells through PP2A/CIP2A axis. Oncotarget 2017, 8, 96710–96724. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claudiani, S.; Mason, C.C.; Milojkovic, D.; Bianchi, A.; Pellegrini, C.; Di Marco, A.; Fiol, C.R.; Robinson, M.; Ponnusamy, K.; Mokretar, K.; et al. Carfilzomib Enhances the Suppressive Effect of Ruxolitinib in Myelofibrosis. Cancers 2021, 13, 4863. https://doi.org/10.3390/cancers13194863
Claudiani S, Mason CC, Milojkovic D, Bianchi A, Pellegrini C, Di Marco A, Fiol CR, Robinson M, Ponnusamy K, Mokretar K, et al. Carfilzomib Enhances the Suppressive Effect of Ruxolitinib in Myelofibrosis. Cancers. 2021; 13(19):4863. https://doi.org/10.3390/cancers13194863
Chicago/Turabian StyleClaudiani, Simone, Clinton C. Mason, Dragana Milojkovic, Andrea Bianchi, Cristina Pellegrini, Antinisca Di Marco, Carme R. Fiol, Mark Robinson, Kanagaraju Ponnusamy, Katya Mokretar, and et al. 2021. "Carfilzomib Enhances the Suppressive Effect of Ruxolitinib in Myelofibrosis" Cancers 13, no. 19: 4863. https://doi.org/10.3390/cancers13194863
APA StyleClaudiani, S., Mason, C. C., Milojkovic, D., Bianchi, A., Pellegrini, C., Di Marco, A., Fiol, C. R., Robinson, M., Ponnusamy, K., Mokretar, K., Chowdhury, A., Albert, M., Reid, A. G., Deininger, M. W., Naresh, K., Apperley, J. F., & Khorashad, J. S. (2021). Carfilzomib Enhances the Suppressive Effect of Ruxolitinib in Myelofibrosis. Cancers, 13(19), 4863. https://doi.org/10.3390/cancers13194863