
Clonal Architecture and Evolutionary Dynamics in Acute Myeloid Leukemias


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Clonal Identity in AML
2.1. Genetic Identity
2.2. Functional Identity
2.3. Multi-Modal Identity
3. Origin of Intraleukemic Heterogeneity and Clonal Evolution
3.1. Characterizing Intratumor Heterogeneity
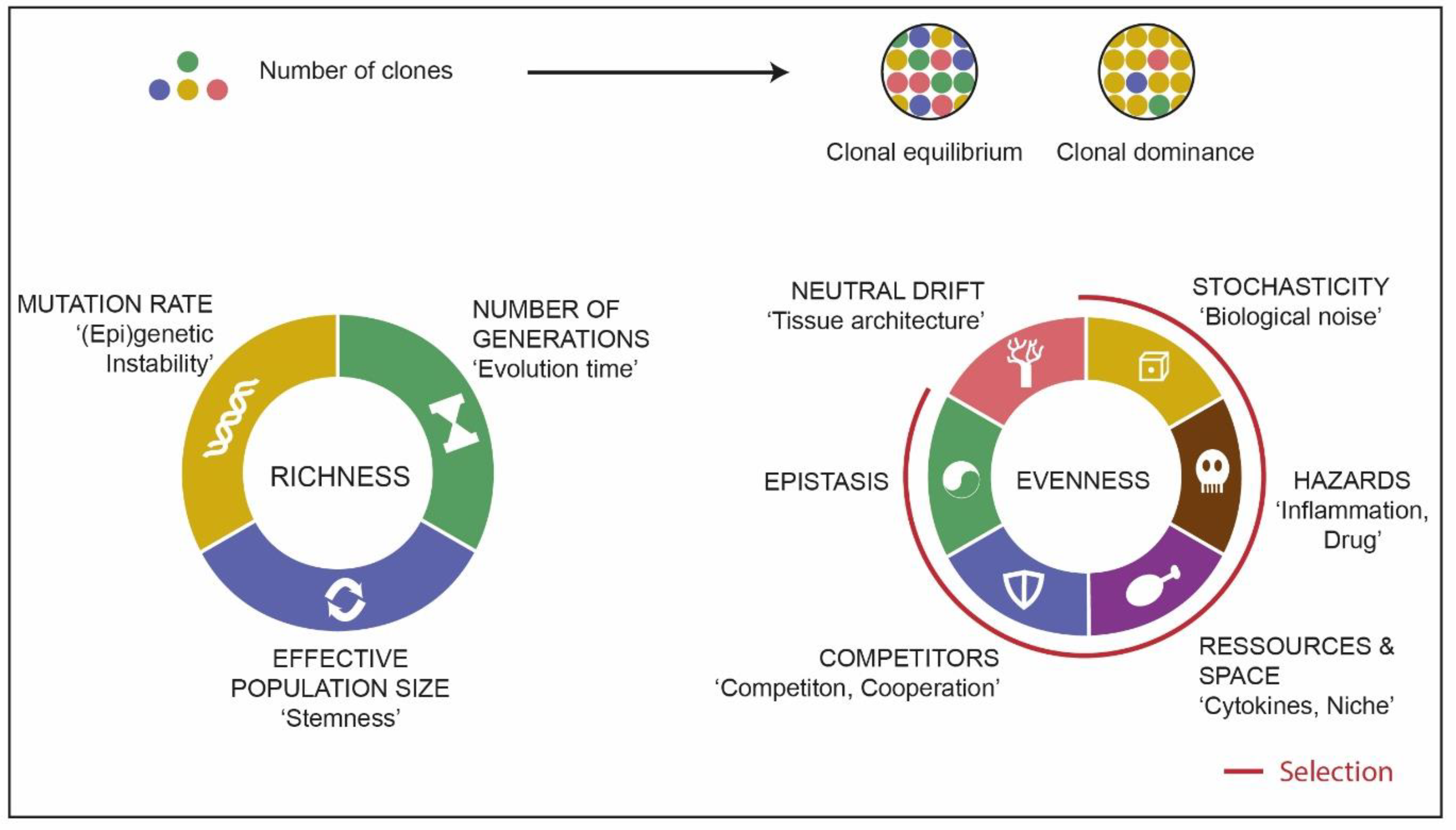
3.2. Source of Clonal Richness
3.3. Clonal Evolution Processes
4. Clinical Impact of Clonal Architecture
4.1. Impact of Clonal Architecture at Diagnosis
4.2. New Treatment Strategies
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Hauschka, T.S. The chromosomes in ontogeny and oncogeny. Cancer Res. 1961, 21, 957–974. [Google Scholar] [PubMed]
- Hausser, J.; Alon, U. Tumour heterogeneity and the evolutionary trade-offs of cancer. Nat. Rev. Cancer 2020, 20, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Jourdain, A.; Duchmann, M.; Willekens, C.; Solary, É.; Perié, L.; Laplane, L. Évolution clonale: Qu’est-ce qu’un clone? In La Biologie au Défi de L’histoire; Chapitre 10; Éditions Matériologiques: Paris, France, 2021; pp. 167–188. [Google Scholar]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osorio, F.G.; Rosendahl Huber, A.; Oka, R.; Verheul, M.; Patel, S.H.; Hasaart, K.; de la Fonteijne, L.; Varela, I.; Camargo, F.D.; van Boxtel, R. Somatic Mutations Reveal Lineage Relationships and Age-Related Mutagenesis in Human Hematopoiesis. Cell Rep. 2018, 25, 2308–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Six, H.; Obro, N.F.; Shepherd, M.S.; Grossmann, S.; Dawson, K.; Belmonte, M.; Osborne, R.J.; Huntly, B.J.P.; Martincorena, I.; Anderson, E.; et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018, 561, 473–478. [Google Scholar] [CrossRef]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Warrell, J.; Li, S.; McGillivray, P.D.; Meyerson, W.; Salichos, L.; Harmanci, A.; Martinez-Fundichely, A.; Chan, C.W.Y.; Nielsen, M.M.; et al. Passenger Mutations in More Than 2,500 Cancer Genomes: Overall Molecular Functional Impact and Consequences. Cell 2020, 180, 915–927.e916. [Google Scholar] [CrossRef] [PubMed]
- Castro-Giner, F.; Ratcliffe, P.; Tomlinson, I. The mini-driver model of polygenic cancer evolution. Nat. Rev. Cancer 2015, 15, 680–685. [Google Scholar] [CrossRef]
- Testa, J.R.; Mintz, U.; Rowley, J.D.; Vardiman, J.W.; Golomb, H.M. Evolution of karyotypes in acute nonlymphocytic leukemia. Cancer Res. 1979, 39, 3619–3627. [Google Scholar]
- Bochtler, T.; Stolzel, F.; Heilig, C.E.; Kunz, C.; Mohr, B.; Jauch, A.; Janssen, J.W.; Kramer, M.; Benner, A.; Bornhauser, M.; et al. Clonal heterogeneity as detected by metaphase karyotyping is an indicator of poor prognosis in acute myeloid leukemia. J. Clin. Oncol. 2013, 31, 3898–3905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medeiros, B.C.; Othus, M.; Fang, M.; Appelbaum, F.R.; Erba, H.P. Cytogenetic heterogeneity negatively impacts outcomes in patients with acute myeloid leukemia. Haematologica 2015, 100, 331–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, A.; Khattra, J.; Yap, D.; Wan, A.; Laks, E.; Biele, J.; Ha, G.; Aparicio, S.; Bouchard-Cote, A.; Shah, S.P. PyClone: Statistical inference of clonal population structure in cancer. Nat. Methods 2014, 11, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Black, J.R.M.; McGranahan, N. Genetic and non-genetic clonal diversity in cancer evolution. Nat. Rev. Cancer 2021, 21, 379–392. [Google Scholar] [CrossRef]
- Cerrano, M.; Duchmann, M.; Kim, R.; Vasseur, L.; Hirsch, P.; Thomas, X.; Quentin, S.; Pasanisi, J.; Passet, M.; Rabian, F.; et al. Clonal dominance is an adverse prognostic factor in acute myeloid leukemia treated with intensive chemotherapy. Leukemia 2020, 35, 712–723. [Google Scholar] [CrossRef]
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; Sträng, E.; Theis, F.; Jahn, N.; Panina, E.; Blätte, T.J.; Herzig, J.; Skambraks, S.; et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood 2021, 137, 3093–3104. [Google Scholar] [CrossRef]
- Greif, P.A.; Hartmann, L.; Vosberg, S.; Stief, S.M.; Mattes, R.; Hellmann, I.; Metzeler, K.; Herold, T.; Bamopoulos, S.; Kerbs, P.; et al. Evolution of Cytogenetically Normal Acute Myeloid Leukemia During Therapy and Relapse: An Exome Sequencing Study of 50 Patients. Clin. Cancer Res. 2018, 24, 1716–1726. [Google Scholar] [CrossRef] [Green Version]
- Cocciardi, S.; Dolnik, A.; Kapp-Schwoerer, S.; Rücker, F.G.; Lux, S.; Blätte, T.J.; Skambraks, S.; Krönke, J.; Heidel, F.H.; Schnöder, T.M.; et al. Clonal evolution patterns in acute myeloid leukemia with NPM1 mutation. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, P.; Zhang, Y.; Tang, R.; Joulin, V.; Boutroux, H.; Pronier, E.; Moatti, H.; Flandrin, P.; Marzac, C.; Bories, D.; et al. Genetic hierarchy and temporal variegation in the clonal history of acute myeloid leukaemia. Nat. Commun. 2016, 7, 12475. [Google Scholar] [CrossRef]
- Klco, J.M.; Spencer, D.H.; Miller, C.A.; Griffith, M.; Lamprecht, T.L.; O’Laughlin, M.; Fronick, C.; Magrini, V.; Demeter, R.T.; Fulton, R.S.; et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 2014, 25, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Quek, L.; David, M.D.; Kennedy, A.; Metzner, M.; Amatangelo, M.; Shih, A.; Stoilova, B.; Quivoron, C.; Heiblig, M.; Willekens, C.; et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat. Med. 2018, 1167–1177. [Google Scholar] [CrossRef]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 5327. [Google Scholar] [CrossRef]
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020, 587, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Duployez, N.; Fasan, A.; Decool, G.; Marceau-Renaut, A.; Meggendorfer, M.; Jourdan, E.; Petit, A.; Lapillonne, H.; Micol, J.B.; et al. Clonal interference of signaling mutations worsens prognosis in core-binding factor acute myeloid leukemia. Blood 2018, 132, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Heffernan, T.P.; Gera, S.; Kovacs, J.J.; et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 2018, 23, 700–713. [Google Scholar] [CrossRef] [Green Version]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Itzykson, R.A.; Fournier, E.; Berthon, C.; Rollig, C.; Braun, T.; Marceau-Renaut, A.; Pautas, C.; Nibourel, O.; Lemasle, E.; Micol, J.B.; et al. Genetic Identification of AML Patients Older than 60 years Achieving Long-term Survival with Intensive Chemotherapy. Blood 2021, 138, 507–519. [Google Scholar] [CrossRef]
- Gerstung, M.; Papaemmanuil, E.; Martincorena, I.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Heuser, M.; Thol, F.; Bolli, N.; Ganly, P.; et al. Precision oncology for acute myeloid leukemia using a knowledge bank approach. Nat. Genet. 2017, 49, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Fenwarth, L.; Thomas, X.; de Botton, S.; Duployez, N.; Bourhis, J.H.; Lesieur, A.; Fortin, G.; Meslin, P.A.; Yakoub-Agha, I.; Sujobert, P.; et al. A personalized approach to guide allogeneic stem cell transplantation in younger adults with acute myeloid leukemia. Blood 2021, 137, 524–532. [Google Scholar] [CrossRef]
- van Galen, P.; Hovestadt, V.; Wadsworth Ii, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; Amir el, A.D.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R.; et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef]
- Zeijlemaker, W.; Grob, T.; Meijer, R.; Hanekamp, D.; Kelder, A.; Carbaat-Ham, J.C.; Oussoren-Brockhoff, Y.J.M.; Snel, A.N.; Veldhuizen, D.; Scholten, W.J.; et al. CD34(+)CD38(-) leukemic stem cell frequency to predict outcome in acute myeloid leukemia. Leukemia 2019, 33, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Acute Myeloid Leukemia Patients. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Kuusanmaki, H.; Leppa, A.M.; Polonen, P.; Kontro, M.; Dufva, O.; Deb, D.; Yadav, B.; Bruck, O.; Kumar, A.; Everaus, H.; et al. Phenotype-based drug screening reveals association between venetoclax response and differentiation stage in acute myeloid leukemia. Haematologica 2020, 105, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L.; et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Chen, X.; Wang, J.; Meydan, C.; Glass, J.L.; Shih, A.H.; Delwel, R.; Levine, R.L.; Mason, C.E.; Melnick, A.M. Somatic Mutations Drive Specific, but Reversible, Epigenetic Heterogeneity States in AML. Cancer Discov. 2020, 10, 1934–1949. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Garrett-Bakelman, F.; Perl, A.E.; Luger, S.M.; Zhang, C.; To, B.L.; Lewis, I.D.; Brown, A.L.; D’Andrea, R.J.; Ross, M.E.; et al. Dynamic evolution of clonal epialleles revealed by methclone. Genome Biol. 2014, 15, 472. [Google Scholar] [CrossRef]
- Brock, A.; Chang, H.; Huang, S. Non-genetic heterogeneity--a mutation-independent driving force for the somatic evolution of tumours. Nat. Rev. Genet. 2009, 10, 336–342. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Emert, B.L.; Reyes Hueros, R.A.; Cote, C.; Harmange, G.; Schaff, D.L.; Sizemore, A.E.; Gupte, R.; Torre, E.; Singh, A.; et al. Memory Sequencing Reveals Heritable Single-Cell Gene Expression Programs Associated with Distinct Cellular Behaviors. Cell 2020, 182, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisco, A.O.; Brock, A.; Zhou, J.; Moor, A.; Mojtahedi, M.; Jackson, D.; Huang, S. Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nat. Commun. 2013, 4, 2467. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Scheiner, S.M. The Baldwin effect: Neglected and misunderstood. Am. Nat. 2014, 184, ii–iii. [Google Scholar] [CrossRef] [PubMed]
- Turati, V.A.; Guerra-Assunção, J.A.; Potter, N.E.; Gupta, R.; Ecker, S.; Daneviciute, A.; Tarabichi, M.; Webster, A.P.; Ding, C.; May, G.; et al. Chemotherapy induces canalization of cell state in childhood B-cell precursor acute lymphoblastic leukemia. Nat. Cancer 2021, 2, 835–852. [Google Scholar] [CrossRef]
- Boyd, A.L.; Aslostovar, L.; Reid, J.; Ye, W.; Tanasijevic, B.; Porras, D.P.; Shapovalova, Z.; Almakadi, M.; Foley, R.; Leber, B.; et al. Identification of Chemotherapy-Induced Leukemic-Regenerating Cells Reveals a Transient Vulnerability of Human AML Recurrence. Cancer Cell 2018, 34, 483–498. [Google Scholar] [CrossRef] [Green Version]
- Duy, C.; Li, M.; Teater, M.; Meydan, C.; Garrett-Bakelman, F.E.; Lee, T.C.; Chin, C.R.; Durmaz, C.; Kawabata, K.C.; Dhimolea, E.; et al. Chemotherapy Induces Senescence-Like Resilient Cells Capable of Initiating AML Recurrence. Cancer Discov. 2021, 11, 1542–1561. [Google Scholar] [CrossRef]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Potter, N.; Miraki-Moud, F.; Ermini, L.; Titley, I.; Vijayaraghavan, G.; Papaemmanuil, E.; Campbell, P.; Gribben, J.; Taussig, D.; Greaves, M. Single cell analysis of clonal architecture in acute myeloid leukaemia. Leukemia 2019, 33, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Sanden, C.; Lilljebjorn, H.; Orsmark Pietras, C.; Henningsson, R.; Saba, K.H.; Landberg, N.; Thorsson, H.; von Palffy, S.; Pena-Martinez, P.; Hogberg, C.; et al. Clonal competition within complex evolutionary hierarchies shapes AML over time. Nat. Commun. 2020, 11, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petti, A.A.; Williams, S.R.; Miller, C.A.; Fiddes, I.T.; Srivatsan, S.N.; Chen, D.Y.; Fronick, C.C.; Fulton, R.S.; Church, D.M.; Ley, T.J. A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat. Commun. 2019, 10, 3660. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Szekely, P.; Bar, N.; Zimmer, A.; Sheftel, H.; Caldas, C.; Alon, U. Tumor diversity and the trade-off between universal cancer tasks. Nat. Commun. 2019, 10, 5423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Meira, A.; Buck, G.; Clark, S.A.; Povinelli, B.J.; Alcolea, V.; Louka, E.; McGowan, S.; Hamblin, A.; Sousos, N.; Barkas, N.; et al. Unravelling Intratumoral Heterogeneity through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol. Cell 2019, 73, 1292–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, R.H. Evolution and measurement of species diversity. Taxon 1972, 21, 213–251. [Google Scholar] [CrossRef] [Green Version]
- Werstein, B.; Dunlap, J.; Cascio, M.J.; Ohgami, R.S.; Fan, G.; Press, R.; Raess, P.W. Molecular Discordance between Myeloid Sarcomas and Concurrent Bone Marrows Occurs in Actionable Genes and Is Associated with Worse Overall Survival. J. Mol. Diagn. 2020, 22, 338–345. [Google Scholar] [CrossRef]
- Duarte, D.; Amarteifio, S.; Ang, H.; Kong, I.Y.; Ruivo, N.; Pruessner, G.; Hawkins, E.D.; Lo Celso, C. Defining the in vivo characteristics of acute myeloid leukemia cells behavior by intravital imaging. Immunol. Cell Biol. 2019, 97, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Maley, C.C.; Aktipis, A.; Graham, T.A.; Sottoriva, A.; Boddy, A.M.; Janiszewska, M.; Silva, A.S.; Gerlinger, M.; Yuan, Y.; Pienta, K.J.; et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer 2017, 17, 605–619. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Barrick, J.E.; Lenski, R.E. Genome dynamics during experimental evolution. Nat. Rev. Genet. 2013, 14, 827–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M.; Ackerman, M.S.; Gout, J.F.; Long, H.; Sung, W.; Thomas, W.K.; Foster, P.L. Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 2016, 17, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Yurchenko, A.A.; Padioleau, I.; Matkarimov, B.T.; Soulier, J.; Sarasin, A.; Nikolaev, S. XPC deficiency increases risk of hematologic malignancies through mutator phenotype and characteristic mutational signature. Nat. Commun. 2020, 11, 5834. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Lindsley, R.C.; Mar, B.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caravagna, G.; Heide, T.; Williams, M.J.; Zapata, L.; Nichol, D.; Chkhaidze, K.; Cross, W.; Cresswell, G.D.; Werner, B.; Acar, A.; et al. Subclonal reconstruction of tumors by using machine learning and population genetics. Nat. Genet. 2020, 52, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Fontana, M.C.; Marconi, G.; Feenstra, J.D.M.; Fonzi, E.; Papayannidis, C.; Ghelli Luserna di Rora, A.; Padella, A.; Solli, V.; Franchini, E.; Ottaviani, E.; et al. Chromothripsis in acute myeloid leukemia: Biological features and impact on survival. Leukemia 2018, 32, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magrì, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Kawecki, T.J.; Lenski, R.E.; Ebert, D.; Hollis, B.; Olivieri, I.; Whitlock, M.C. Experimental evolution. Trends Ecol. Evol. 2012, 27, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Grajzel, D.; Derenyi, I.; Szollosi, G.J. A compartment size-dependent selective threshold limits mutation accumulation in hierarchical tissues. Proc. Natl. Acad. Sci. USA 2020, 117, 1606–1611. [Google Scholar] [CrossRef] [Green Version]
- Cairns, J. Mutation selection and the natural history of cancer. Nature 1975, 255, 197–200. [Google Scholar] [CrossRef]
- Greaves, M. Cancer stem cells as ‘units of selection’. Evol. Appl. 2013, 6, 102–108. [Google Scholar] [CrossRef]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; de Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef] [PubMed]
- de Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, M.D.; Ghisi, M.; Oxley, E.P.; Ngo, S.; Cimmino, L.; Esnault, C.; Liu, R.; Salmon, J.M.; Bell, C.C.; Ahmed, N.; et al. Interconversion between Tumorigenic and Differentiated States in Acute Myeloid Leukemia. Cell Stem Cell 2019, 25, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, J.; Schenck, R.O.; Gatenbee, C.; Robertson-Tessi, M.; Anderson, A.R.A. Normal tissue architecture determines the evolutionary course of cancer. Nat. Commun. 2021, 12, 2060. [Google Scholar] [CrossRef]
- Lyne, A.M.; Laplane, L.; Perie, L. To portray clonal evolution in blood cancer, count your stem cells. Blood 2021, 137, 1862–1870. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2012, 44, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Henry, C.J.; Marusyk, A.; Zaberezhnyy, V.; Adane, B.; DeGregori, J. Declining lymphoid progenitor fitness promotes aging-associated leukemogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 21713–21718. [Google Scholar] [CrossRef] [Green Version]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef] [Green Version]
- Beerman, I.; Bhattacharya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D.; Rossi, D.J. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 5465–5470. [Google Scholar] [CrossRef] [Green Version]
- Kuranda, K.; Vargaftig, J.; de la Rochere, P.; Dosquet, C.; Charron, D.; Bardin, F.; Tonnelle, C.; Bonnet, D.; Goodhardt, M. Age-related changes in human hematopoietic stem/progenitor cells. Aging Cell 2011, 10, 542–546. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; A Sekeres, M.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2016, 49, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Archetti, M. Cooperation between cancer cells. Evol. Med. Public Health 2018, 2018, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Goparaju, R.; Schwarzer, A.; Gorlich, K.; Schottmann, R.; Struys, E.A.; Jansen, E.E.; et al. Enantiomer-specific and paracrine leukemogenicity of mutant IDH metabolite 2-hydroxyglutarate. Leukemia 2016, 30, 1708–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madan, E.; Gogna, R.; Moreno, E. Cell competition in development: Information from flies and vertebrates. Curr. Opin. Cell Biol. 2018, 55, 150–157. [Google Scholar] [CrossRef]
- Miething, C.; Scuoppo, C.; Bosbach, B.; Appelmann, I.; Nakitandwe, J.; Ma, J.; Wu, G.; Lintault, L.; Auer, M.; Premsrirut, P.K.; et al. PTEN action in leukaemia dictated by the tissue microenvironment. Nature 2014, 510, 402–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kon, S.; Ishibashi, K.; Katoh, H.; Kitamoto, S.; Shirai, T.; Tanaka, S.; Kajita, M.; Ishikawa, S.; Yamauchi, H.; Yako, Y.; et al. Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat. Cell Biol. 2017, 19, 530–541. [Google Scholar] [CrossRef]
- Madan, E.; Pelham, C.J.; Nagane, M.; Parker, T.M.; Canas-Marques, R.; Fazio, K.; Shaik, K.; Yuan, Y.; Henriques, V.; Galzerano, A.; et al. Flower isoforms promote competitive growth in cancer. Nature 2019, 572, 260–264. [Google Scholar] [CrossRef]
- Bondar, T.; Medzhitov, R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell 2010, 6, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, A.L.; Campbell, C.J.; Hopkins, C.I.; Fiebig-Comyn, A.; Russell, J.; Ulemek, J.; Foley, R.; Leber, B.; Xenocostas, A.; Collins, T.J.; et al. Niche displacement of human leukemic stem cells uniquely allows their competitive replacement with healthy HSPCs. J. Exp. Med. 2014, 211, 1925–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glait-Santar, C.; Desmond, R.; Feng, X.; Bat, T.; Chen, J.; Heuston, E.; Mizukawa, B.; Mulloy, J.C.; Bodine, D.M.; Larochelle, A.; et al. Functional Niche Competition Between Normal Hematopoietic Stem and Progenitor Cells and Myeloid Leukemia Cells. Stem Cells 2015, 33, 3635–3642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guezguez, B.; Campbell, C.J.; Boyd, A.L.; Karanu, F.; Casado, F.L.; Di Cresce, C.; Collins, T.J.; Shapovalova, Z.; Xenocostas, A.; Bhatia, M. Regional localization within the bone marrow influences the functional capacity of human HSCs. Cell Stem Cell 2013, 13, 175–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colvin, G.A.; Lambert, J.F.; Abedi, M.; Hsieh, C.C.; Carlson, J.E.; Stewart, F.M.; Quesenberry, P.J. Murine marrow cellularity and the concept of stem cell competition: Geographic and quantitative determinants in stem cell biology. Leukemia 2004, 18, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Czechowicz, A.; Kraft, D.; Weissman, I.L.; Bhattacharya, D. Efficient transplantation via antibody-based clearance of hematopoietic stem cell niches. Science 2007, 318, 1296–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Duarte, D.; Hawkins, E.D.; Akinduro, O.; Ang, H.; De Filippo, K.; Kong, I.Y.; Haltalli, M.; Ruivo, N.; Straszkowski, L.; Vervoort, S.J.; et al. Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Cell Stem Cell 2018, 22, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932. [Google Scholar] [CrossRef]
- Arranz, L.; Sanchez-Aguilera, A.; Martin-Perez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntion, S.; Tzeng, Y.S.; Lai, D.M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.J.; Neo, W.H.; Barkas, N.; Matsuoka, S.; Giustacchini, A.; Facchini, R.; Thongjuea, S.; Jamieson, L.; Booth, C.A.G.; Fordham, N.; et al. Niche-mediated depletion of the normal hematopoietic stem cell reservoir by Flt3-ITD-induced myeloproliferation. J. Exp. Med. 2017, 214, 2005–2021. [Google Scholar] [CrossRef]
- Kiyoi, H.; Naoe, T.; Yokota, S.; Nakao, M.; Minami, S.; Kuriyama, K.; Takeshita, A.; Saito, K.; Hasegawa, S.; Shimodaira, S.; et al. Internal tandem duplication of FLT3 associated with leukocytosis in acute promyelocytic leukemia. Leukemia Study Group of the Ministry of Health and Welfare (Kohseisho). Leukemia 1997, 11, 1447–1452. [Google Scholar] [CrossRef] [Green Version]
- Meisel, M.; Hinterleitner, R.; Pacis, A.; Chen, L.; Earley, Z.M.; Mayassi, T.; Pierre, J.F.; Ernest, J.D.; Galipeau, H.J.; Thuille, N.; et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018, 557, 580–584. [Google Scholar] [CrossRef]
- Higa, K.C.; Goodspeed, A.; Chavez, J.S.; De Dominici, M.; Danis, E.; Zaberezhnyy, V.; Rabe, J.L.; Tenen, D.G.; Pietras, E.M.; DeGregori, J. Chronic interleukin-1 exposure triggers selection for Cebpa-knockout multipotent hematopoietic progenitors. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Hormaechea-Agulla, D.; Matatall, K.A.; Le, D.T.; Kain, B.; Long, X.; Kus, P.; Jaksik, R.; Challen, G.A.; Kimmel, M.; King, K.Y. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNgamma signaling. Cell Stem Cell 2021, 28, 1428–1442. [Google Scholar] [CrossRef]
- Marusyk, A.; Porter, C.C.; Zaberezhnyy, V.; DeGregori, J. Irradiation selects for p53-deficient hematopoietic progenitors. PLoS Biol. 2010, 8, e1000324. [Google Scholar] [CrossRef] [Green Version]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Christen, F.; Hoyer, K.; Yoshida, K.; Hou, H.-A.; Waldhueter, N.; Heuser, M.; Hills, R.K.; Chan, W.; Hablesreiter, R.; Blau, O.; et al. Genomic landscape and clonal evolution of acute myeloid leukemia with t(8;21): An international study on 331 patients. Blood 2019, 133, 1140–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.D.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenburg, A.S.; Albanese, S.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, F.; Yoshida, S.; Saito, Y.; Hijikata, A.; Kitamura, H.; Tanaka, S.; Nakamura, R.; Tanaka, T.; Tomiyama, H.; Saito, N.; et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 2007, 25, 1315–1321. [Google Scholar] [CrossRef]
- Fong, C.Y.; Gilan, O.; Ni Lam, E.Y.; Rubin, A.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Werner, B.; Heide, T.; Curtis, C.; Barnes, C.P.; Sottoriva, A.; Graham, T.A. Quantification of subclonal selection in cancer from bulk sequencing data. Nat. Genet. 2018, 50, 895–903. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, X.; Wang, X.Q.D.; Su, J.; Putluri, N.; Zhou, T.; Qu, Y.; Jeong, M.; Guzman, A.; Rosas, C.; et al. Dnmt3a loss and Idh2 neomorphic mutations mutually potentiate malignant hematopoiesis. Blood 2020, 135, 845–856. [Google Scholar] [CrossRef]
- Duchmann, M.; Micol, J.B.; Duployez, N.; Raffoux, E.; Thomas, X.; Marolleau, J.P.; Braun, T.; Ades, L.; Chantepie, S.; Lemasle, E.; et al. Prognostic significance of concurrent gene mutations in intensively treated patients with IDH-mutated AML: An ALFA study. Blood 2021, 137, 2827–2837. [Google Scholar] [CrossRef]
- McFarland, C.D.; Korolev, K.S.; Kryukov, G.V.; Sunyaev, S.R.; Mirny, L.A. Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci. USA 2013, 110, 2910–2915. [Google Scholar] [CrossRef] [Green Version]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2,658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Pastor-Pareja, J.C.; Xu, T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature 2010, 463, 545–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleary, A.S.; Leonard, T.L.; Gestl, S.A.; Gunther, E.J. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 2014, 508, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Tabassum, D.P.; Altrock, P.M.; Almendro, V.; Michor, F.; Polyak, K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 2014, 514, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Tammela, T.; Sanchez-Rivera, F.J.; Cetinbas, N.M.; Wu, K.; Joshi, N.S.; Helenius, K.; Park, Y.; Azimi, R.; Kerper, N.R.; Wesselhoeft, R.A.; et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 2017, 545, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.S.; Ibaseta, A.; Fischer, M.M.; Cancilla, B.; O’Young, G.; Cristea, S.; Luca, V.C.; Yang, D.; Jahchan, N.S.; Hamard, C.; et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 2017, 545, 360–364. [Google Scholar] [CrossRef]
- Aktipis, C.A.; Boddy, A.M.; Gatenby, R.A.; Brown, J.S.; Maley, C.C. Life history trade-offs in cancer evolution. Nat. Rev. Cancer 2013, 13, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Akinduro, O.; Weber, T.S.; Ang, H.; Haltalli, M.L.R.; Ruivo, N.; Duarte, D.; Rashidi, N.M.; Hawkins, E.D.; Duffy, K.R.; Lo Celso, C. Proliferation dynamics of acute myeloid leukaemia and haematopoietic progenitors competing for bone marrow space. Nat. Commun. 2018, 9, 519. [Google Scholar] [CrossRef] [Green Version]
- Metzeler, K.H.; Herold, T.; Rothenberg-Thurley, M.; Amler, S.; Sauerland, M.C.; Gorlich, D.; Schneider, S.; Konstandin, N.P.; Dufour, A.; Braundl, K.; et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 2016, 128, 686–698. [Google Scholar] [CrossRef] [Green Version]
- Wakita, S.; Yamaguchi, H.; Ueki, T.; Usuki, K.; Kurosawa, S.; Kobayashi, Y.; Kawata, E.; Tajika, K.; Gomi, S.; Koizumi, M.; et al. Complex molecular genetic abnormalities involving three or more genetic mutations are important prognostic factors for acute myeloid leukemia. Leukemia 2016, 30, 545–554. [Google Scholar] [CrossRef]
- Schlenk, R.F.; Kayser, S.; Bullinger, L.; Kobbe, G.; Casper, J.; Ringhoffer, M.; Held, G.; Brossart, P.; Lubbert, M.; Salih, H.R.; et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood 2014, 124, 3441–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rucker, F.G.; Du, L.; Luck, T.J.; Benner, A.; Krzykalla, J.; Gathmann, I.; Voso, M.T.; Amadori, S.; Prior, T.W.; Brandwein, J.M.; et al. Molecular landscape and prognostic impact of FLT3-ITD insertion site in acute myeloid leukemia: RATIFY study results. Leukemia 2021, 1–10. [Google Scholar] [CrossRef]
- Hughes, A.L. The origin of adaptive phenotypes. Proc. Natl. Acad. Sci. USA 2008, 105, 13193–13194. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Voss, M.H.; Hakimi, A.A.; Pham, C.G.; Brannon, A.R.; Chen, Y.B.; Cunha, L.F.; Akin, O.; Liu, H.; Takeda, S.; Scott, S.N.; et al. Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clin. Cancer Res. 2014, 20, 1955–1964. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.D.; Marteyn, B.; Farnoud, N.R.; de Botton, S.; Bernard, O.A.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef]
- Keller, R.R.; Gunther, E.J. Evolution of Relapse-Proficient Subclones Constrained by Collateral Sensitivity to Oncogene Overdose in Wnt-Driven Mammary Cancer. Cell Rep. 2019, 26, 893–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choe, S.; Wang, H.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Martelotto, L.; Baslan, T.; Vides, A.; Solomon, M.; Mai, T.T.; Chaudhary, N.; Riely, G.J.; Li, B.T.; Scott, K.; et al. An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat. Med. 2017, 23, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Tzoneva, G.; Dieck, C.L.; Oshima, K.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Madubata, C.J.; Khiabanian, H.; Yu, J.; Waanders, E.; Iacobucci, I.; et al. Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature 2018, 553, 511–514. [Google Scholar] [CrossRef]
- Boshuizen, J.; Peeper, D.S. Rational Cancer Treatment Combinations: An Urgent Clinical Need. Mol. Cell 2020, 78, 1002–1018. [Google Scholar] [CrossRef]
- Lin, K.H.; Rutter, J.C.; Xie, A.; Pardieu, B.; Winn, E.T.; Bello, R.D.; Forget, A.; Itzykson, R.; Ahn, Y.R.; Dai, Z.; et al. Using antagonistic pleiotropy to design a chemotherapy-induced evolutionary trap to target drug resistance in cancer. Nat. Genet. 2020, 52, 408–417. [Google Scholar] [CrossRef]
- Acar, A.; Nichol, D.; Fernandez-Mateos, J.; Cresswell, G.D.; Barozzi, I.; Hong, S.P.; Trahearn, N.; Spiteri, I.; Stubbs, M.; Burke, R.; et al. Exploiting evolutionary steering to induce collateral drug sensitivity in cancer. Nat. Commun. 2020, 11, 1923. [Google Scholar] [CrossRef] [Green Version]
- Le Tourneau, C.; Delord, J.P.; Goncalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Tredan, O.; Massiani, M.A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Pemovska, T.; Kontro, M.; Yadav, B.; Edgren, H.; Eldfors, S.; Szwajda, A.; Almusa, H.; Bespalov, M.M.; Ellonen, P.; Elonen, E.; et al. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 2013, 3, 1416–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijder, B.; Vladimer, G.I.; Krall, N.; Miura, K.; Schmolke, A.S.; Kornauth, C.; Lopez de la Fuente, O.; Choi, H.S.; van der Kouwe, E.; Gultekin, S.; et al. Image-based ex-vivo drug screening for patients with aggressive haematological malignancies: Interim results from a single-arm, open-label, pilot study. Lancet Haematol. 2017, 4, e595–e606. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Pritchard, J.R.; Lauffenburger, D.A.; Hemann, M.T. Addressing genetic tumor heterogeneity through computationally predictive combination therapy. Cancer Discov. 2014, 4, 166–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiras, R.D.B.; Pasanisi, J.; Joudinaud, R.; Duchmann, M.; Sodaro, G.; Chauvel, C.; Vasseur, L.; Pardieu, B.; Ling, F.; Pacchiardi, K.; et al. Niche-like Ex Vivo High Throughput (NEXT) Drug Screening Platform in Acute Myeloid Leukemia. Blood 2020, 136, 12–13. [Google Scholar] [CrossRef]
- Ianevski, A.; Lahtela, J.; Javarappa, K.K.; Sergeev, P.; Ghimire, B.R.; Gautam, P.; Vähä-Koskela, M.; Turunen, L.; Linnavirta, N.; Kuusanmäki, H.; et al. Patient-tailored design for selective co-inhibition of leukemic cell subpopulations. Sci. Adv. 2021, 7, eabe4038. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Silva, A.S.; Gillies, R.J.; Frieden, B.R. Adaptive therapy. Cancer Res. 2009, 69, 4894–4903. [Google Scholar] [CrossRef] [Green Version]
- Gallaher, J.A.; Enriquez-Navas, P.M.; Luddy, K.A.; Gatenby, R.A.; Anderson, A.R.A. Spatial Heterogeneity and Evolutionary Dynamics Modulate Time to Recurrence in Continuous and Adaptive Cancer Therapies. Cancer Res. 2018, 78, 2127–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enriquez-Navas, P.M.; Kam, Y.; Das, T.; Hassan, S.; Silva, A.; Foroutan, P.; Ruiz, E.; Martinez, G.; Minton, S.; Gillies, R.J.; et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci. Transl. Med. 2016, 8, 327ra324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Cunningham, J.J.; Brown, J.S.; Gatenby, R.A. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat. Commun. 2017, 8, 1816. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Driscoll, W.W.; Pepper, J.W. Theory for the evolution of diffusible external goods. Evol. Int. J. Org. Evol. 2010, 64, 2682–2687. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Jordan, C.T. Therapeutic targeting of acute myeloid leukemia stem cells. Blood 2017, 129, 1627–1635. [Google Scholar] [CrossRef]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hutter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 1–7. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duchmann, M.; Laplane, L.; Itzykson, R. Clonal Architecture and Evolutionary Dynamics in Acute Myeloid Leukemias. Cancers 2021, 13, 4887. https://doi.org/10.3390/cancers13194887
Duchmann M, Laplane L, Itzykson R. Clonal Architecture and Evolutionary Dynamics in Acute Myeloid Leukemias. Cancers. 2021; 13(19):4887. https://doi.org/10.3390/cancers13194887
Chicago/Turabian StyleDuchmann, Matthieu, Lucie Laplane, and Raphael Itzykson. 2021. "Clonal Architecture and Evolutionary Dynamics in Acute Myeloid Leukemias" Cancers 13, no. 19: 4887. https://doi.org/10.3390/cancers13194887
APA StyleDuchmann, M., Laplane, L., & Itzykson, R. (2021). Clonal Architecture and Evolutionary Dynamics in Acute Myeloid Leukemias. Cancers, 13(19), 4887. https://doi.org/10.3390/cancers13194887