
Remodeling the Tumor Myeloid Landscape to Enhance Antitumor Antibody Immunotherapies



Abstract
:Simple Summary
Abstract
1. Introduction
2. Tumor-Associated Macrophages
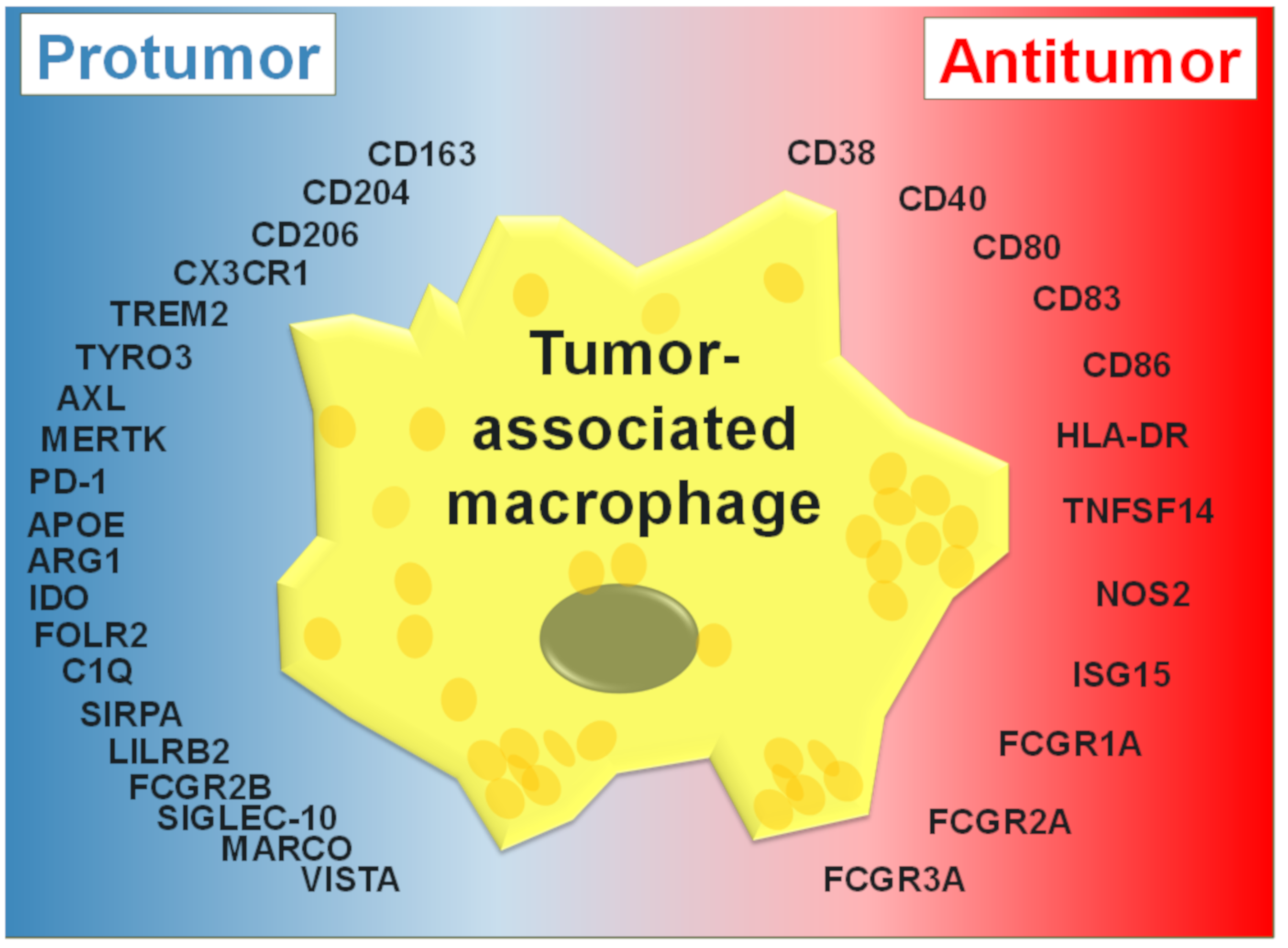
3. TAM Activation States
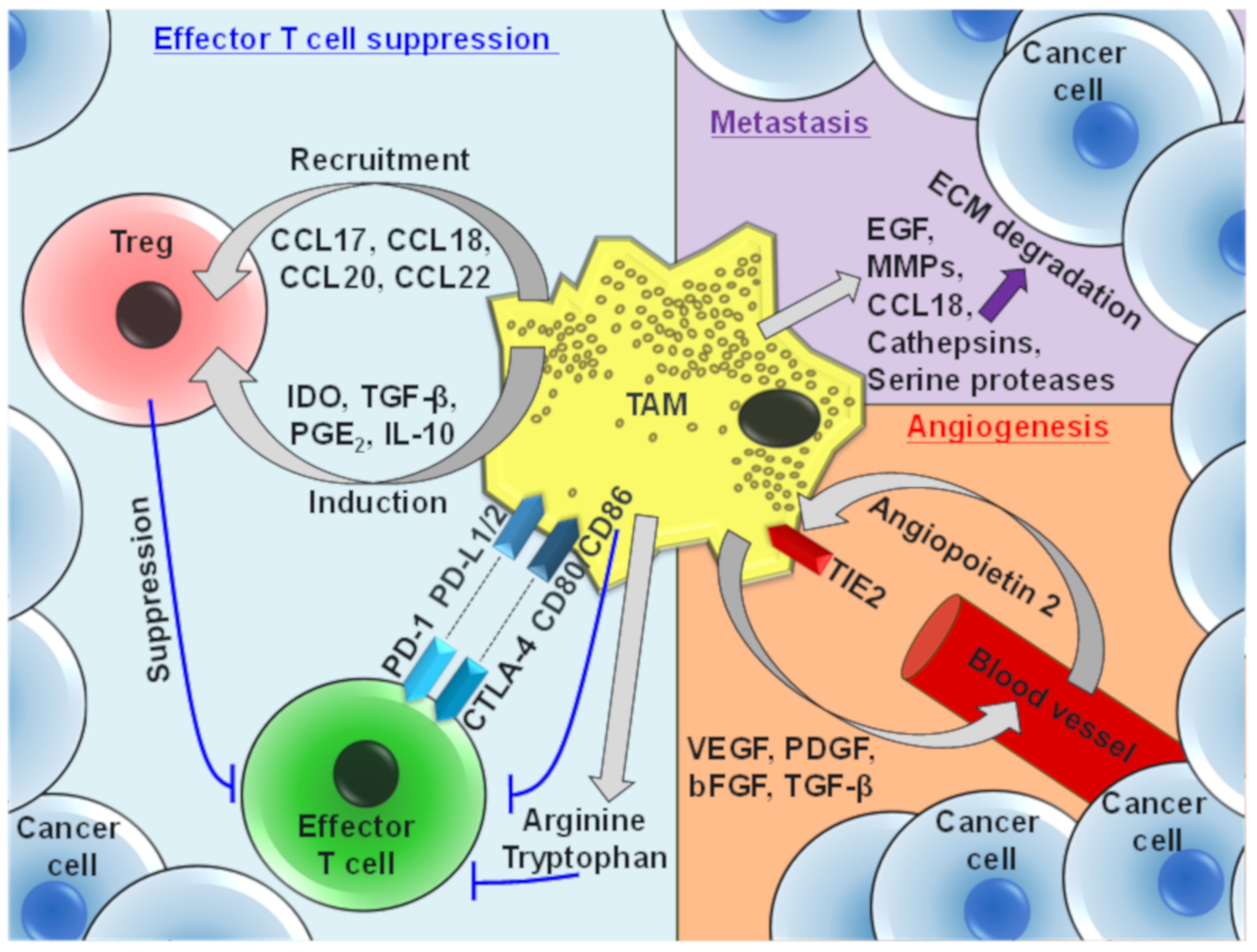
4. Protumor Functions of TAMs
5. TAM-Mediated Depletion of Cancer Cells
6. Antibody-Mediated Modulation of TAM Recruitment, Survival, and Effector Functions
7. TAM Recruitment and Survival
7.1. CSF-1R
7.2. CCR2/CCL2
8. TAM Repolarization
8.1. CD40
8.2. PD-1
8.3. Macrophage Receptor with Collagenous Structure (MARCO)
8.4. V-domain Ig Suppressor of T Cell Activation (VISTA)
8.5. Triggering-Receptor-Expressed on Myeloid Cells 2 (TREM2)
8.6. CD204
8.7. Leucocyte Immunoglobulin-Like Receptor B 2 (LILRB2)
8.8. Tyro3, Axl, and MerTK (TAM) Receptors
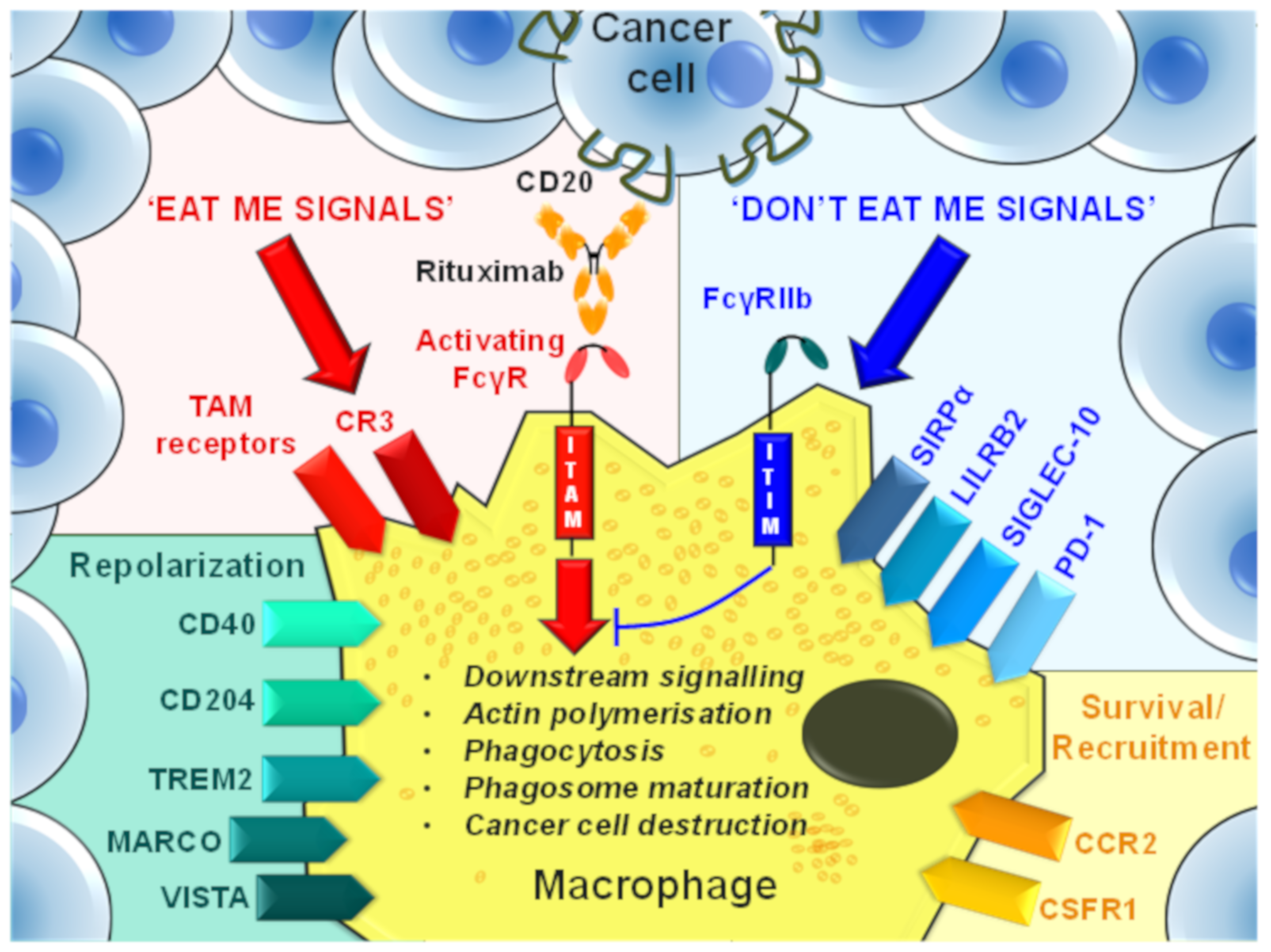
9. Phagocytosis Checkpoints
9.1. Activating FcγRs
9.2. FcγRIIb
9.3. CD47
9.4. CD24
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lauder, I.; Aherne, W.; Stewart, J.; Sainsbury, R. Macrophage infiltration of breast tumours: A prospective study. J. Clin. Pathol. 1977, 30, 563–568. [Google Scholar] [CrossRef]
- Monis, B.; Weinberg, T. Cytochemical study of esterase activity of human neoplasms and stromal macrophages. Cancer 1961, 14, 369–377. [Google Scholar] [CrossRef]
- Underwood, J.C.; Carr, I. The lymphoreticular infiltrate in human tumours studied by electron microscopy and tissue culture. J. Pathol. 1972, 107, Pv. [Google Scholar] [PubMed]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Laoui, D.; Van Overmeire, E.; Movahedi, K.; Van den Bossche, J.; Schouppe, E.; Mommer, C.; Nikolaou, A.; Morias, Y.; De Baetselier, P.; Van Ginderachter, J.A. Mononuclear phagocyte heterogeneity in cancer: Different subsets and activation states reaching out at the tumor site. Immunobiology 2011, 216, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Angell, H.K.; Bedognetti, D.; Marincola, F.M. The continuum of cancer immunosurveillance: Prognostic, predictive, and mechanistic signatures. Immunity 2013, 39, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Cheson, B.D.; Leonard, J.P. Monoclonal antibody therapy for B-cell non-Hodgkin’s lymphoma. N. Engl. J. Med. 2008, 359, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradishar, W.J. HER2 therapy--an abundance of riches. N. Engl. J. Med. 2012, 366, 176–178. [Google Scholar] [CrossRef] [Green Version]
- Feugier, P. A review of rituximab, the first anti-CD20 monoclonal antibody used in the treatment of B non-Hodgkin’s lymphomas. Future Oncol. 2015, 11, 1327–1342. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Torka, P.; Barth, M.; Ferdman, R.; Hernandez-Ilizaliturri, F.J. Mechanisms of Resistance to Monoclonal Antibodies (mAbs) in Lymphoid Malignancies. Curr. Hematol. Malig. Rep. 2019, 14, 426–438. [Google Scholar] [CrossRef]
- Karlsen, E.A.; Kahler, S.; Tefay, J.; Joseph, S.R.; Simpson, F. Epidermal Growth Factor Receptor Expression and Resistance Patterns to Targeted Therapy in Non-Small Cell Lung Cancer: A Review. Cells 2021, 10, 1206. [Google Scholar] [CrossRef]
- Zou, L.; Song, G.; Gu, S.; Kong, L.; Sun, S.; Yang, L.; Cho, W.C. Mechanism and Treatment of Rituximab Resistance in Diffuse Large Bcell Lymphoma. Curr. Cancer Drug Targets 2019, 19, 681–687. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Translating basic mechanisms of IgG effector activity into next generation cancer therapies. Cancer Immun. 2012, 12, 13. [Google Scholar] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.; Plantinga, T.S.; Tuit, S.; Ulas, T.; Heinhuis, B.; Tesselaar, M.; Sloot, Y.; Adema, G.J.; Joosten, L.A.; Smit, J.W.; et al. Transcriptional and metabolic reprogramming induce an inflammatory phenotype in non-medullary thyroid carcinoma-induced macrophages. Oncoimmunology 2016, 5, e1229725. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Dahal, L.N.; Dou, L.; Hussain, K.; Liu, R.; Earley, A.; Cox, K.L.; Murinello, S.; Tracy, I.; Forconi, F.; Steele, A.J.; et al. STING Activation Reverses Lymphoma-Mediated Resistance to Antibody Immunotherapy. Cancer Res. 2017, 77, 3619–3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- te Velde, A.A.; Rousset, F.; Peronne, C.; De Vries, J.E.; Figdor, C.G. IFN-alpha and IFN-gamma have different regulatory effects on IL-4-induced membrane expression of Fc epsilon RIIb and release of soluble Fc epsilon RIIb by human monocytes. J. Immunol. 1990, 144, 3052–3059. [Google Scholar] [PubMed]
- te Velde, A.A.; Huijbens, R.J.; de Vries, J.E.; Figdor, C.G. IL-4 decreases Fc gamma R membrane expression and Fc gamma R-mediated cytotoxic activity of human monocytes. J. Immunol. 1990, 144, 3046–3051. [Google Scholar] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, A.; Brandenburg, S.; Turkowski, K.; Muller, S.; Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 2015, 137, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arwert, E.N.; Harney, A.S.; Entenberg, D.; Wang, Y.; Sahai, E.; Pollard, J.W.; Condeelis, J.S. A Unidirectional Transition from Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Rep. 2018, 23, 1239–1248. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Brown, C.E.; Vishwanath, R.P.; Aguilar, B.; Starr, R.; Najbauer, J.; Aboody, K.S.; Jensen, M.C. Tumor-derived chemokine MCP-1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J. Immunol. 2007, 179, 3332–3341. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Macrophages define the invasive microenvironment in breast cancer. J. Leukoc. Biol. 2008, 84, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Lu, Y.; Pienta, K.J. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J. Natl. Cancer Inst. 2010, 102, 522–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiola, I.; Tomay, F.; De Pizzol, M.; Silva-Gomes, R.; Savino, B.; Gulic, T.; Doni, A.; Lonardi, S.; Astrid Boutet, M.; Nerviani, A.; et al. The macrophage tetraspan MS4A4A enhances dectin-1-dependent NK cell-mediated resistance to metastasis. Nat. Immunol. 2019, 20, 1012–1022. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Blando, J.; Sharma, A.; Higa, M.G.; Zhao, H.; Vence, L.; Yadav, S.S.; Kim, J.; Sepulveda, A.M.; Sharp, M.; Maitra, A.; et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 1692–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Okamura, R.; Kumaki, Y.; Ikeda, S.; Nikanjam, M.; Eskander, R.; Goodman, A.; Lee, S.; Glenn, S.T.; Dressman, D.; et al. Expression of TIM3/VISTA checkpoints and the CD68 macrophage-associated marker correlates with anti-PD1/PDL1 resistance: Implications of immunogram heterogeneity. Oncoimmunology 2020, 9, 1708065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dander, E.; Fallati, A.; Gulic, T.; Pagni, F.; Gaspari, S.; Silvestri, D.; Cricri, G.; Bedini, G.; Portale, F.; Buracchi, C.; et al. Monocyte-macrophage polarization and recruitment pathways in the tumour microenvironment of B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2021, 193, 1157–1171. [Google Scholar] [CrossRef] [PubMed]
- Molgora, M.; Colonna, M. Turning enemies into allies-reprogramming tumor-associated macrophages for cancer therapy. Med 2021, 2, 666–681. [Google Scholar] [CrossRef]
- Pace, J.L.; Russell, S.W.; Schreiber, R.D.; Altman, A.; Katz, D.H. Macrophage activation: Priming activity from a T-cell hybridoma is attributable to interferon-gamma. Proc. Natl. Acad. Sci. USA 1983, 80, 3782–3786. [Google Scholar] [CrossRef] [Green Version]
- Celada, A.; Gray, P.W.; Rinderknecht, E.; Schreiber, R.D. Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J. Exp. Med. 1984, 160, 55–74. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Komohara, Y.; Jinushi, M.; Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014, 105, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taub, D.D.; Cox, G.W. Murine Th1 and Th2 cell clones differentially regulate macrophage nitric oxide production. J. Leukoc. Biol. 1995, 58, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, R.; Roth-Walter, F.; Ohradanova-Repic, A.; Flicker, S.; Hufnagl, K.; Fischer, M.B.; Stockinger, H.; Jensen-Jarolim, E. IgG4 drives M2a macrophages to a regulatory M2b-like phenotype: Potential implication in immune tolerance. Allergy 2019, 74, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bumgardner, S.A.; Zhang, L.; LaVoy, A.S.; Andre, B.; Frank, C.B.; Kajikawa, A.; Klaenhammer, T.R.; Dean, G.A. Nod2 is required for antigen-specific humoral responses against antigens orally delivered using a recombinant Lactobacillus vaccine platform. PLoS ONE 2018, 13, e0196950. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, D.M.; Song, Z.B.; Hou, Y.Q.; Bao, Y.L.; Sun, L.G.; Yu, C.L.; Li, Y.X. Protumoral TSP50 Regulates Macrophage Activities and Polarization via Production of TNF-alpha and IL-1beta, and Activation of the NF-kappaB Signaling Pathway. PLoS ONE 2015, 10, e0145095. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [Green Version]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusinkveld, M.; van der Burg, S.H. Identification and manipulation of tumor associated macrophages in human cancers. J. Transl. Med. 2011, 9, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.W.; Joyce, J.A. Alternative activation of tumor-associated macrophages by IL-4: Priming for protumoral functions. Cell Cycle 2010, 9, 4824–4835. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Zhu, S.; Qu, K.; Meeth, K.; Cheng, J.; He, K.; Ma, H.; Liao, Y.; Wen, X.; Roden, C.; et al. The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor-Infiltrating Myeloid Cells to Promote Melanoma Progression. Immunity 2017, 47, 284–297.e5. [Google Scholar] [CrossRef] [PubMed]
- Roca, H.; Varsos, Z.S.; Sud, S.; Craig, M.J.; Ying, C.; Pienta, K.J. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J. Biol. Chem. 2009, 284, 34342–34354. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, A.P.; Pinto, M.L.; Castro, F.; Costa, A.M.; Marques-Magalhaes, A.; Canha-Borges, A.; Cruz, T.; Velho, S.; Oliveira, M.J. The immunosuppressive and pro-tumor functions of CCL18 at the tumor microenvironment. Cytokine Growth Factor Rev. 2021, 60, 107–119. [Google Scholar] [CrossRef]
- Zhang, A.; Xu, Y.; Xu, H.; Ren, J.; Meng, T.; Ni, Y.; Zhu, Q.; Zhang, W.B.; Pan, Y.B.; Jin, J.; et al. Lactate-induced M2 polarization of tumor-associated macrophages promotes the invasion of pituitary adenoma by secreting CCL17. Theranostics 2021, 11, 3839–3852. [Google Scholar] [CrossRef]
- Tulotta, C.; Stefanescu, C.; Chen, Q.; Torraca, V.; Meijer, A.H.; Snaar-Jagalska, B.E. CXCR4 signaling regulates metastatic onset by controlling neutrophil motility and response to malignant cells. Sci. Rep. 2019, 9, 2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.; Lewis, C.E. Macrophage migration and gene expression in response to tumor hypoxia. Int. J. Cancer 2005, 117, 701–708. [Google Scholar] [CrossRef]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Sica, A.; Lewis, C.E. Plasticity of macrophage function during tumor progression: Regulation by distinct molecular mechanisms. J. Immunol. 2008, 180, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shay, J.E.; Celeste Simon, M. Hypoxia-inducible factors: Crosstalk between inflammation and metabolism. Semin. Cell Dev. Biol. 2012, 23, 389–394. [Google Scholar] [CrossRef]
- Arteel, G.E.; Thurman, R.G.; Yates, J.M.; Raleigh, J.A. Evidence that hypoxia markers detect oxygen gradients in liver: Pimonidazole and retrograde perfusion of rat liver. Br. J. Cancer 1995, 72, 889–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awwad, H.K.; El Merzabani, M.M.; El Badawy, S.; Ezzat, S.; Akoush, H.; Abd El Moneim, H.; Saiid, A.; Soliman, O.; Khafagy, M.; Burgers, M.V. Misonidazole in the preoperative and radical radiotherapy of bladder cancer. Cancer Clin. Trials 1980, 3, 275–280. [Google Scholar]
- Becker, A.; Hansgen, G.; Bloching, M.; Weigel, C.; Lautenschlager, C.; Dunst, J. Oxygenation of squamous cell carcinoma of the head and neck: Comparison of primary tumors, neck node metastases, and normal tissue. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 35–41. [Google Scholar] [CrossRef]
- Koong, A.C.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.M.; Bastidas, A.J.; Vierra, M. Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 919–922. [Google Scholar] [CrossRef]
- Rampling, R.; Cruickshank, G.; Lewis, A.D.; Fitzsimmons, S.A.; Workman, P. Direct measurement of pO2 distribution and bioreductive enzymes in human malignant brain tumors. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 427–431. [Google Scholar] [CrossRef]
- Thomlinson, R.H.; Gray, L.H. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Briest, S.; Hockel, M. Hypoxia in breast cancer: Pathogenesis, characterization and biological/therapeutic implications. Wien. Med. Wochenschr. 2002, 152, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Vaupel, P.; Mayer, A.; Hockel, M. Tumor hypoxia and malignant progression. Methods Enzymol. 2004, 381, 335–354. [Google Scholar] [CrossRef]
- Molgora, M.; Supino, D.; Mavilio, D.; Santoni, A.; Moretta, L.; Mantovani, A.; Garlanda, C. The yin-yang of the interaction between myelomonocytic cells and NK cells. Scand. J. Immunol. 2018, 88, e12705. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Martin, E.M.; Mellows, T.W.P.; Clarke, J.; Ganesan, A.P.; Wood, O.; Cazaly, A.; Seumois, G.; Chee, S.J.; Alzetani, A.; King, E.V.; et al. M1(hot) tumor-associated macrophages boost tissue-resident memory T cells infiltration and survival in human lung cancer. J. Immunother. Cancer 2020, 8, e000778. [Google Scholar] [CrossRef]
- Leuverink, E.M.; Brennan, B.A.; Crook, M.L.; Doherty, D.A.; Hammond, I.G.; Ruba, S.; Stewart, C.J. Prognostic value of mitotic counts and Ki-67 immunoreactivity in adult-type granulosa cell tumour of the ovary. J. Clin. Pathol. 2008, 61, 914–919. [Google Scholar] [CrossRef]
- Watanabe, M.; Kanao, K.; Suzuki, S.; Muramatsu, H.; Morinaga, S.; Kajikawa, K.; Kobayashi, I.; Nishikawa, G.; Kato, Y.; Zennami, K.; et al. Increased infiltration of CCR4-positive regulatory T cells in prostate cancer tissue is associated with a poor prognosis. Prostate 2019, 79, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Oleinika, K.; Nibbs, R.J.; Graham, G.J.; Fraser, A.R. Suppression, subversion and escape: The role of regulatory T cells in cancer progression. Clin. Exp. Immunol. 2013, 171, 36–45. [Google Scholar] [CrossRef]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.Y.; Chen, W.; Bai, X.L.; Xu, X.Y.; Zhang, Q.; Xia, X.F.; Sun, X.; Li, G.G.; Hu, Q.D.; Fu, Q.H.; et al. Hypoxia-Induced Epithelial-to-Mesenchymal Transition in Hepatocellular Carcinoma Induces an Immunosuppressive Tumor Microenvironment to Promote Metastasis. Cancer Res. 2016, 76, 818–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henze, A.T.; Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 2016, 126, 3672–3679. [Google Scholar] [CrossRef]
- Pelosi, A.; Fiore, P.F.; Di Matteo, S.; Veneziani, I.; Caruana, I.; Ebert, S.; Munari, E.; Moretta, L.; Maggi, E.; Azzarone, B. Pediatric Tumors-Mediated Inhibitory Effect on NK Cells: The Case of Neuroblastoma and Wilms’ Tumors. Cancers 2021, 13, 2374. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Sangaletti, S.; Di Carlo, E.; Gariboldi, S.; Miotti, S.; Cappetti, B.; Parenza, M.; Rumio, C.; Brekken, R.A.; Chiodoni, C.; Colombo, M.P. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008, 68, 9050–9059. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yao, Y.; Gong, C.; Yu, F.; Su, S.; Chen, J.; Liu, B.; Deng, H.; Wang, F.; Lin, L.; et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 2011, 19, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Leek, R.D.; Lewis, C.E.; Whitehouse, R.; Greenall, M.; Clarke, J.; Harris, A.L. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996, 56, 4625–4629. [Google Scholar]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [Green Version]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef]
- De Palma, M.; Venneri, M.A.; Galli, R.; Sergi Sergi, L.; Politi, L.S.; Sampaolesi, M.; Naldini, L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005, 8, 211–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffelt, S.B.; Tal, A.O.; Scholz, A.; De Palma, M.; Patel, S.; Urbich, C.; Biswas, S.K.; Murdoch, C.; Plate, K.H.; Reiss, Y.; et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010, 70, 5270–5280. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Chiba, S.; Yoshiyama, H.; Masutomi, K.; Kinoshita, I.; Dosaka-Akita, H.; Yagita, H.; Takaoka, A.; Tahara, H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12425–12430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakes, F.M.; Chinratanalab, W.; Ritter, C.A.; King, W.; Seelig, S.; Arteaga, C.L. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002, 62, 4132–4141. [Google Scholar] [PubMed]
- Gong, Q.; Ou, Q.; Ye, S.; Lee, W.P.; Cornelius, J.; Diehl, L.; Lin, W.Y.; Hu, Z.; Lu, Y.; Chen, Y.; et al. Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J. Immunol. 2005, 174, 817–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, J.; Hamaguchi, Y.; Oliver, J.A.; Ravetch, J.V.; Poe, J.C.; Haas, K.M.; Tedder, T.F. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J. Exp. Med. 2004, 199, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Beers, S.A.; French, R.R.; Chan, H.T.; Lim, S.H.; Jarrett, T.C.; Vidal, R.M.; Wijayaweera, S.S.; Dixon, S.V.; Kim, H.; Cox, K.L.; et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: Implications for antibody selection. Blood 2010, 115, 5191–5201. [Google Scholar] [CrossRef]
- Biburger, M.; Aschermann, S.; Schwab, I.; Lux, A.; Albert, H.; Danzer, H.; Woigk, M.; Dudziak, D.; Nimmerjahn, F. Monocyte subsets responsible for immunoglobulin G-dependent effector functions in vivo. Immunity 2011, 35, 932–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.; Biburger, M.; Bruckner, C.; Ipsen-Escobedo, A.; Gordan, S.; Lehmann, C.; Voehringer, D.; Winkler, T.; Schaft, N.; Dudziak, D.; et al. Tumor location determines tissue-specific recruitment of tumor-associated macrophages and antibody-dependent immunotherapy response. Sci. Immunol. 2017, 2, eaah6413. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef] [Green Version]
- Arce Vargas, F.; Furness, A.J.S.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018, 33, 649–663.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef] [Green Version]
- Monks, J.; Rosner, D.; Geske, F.J.; Lehman, L.; Hanson, L.; Neville, M.C.; Fadok, V.A. Epithelial cells as phagocytes: Apoptotic epithelial cells are engulfed by mammary alveolar epithelial cells and repress inflammatory mediator release. Cell Death Differ. 2005, 12, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Tso, G.H.; Law, H.K.; Tu, W.; Chan, G.C.; Lau, Y.L. Phagocytosis of apoptotic cells modulates mesenchymal stem cells osteogenic differentiation to enhance IL-17 and RANKL expression on CD4+ T cells. Stem Cells 2010, 28, 939–954. [Google Scholar] [CrossRef]
- Seeberg, J.C.; Loibl, M.; Moser, F.; Schwegler, M.; Buttner-Herold, M.; Daniel, C.; Engel, F.B.; Hartmann, A.; Schlotzer-Schrehardt, U.; Goppelt-Struebe, M.; et al. Non-professional phagocytosis: A general feature of normal tissue cells. Sci. Rep. 2019, 9, 11875. [Google Scholar] [CrossRef] [Green Version]
- Weiskopf, K.; Weissman, I.L. Macrophages are critical effectors of antibody therapies for cancer. mAbs 2015, 7, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Taylor, R.P. The Role of Complement in the Mechanism of Action of Therapeutic Anti-Cancer mAbs. Antibodies 2020, 9, 58. [Google Scholar] [CrossRef]
- Nagelkerke, S.Q.; Tacke, C.E.; Breunis, W.B.; Tanck, M.W.T.; Geissler, J.; Png, E.; Hoang, L.T.; van der Heijden, J.; Naim, A.N.M.; Yeung, R.S.M.; et al. Extensive Ethnic Variation and Linkage Disequilibrium at the FCGR2/3 Locus: Different Genetic Associations Revealed in Kawasaki Disease. Front. Immunol. 2019, 10, 185. [Google Scholar] [CrossRef] [Green Version]
- White, A.L.; Dou, L.; Chan, H.T.; Field, V.L.; Mockridge, C.I.; Moss, K.; Williams, E.L.; Booth, S.G.; French, R.R.; Potter, E.A.; et al. Fcgamma receptor dependency of agonistic CD40 antibody in lymphoma therapy can be overcome through antibody multimerization. J. Immunol. 2014, 193, 1828–1835. [Google Scholar] [CrossRef] [Green Version]
- Hamaguchi, Y.; Xiu, Y.; Komura, K.; Nimmerjahn, F.; Tedder, T.F. Antibody isotype-specific engagement of Fcgamma receptors regulates B lymphocyte depletion during CD20 immunotherapy. J. Exp. Med. 2006, 203, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.L.; Mason, D.; Schreiber, A.D.; Grinstein, S. Quantitative analysis of membrane remodeling at the phagocytic cup. Mol. Biol. Cell 2007, 18, 2883–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, F.M., Jr.; Griffin, J.A.; Leider, J.E.; Silverstein, S.C. Studies on the mechanism of phagocytosis. I. Requirements for circumferential attachment of particle-bound ligands to specific receptors on the macrophage plasma membrane. J. Exp. Med. 1975, 142, 1263–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, A.; Mayorga, L.S.; Stahl, P.D.; Schwartz, A.L. Alterations in the protein composition of maturing phagosomes. J. Clin. Investig. 1992, 90, 1978–1983. [Google Scholar] [CrossRef] [PubMed]
- Gul, N.; Babes, L.; Siegmund, K.; Korthouwer, R.; Bogels, M.; Braster, R.; Vidarsson, G.; ten Hagen, T.L.; Kubes, P.; van Egmond, M. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J. Clin. Investig. 2014, 124, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Jaumouille, V.; Grinstein, S. The cell biology of phagocytosis. Annu. Rev. Pathol. 2012, 7, 61–98. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, J.; Carter, R.H.; Edberg, J.C.; Su, K.; Cooper, G.S.; Kimberly, R.P. A novel polymorphism in the Fcgamma receptor IIB (CD32B) transmembrane region alters receptor signaling. Arthritis Rheum. 2003, 48, 3242–3252. [Google Scholar] [CrossRef]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Montalvao, F.; Garcia, Z.; Celli, S.; Breart, B.; Deguine, J.; Van Rooijen, N.; Bousso, P. The mechanism of anti-CD20-mediated B cell depletion revealed by intravital imaging. J. Clin. Investig. 2013, 123, 5098–5103. [Google Scholar] [CrossRef] [Green Version]
- Grandjean, C.L.; Montalvao, F.; Celli, S.; Michonneau, D.; Breart, B.; Garcia, Z.; Perro, M.; Freytag, O.; Gerdes, C.A.; Bousso, P. Intravital imaging reveals improved Kupffer cell-mediated phagocytosis as a mode of action of glycoengineered anti-CD20 antibodies. Sci. Rep. 2016, 6, 34382. [Google Scholar] [CrossRef] [PubMed]
- Minard-Colin, V.; Xiu, Y.; Poe, J.C.; Horikawa, M.; Magro, C.M.; Hamaguchi, Y.; Haas, K.M.; Tedder, T.F. Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcgammaRI, FcgammaRIII, and FcgammaRIV. Blood 2008, 112, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Bonjardim, C.A.; Ferreira, P.C.; Kroon, E.G. Interferons: Signaling, antiviral and viral evasion. Immunol. Lett. 2009, 122, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sivick, K.E.; Desbien, A.L.; Glickman, L.H.; Reiner, G.L.; Corrales, L.; Surh, N.H.; Hudson, T.E.; Vu, U.T.; Francica, B.J.; Banda, T.; et al. Magnitude of Therapeutic STING Activation Determines CD8(+) T Cell-Mediated Anti-tumor Immunity. Cell Rep. 2018, 25, 3074–3085.e5. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Somiya, M.; Kuroda, S. Enhancing antibody-dependent cellular phagocytosis by Re-education of tumor-associated macrophages with resiquimod-encapsulated liposomes. Biomaterials 2021, 268, 120601. [Google Scholar] [CrossRef]
- Kash, N.; Silapunt, S. A review of emerging and non-US FDA-approved topical agents for the treatment of basal cell carcinoma. Future Oncol. 2021, 17, 3111–3132. [Google Scholar] [CrossRef]
- Smith, M.; Garcia-Martinez, E.; Pitter, M.R.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. Oncoimmunology 2018, 7, e1526250. [Google Scholar] [CrossRef] [PubMed]
- Martins, K.A.; Bavari, S.; Salazar, A.M. Vaccine adjuvant uses of poly-IC and derivatives. Expert Rev. Vaccines 2015, 14, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Devaraja, K. Current Prospects of Molecular Therapeutics in Head and Neck Squamous Cell Carcinoma. Pharm. Med. 2019, 33, 269–289. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, J.; Zitvogel, L.; Galluzzi, L.; Vacchelli, E.; Kroemer, G. Trial watch: STING agonists in cancer therapy. Oncoimmunology 2020, 9, 1777624. [Google Scholar] [CrossRef]
- Lenzo, J.C.; Turner, A.L.; Cook, A.D.; Vlahos, R.; Anderson, G.P.; Reynolds, E.C.; Hamilton, J.A. Control of macrophage lineage populations by CSF-1 receptor and GM-CSF in homeostasis and inflammation. Immunol. Cell Biol. 2012, 90, 429–440. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [Green Version]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS ONE 2011, 6, e26317. [Google Scholar] [CrossRef] [Green Version]
- Strachan, D.C.; Ruffell, B.; Oei, Y.; Bissell, M.J.; Coussens, L.M.; Pryer, N.; Daniel, D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology 2013, 2, e26968. [Google Scholar] [CrossRef] [Green Version]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Roca, C.A.; Italiano, A.; Le Tourneau, C.; Cassier, P.A.; Toulmonde, M.; D’Angelo, S.P.; Campone, M.; Weber, K.L.; Loirat, D.; Cannarile, M.A.; et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann. Oncol. 2019, 30, 1381–1392. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Lynn, R.C.; Poussin, M.; Eiva, M.A.; Shaw, L.C.; O’Connor, R.S.; Minutolo, N.G.; Casado-Medrano, V.; Lopez, G.; Matsuyama, T.; et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat. Commun. 2021, 12, 877. [Google Scholar] [CrossRef]
- Leseigneur, C.; Le-Bury, P.; Pizarro-Cerda, J.; Dussurget, O. Emerging Evasion Mechanisms of Macrophage Defenses by Pathogenic Bacteria. Front. Cell Infect. Microbiol. 2020, 10, 577559. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, J.; Lekstrom-Himes, J.; Donaldson, D.; Lee, Y.; Hu, M.; Xu, J.; Wyant, T.; Davidson, M.; Group, M.L.N.S. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am. J. Cardiol. 2011, 107, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, H.; Sangai, T.; Ishii, G.; Ikehara, A.; Nagashima, T.; Miyazaki, M.; Ochiai, A. Stromal MCP-1 in mammary tumors induces tumor-associated macrophage infiltration and contributes to tumor progression. Int. J. Cancer 2009, 125, 1276–1284. [Google Scholar] [CrossRef]
- Brana, I.; Calles, A.; LoRusso, P.M.; Yee, L.K.; Puchalski, T.A.; Seetharam, S.; Zhong, B.; de Boer, C.J.; Tabernero, J.; Calvo, E. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: An open-label, multicenter phase 1b study. Target. Oncol. 2015, 10, 111–123. [Google Scholar] [CrossRef]
- Argyle, D.; Kitamura, T. Targeting Macrophage-Recruiting Chemokines as a Novel Therapeutic Strategy to Prevent the Progression of Solid Tumors. Front. Immunol. 2018, 9, 2629. [Google Scholar] [CrossRef]
- Li, F.; Ravetch, J.V. Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science 2011, 333, 1030–1034. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [Green Version]
- Djureinovic, D.; Wang, M.; Kluger, H.M. Agonistic CD40 Antibodies in Cancer Treatment. Cancers 2021, 13, 1302. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.P.; van Montfoort, N.; Kinderman, P.; Lukkes, M.; Klaase, L.; van Nimwegen, M.; van Gulijk, M.; Dumas, J.; Mustafa, D.A.M.; Lievense, S.L.A.; et al. Dendritic cell vaccination and CD40-agonist combination therapy licenses T cell-dependent antitumor immunity in a pancreatic carcinoma murine model. J. Immunother. Cancer 2020, 8, e000772. [Google Scholar] [CrossRef]
- Byrne, K.T.; Betts, C.B.; Mick, R.; Sivagnanam, S.; Bajor, D.L.; Laheru, D.A.; Chiorean, E.G.; O’Hara, M.H.; Liudahl, S.M.; Newcomb, C.W.; et al. Neoadjuvant selicrelumab, an agonist CD40 antibody, induces changes in the tumor microenvironment in patients with resectable pancreatic cancer. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Luheshi, N.M.; Coates-Ulrichsen, J.; Harper, J.; Mullins, S.; Sulikowski, M.G.; Martin, P.; Brown, L.; Lewis, A.; Davies, G.; Morrow, M.; et al. Transformation of the tumour microenvironment by a CD40 agonist antibody correlates with improved responses to PD-L1 blockade in a mouse orthotopic pancreatic tumour model. Oncotarget 2016, 7, 18508–18520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenberger, S.P.; Toes, R.E.; van der Voort, E.I.; Offringa, R.; Melief, C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef]
- Bonnans, C.; Thomas, G.; He, W.; Jung, B.; Chen, W.; Liao, M.; Heyen, J.; Buetow, B.; Pillai, S.; Matsumoto, D.; et al. CD40 agonist-induced IL-12p40 potentiates hepatotoxicity. J. Immunother. Cancer 2020, 8, e000624. [Google Scholar] [CrossRef] [PubMed]
- Rech, A.J.; Dada, H.; Kotzin, J.J.; Henao-Mejia, J.; Minn, A.J.; Twyman-Saint Victor, C.; Vonderheide, R.H. Radiotherapy and CD40 Activation Separately Augment Immunity to Checkpoint Blockade in Cancer. Cancer Res. 2018, 78, 4282–4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, T.A.; Heath, A.W. Functional activity of CD40 antibodies correlates to the position of binding relative to CD154. Immunology 2001, 102, 39–43. [Google Scholar] [CrossRef]
- Richman, L.P.; Vonderheide, R.H. Role of crosslinking for agonistic CD40 monoclonal antibodies as immune therapy of cancer. Cancer Immunol. Res. 2014, 2, 19–26. [Google Scholar] [CrossRef] [Green Version]
- White, A.L.; Chan, H.T.; French, R.R.; Willoughby, J.; Mockridge, C.I.; Roghanian, A.; Penfold, C.A.; Booth, S.G.; Dodhy, A.; Polak, M.E.; et al. Conformation of the human immunoglobulin G2 hinge imparts superagonistic properties to immunostimulatory anticancer antibodies. Cancer Cell 2015, 27, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; James, S.; Felce, J.H.; Kellermayer, B.; Johnston, D.A.; Chan, H.T.C.; Penfold, C.A.; Kim, J.; Inzhelevskaya, T.; Mockridge, C.I.; et al. TNF receptor agonists induce distinct receptor clusters to mediate differential agonistic activity. Commun. Biol. 2021, 4, 772. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Spigel, D.R.; Vokes, E.E.; Holgado, E.; Ready, N.; Steins, M.; Poddubskaya, E.; Borghaei, H.; Felip, E.; Paz-Ares, L.; et al. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). J. Clin. Oncol. 2017, 35, 3924–3933. [Google Scholar] [CrossRef] [PubMed]
- Dyhl-Polk, A.; Mikkelsen, M.K.; Ladekarl, M.; Nielsen, D.L. Clinical Trials of Immune Checkpoint Inhibitors in Hepatocellular Carcinoma. J. Clin. Med. 2021, 10, 2662. [Google Scholar] [CrossRef]
- Leighl, N.B.; Hellmann, M.D.; Hui, R.; Carcereny, E.; Felip, E.; Ahn, M.J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Pembrolizumab in patients with advanced non-small-cell lung cancer (KEYNOTE-001): 3-year results from an open-label, phase 1 study. Lancet Respir. Med. 2019, 7, 347–357. [Google Scholar] [CrossRef]
- Huang, X.; Venet, F.; Wang, Y.L.; Lepape, A.; Yuan, Z.; Chen, Y.; Swan, R.; Kherouf, H.; Monneret, G.; Chung, C.S.; et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl. Acad. Sci. USA 2009, 106, 6303–6308. [Google Scholar] [CrossRef] [Green Version]
- Dhupkar, P.; Gordon, N.; Stewart, J.; Kleinerman, E.S. Anti-PD-1 therapy redirects macrophages from an M2 to an M1 phenotype inducing regression of OS lung metastases. Cancer Med. 2018, 7, 2654–2664. [Google Scholar] [CrossRef]
- Bally, A.P.; Lu, P.; Tang, Y.; Austin, J.W.; Scharer, C.D.; Ahmed, R.; Boss, J.M. NF-kappaB regulates PD-1 expression in macrophages. J. Immunol. 2015, 194, 4545–4554. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.J.; Ni, L.; Zhang, Y.; Zhang, C.L.; Wu, X.Y.; Atia, A.N.; Thayer, P.; Moorman, J.P.; Yao, Z.Q. PD-1 negatively regulates interleukin-12 expression by limiting STAT-1 phosphorylation in monocytes/macrophages during chronic hepatitis C virus infection. Immunology 2011, 132, 421–431. [Google Scholar] [CrossRef]
- Arredouani, M.S. Is the scavenger receptor MARCO a new immune checkpoint? Oncoimmunology 2014, 3, e955709. [Google Scholar] [CrossRef] [Green Version]
- La Fleur, L.; Botling, J.; He, F.; Pelicano, C.; Zhou, C.; He, C.; Palano, G.; Mezheyeuski, A.; Micke, P.; Ravetch, J.V.; et al. Targeting MARCO and IL37R on Immunosuppressive Macrophages in Lung Cancer Blocks Regulatory T Cells and Supports Cytotoxic Lymphocyte Function. Cancer Res. 2021, 81, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Georgoudaki, A.M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Ostling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisinger, S.; Sarhan, D.; Boura, V.F.; Ibarlucea-Benitez, I.; Tyystjarvi, S.; Oliynyk, G.; Arsenian-Henriksson, M.; Lane, D.; Wikstrom, S.L.; Kiessling, R.; et al. Targeting a scavenger receptor on tumor-associated macrophages activates tumor cell killing by natural killer cells. Proc. Natl. Acad. Sci. USA 2020, 117, 32005–32016. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef]
- Lines, J.L.; Pantazi, E.; Mak, J.; Sempere, L.F.; Wang, L.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Huang, J.; Qiao, Q.; Zang, W.; Hong, S.; Tan, H.; Dong, C.; Yang, Z.; Ni, L. Expression of the inhibitory B7 family molecule VISTA in human colorectal carcinoma tumors. Cancer Immunol. Immunother. 2018, 67, 1685–1694. [Google Scholar] [CrossRef]
- ElTanbouly, M.A.; Schaafsma, E.; Smits, N.C.; Shah, P.; Cheng, C.; Burns, C.; Blazar, B.R.; Noelle, R.J.; Mabaera, R. VISTA Re-programs Macrophage Biology Through the Combined Regulation of Tolerance and Anti-inflammatory Pathways. Front. Immunol. 2020, 11, 580187. [Google Scholar] [CrossRef]
- Deczkowska, A.; Weiner, A.; Amit, I. The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 2020, 181, 1207–1217. [Google Scholar] [CrossRef]
- Katzenelenbogen, Y.; Sheban, F.; Yalin, A.; Yofe, I.; Svetlichnyy, D.; Jaitin, D.A.; Bornstein, C.; Moshe, A.; Keren-Shaul, H.; Cohen, M.; et al. Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell 2020, 182, 872–885.e19. [Google Scholar] [CrossRef]
- Molgora, M.; Esaulova, E.; Vermi, W.; Hou, J.; Chen, Y.; Luo, J.; Brioschi, S.; Bugatti, M.; Omodei, A.S.; Ricci, B.; et al. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 2020, 182, 886–900.e17. [Google Scholar] [CrossRef]
- Becker, M.; Cotena, A.; Gordon, S.; Platt, N. Expression of the class A macrophage scavenger receptor on specific subpopulations of murine dendritic cells limits their endotoxin response. Eur. J. Immunol. 2006, 36, 950–960. [Google Scholar] [CrossRef]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef]
- Krieger, M. The other side of scavenger receptors: Pattern recognition for host defense. Curr. Opin. Lipidol. 1997, 8, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Ricci, R.; Sumara, G.; Sumara, I.; Rozenberg, I.; Kurrer, M.; Akhmedov, A.; Hersberger, M.; Eriksson, U.; Eberli, F.R.; Becher, B.; et al. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science 2004, 306, 1558–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Kurihara, Y.; Takeya, M.; Kamada, N.; Kataoka, M.; Jishage, K.; Ueda, O.; Sakaguchi, H.; Higashi, T.; Suzuki, T.; et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature 1997, 386, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Cotena, A.; Gordon, S.; Platt, N. The class A macrophage scavenger receptor attenuates CXC chemokine production and the early infiltration of neutrophils in sterile peritonitis. J. Immunol. 2004, 173, 6427–6432. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, T.; Naito, M.; Yamamoto, T.; Hasegawa, G.; Gejyo, F.; Mitsuyama, M.; Suzuki, H.; Kodama, T. Role of macrophage scavenger receptors in response to Listeria monocytogenes infection in mice. Am. J. Pathol. 2001, 158, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.A.; Li, Y.; Kodama, T.; Suzuki, H.; Silverstein, S.C.; El Khoury, J. Protection from lethal gram-positive infection by macrophage scavenger receptor-dependent phagocytosis. J. Exp. Med. 2000, 191, 147–156. [Google Scholar] [CrossRef]
- Haworth, R.; Platt, N.; Keshav, S.; Hughes, D.; Darley, E.; Suzuki, H.; Kurihara, Y.; Kodama, T.; Gordon, S. The macrophage scavenger receptor type A is expressed by activated macrophages and protects the host against lethal endotoxic shock. J. Exp. Med. 1997, 186, 1431–1439. [Google Scholar] [CrossRef]
- Seimon, T.A.; Obstfeld, A.; Moore, K.J.; Golenbock, D.T.; Tabas, I. Combinatorial pattern recognition receptor signaling alters the balance of life and death in macrophages. Proc. Natl. Acad. Sci. USA 2006, 103, 19794–19799. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.Y.; Facciponte, J.; Chen, X.; Subjeck, J.R.; Repasky, E.A. Scavenger receptor-A negatively regulates antitumor immunity. Cancer Res. 2007, 67, 4996–5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komohara, Y.; Takemura, K.; Lei, X.F.; Sakashita, N.; Harada, M.; Suzuki, H.; Kodama, T.; Takeya, M. Delayed growth of EL4 lymphoma in SR-A-deficient mice is due to upregulation of nitric oxide and interferon-gamma production by tumor-associated macrophages. Cancer Sci. 2009, 100, 2160–2166. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Samaridis, J.; Cella, M.; Angman, L.; Allen, R.L.; O’Callaghan, C.A.; Dunbar, R.; Ogg, G.S.; Cerundolo, V.; Rolink, A. Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J. Immunol. 1998, 160, 3096–3100. [Google Scholar] [PubMed]
- Jones, D.C.; Kosmoliaptsis, V.; Apps, R.; Lapaque, N.; Smith, I.; Kono, A.; Chang, C.; Boyle, L.H.; Taylor, C.J.; Trowsdale, J.; et al. HLA class I allelic sequence and conformation regulate leukocyte Ig-like receptor binding. J. Immunol. 2011, 186, 2990–2997. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Umikawa, M.; Cui, C.; Li, J.; Chen, X.; Zhang, C.; Huynh, H.; Kang, X.; Silvany, R.; Wan, X.; et al. Inhibitory receptors bind ANGPTLs and support blood stem cells and leukaemia development. Nature 2012, 485, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Shiroishi, M.; Tsumoto, K.; Amano, K.; Shirakihara, Y.; Colonna, M.; Braud, V.M.; Allan, D.S.; Makadzange, A.; Rowland-Jones, S.; Willcox, B.; et al. Human inhibitory receptors Ig-like transcript 2 (ILT2) and ILT4 compete with CD8 for MHC class I binding and bind preferentially to HLA-G. Proc. Natl. Acad. Sci. USA 2003, 100, 8856–8861. [Google Scholar] [CrossRef] [Green Version]
- Willcox, B.E.; Thomas, L.M.; Bjorkman, P.J. Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat. Immunol. 2003, 4, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Cella, M.; Nakajima, H.; Facchetti, F.; Hoffmann, T.; Colonna, M. ILT receptors at the interface between lymphoid and myeloid cells. In Lymphoid Organogenesis; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2000; Volume 251, pp. 161–166. [Google Scholar] [CrossRef]
- Shakhawat, A.; Shaikly, V.; Elzatma, E.; Mavrakos, E.; Jabeen, A.; Fernandez, N. Interaction between HLA-G and monocyte/macrophages in human pregnancy. J. Reprod. Immunol. 2010, 85, 40–46. [Google Scholar] [CrossRef]
- Barkal, A.A.; Weiskopf, K.; Kao, K.S.; Gordon, S.R.; Rosental, B.; Yiu, Y.Y.; George, B.M.; Markovic, M.; Ring, N.G.; Tsai, J.M.; et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 2018, 19, 76–84. [Google Scholar] [CrossRef]
- Chen, H.M.; van der Touw, W.; Wang, Y.S.; Kang, K.; Mai, S.; Zhang, J.; Alsina-Beauchamp, D.; Duty, J.A.; Mungamuri, S.K.; Zhang, B.; et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J. Clin. Investig. 2018, 128, 5647–5662. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Meer, J.H.; van der Poll, T.; van ‘t Veer, C. TAM receptors, Gas6, and protein S: Roles in inflammation and hemostasis. Blood 2014, 123, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Zagorska, A.; Traves, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001, 411, 207–211. [Google Scholar] [CrossRef]
- A-Gonzalez, N.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramirez, C.; Diaz, M.; Gallardo, G.; et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009, 31, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Shibata, T.; Habiel, D.M.; Coelho, A.L.; Kunkel, S.L.; Lukacs, N.W.; Hogaboam, C.M. Axl receptor blockade ameliorates pulmonary pathology resulting from primary viral infection and viral exacerbation of asthma. J. Immunol. 2014, 192, 3569–3581. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.H.; Kim, D.; Lee, D.; Lee, H.M.; Song, D.G.; Jung, J.W.; Kim, J.E.; Kim, H.J.; Kwon, N.H.; Jo, E.K.; et al. Lysyl-tRNA synthetase-expressing colon spheroids induce M2 macrophage polarization to promote metastasis. J. Clin. Investig. 2018, 128, 5034–5055. [Google Scholar] [CrossRef]
- Curnow, R.T. Clinical experience with CD64-directed immunotherapy. An overview. Cancer Immunol. Immunother. 1997, 45, 210–215. [Google Scholar] [CrossRef]
- Beutier, H.; Hechler, B.; Godon, O.; Wang, Y.; Gillis, C.M.; de Chaisemartin, L.; Gouel-Cheron, A.; Magnenat, S.; Macdonald, L.E.; Murphy, A.J.; et al. Platelets expressing IgG receptor FcgammaRIIA/CD32A determine the severity of experimental anaphylaxis. Sci. Immunol. 2018, 3, eaan5997. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T.; Robles-Carrillo, L.; Davila, M.; Brodie, M.; Desai, H.; Rivera-Amaya, M.; Francis, J.L.; Amirkhosravi, A. CD32a antibodies induce thrombocytopenia and type II hypersensitivity reactions in FCGR2A mice. Blood 2015, 126, 2230–2238. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Vousden, K.A.; Naiman, B.; Turman, S.; Sun, H.; Wang, S.; Vinall, L.M.K.; Kemp, B.P.; Kasturiangan, S.; Rees, D.G.; et al. Humanised effector-null FcgammaRIIA antibody inhibits immune complex-mediated proinflammatory responses. Ann. Rheum. Dis. 2019, 78, 228–237. [Google Scholar] [CrossRef] [Green Version]
- Soubrane, C.; Tourani, J.M.; Andrieu, J.M.; Visonneau, S.; Beldjord, K.; Israel-Biet, D.; Mouawad, R.; Bussel, J.; Weil, M.; Khayat, D. Biologic response to anti-CD16 monoclonal antibody therapy in a human immunodeficiency virus-related immune thrombocytopenic purpura patient. Blood 1993, 81, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Clarkson, S.B.; Bussel, J.B.; Kimberly, R.P.; Valinsky, J.E.; Nachman, R.L.; Unkeless, J.C. Treatment of refractory immune thrombocytopenic purpura with an anti-Fc gamma-receptor antibody. N. Engl. J. Med. 1986, 314, 1236–1239. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, M.M.; MacLachlan, T.K.; Troutt, M.; Magee, T.; Tuaillon, N.; Johnson, S.; Stein, K.E.; Bonvini, E.; Garman, R.; Andrews, L. Nonclinical evaluation of GMA161--an antihuman CD16 (FcgammaRIII) monoclonal antibody for treatment of autoimmune disorders in CD16 transgenic mice. Toxicol. Sci. 2012, 125, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallasch, C.P.; Leskov, I.; Braun, C.J.; Vorholt, D.; Drake, A.; Soto-Feliciano, Y.M.; Bent, E.H.; Schwamb, J.; Iliopoulou, B.; Kutsch, N.; et al. Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell 2014, 156, 590–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roghanian, A.; Teige, I.; Martensson, L.; Cox, K.L.; Kovacek, M.; Ljungars, A.; Mattson, J.; Sundberg, A.; Vaughan, A.T.; Shah, V.; et al. Antagonistic human FcgammaRIIB (CD32B) antibodies have anti-tumor activity and overcome resistance to antibody therapy in vivo. Cancer Cell 2015, 27, 473–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, C.T.; Veri, M.C.; Gorlatov, S.; Tuaillon, N.; Burke, S.; Huang, L.; Inzunza, H.D.; Li, H.; Thomas, S.; Johnson, S.; et al. CD32B, the human inhibitory Fc-gamma receptor IIB, as a target for monoclonal antibody therapy of B-cell lymphoma. Blood 2006, 108, 2384–2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, E.L.; Tutt, A.L.; Beers, S.A.; French, R.R.; Chan, C.H.; Cox, K.L.; Roghanian, A.; Penfold, C.A.; Butts, C.L.; Boross, P.; et al. Immunotherapy Targeting Inhibitory Fcgamma Receptor IIB (CD32b) in the Mouse Is Limited by Monoclonal Antibody Consumption and Receptor Internalization. J. Immunol. 2013, 191, 4130–4140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teige, I.; Martensson, L.; Frendeus, B.L. Targeting the Antibody Checkpoints to Enhance Cancer Immunotherapy-Focus on FcgammaRIIB. Front. Immunol. 2019, 10, 481. [Google Scholar] [CrossRef] [Green Version]
- Veillette, A.; Chen, J. SIRPalpha-CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, K.; Buque, A.; Mondragon, L.; Xie, W.; Levesque, S.; Pol, J.; Zitvogel, L.; Kepp, O.; Kroemer, G. Anticancer effects of anti-CD47 immunotherapy in vivo. Oncoimmunology 2019, 8, 1550619. [Google Scholar] [CrossRef] [Green Version]
- Okazawa, H.; Motegi, S.; Ohyama, N.; Ohnishi, H.; Tomizawa, T.; Kaneko, Y.; Oldenborg, P.A.; Ishikawa, O.; Matozaki, T. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J. Immunol. 2005, 174, 2004–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, S.A.; Grinstein, S. Phagocytosis: Receptors, signal integration, and the cytoskeleton. Immunol. Rev. 2014, 262, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Hutter, G.; Kahn, S.A.; Azad, T.D.; Gholamin, S.; Xu, C.Y.; Liu, J.; Achrol, A.S.; Richard, C.; Sommerkamp, P.; et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PLoS ONE 2016, 11, e0153550. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Petrova, P.S.; Viller, N.N.; Wong, M.; Pang, X.; Lin, G.H.; Dodge, K.; Chai, V.; Chen, H.; Lee, V.; House, V.; et al. TTI-621 (SIRPalphaFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin. Cancer Res. 2017, 23, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Logtenberg, M.E.W.; Jansen, J.H.M.; Raaben, M.; Toebes, M.; Franke, K.; Brandsma, A.M.; Matlung, H.L.; Fauster, A.; Gomez-Eerland, R.; Bakker, N.A.M.; et al. Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPalpha axis and a target for cancer immunotherapy. Nat. Med. 2019, 25, 612–619. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.; Volkmer, J.P.; Levin, A.M.; Volkmer, A.K.; Ozkan, E.; Fernhoff, N.B.; van de Rijn, M.; Weissman, I.L.; et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013, 341, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Dheilly, E.; Moine, V.; Broyer, L.; Salgado-Pires, S.; Johnson, Z.; Papaioannou, A.; Cons, L.; Calloud, S.; Majocchi, S.; Nelson, R.; et al. Selective Blockade of the Ubiquitous Checkpoint Receptor CD47 Is Enabled by Dual-Targeting Bispecific Antibodies. Mol. Ther. 2017, 25, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Pirruccello, S.J.; LeBien, T.W. The human B cell-associated antigen CD24 is a single chain sialoglycoprotein. J. Immunol. 1986, 136, 3779–3784. [Google Scholar] [PubMed]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Zhu, X.; Li, K.; Chen, Y.; Liu, X.; Xu, B.; Lei, M.; Xu, J.; Sun, H.C. Blocking siglec-10(hi) tumor-associated macrophages improves anti-tumor immunity and enhances immunotherapy for hepatocellular carcinoma. Exp. Hematol. Oncol. 2021, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Han, C.; Xie, B.; Hu, X.; Yu, Q.; Shi, L.; Wang, Q.; Li, D.; Wang, J.; Zheng, P.; et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 2013, 152, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.Y.; Chen, X.; King, S.; Cavassani, K.A.; Cheng, J.; Zheng, X.; Cao, H.; Yu, H.; Qu, J.; Fang, D.; et al. Amelioration of sepsis by inhibiting sialidase-mediated disruption of the CD24-SiglecG interaction. Nat. Biotechnol. 2011, 29, 428–435. [Google Scholar] [CrossRef]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target | Compound | Sponsor | Phase | Indication | Status | ClinicalTrials.gov identifier |
|---|---|---|---|---|---|---|
| CCR2 | Plozalizumab | Southwest Oncology Group | II | Metastatic cancer, unspecified adult solid tumor | Completed | NCT01015560 |
| CCL2 | Carlumab | Centocor Research & Development, Inc. | II | Prostate cancer | Completed | NCT00992186 |
| CSF-1R | AMG820 | Amgen | I | Solid tumors | Completed | NCT0144404 |
| Emactuzumab (RG7155) | Roche | I | Solid tumors | Completed | NCT01494688 | |
| IMC-SC4 | Eli Lilly | I | Breast and prostate cancer | Active | NCT02265536 | |
| CD40 | SEA-CD40 | Seagen Inc. | I | Non-small-cell lung carcinoma, squamous solid tumors | Active | NCT02376699 |
| LVGN7409 | Lyvgen Biopharma Holdings Limited | I | Solid tumors | Active | NCT04635995 | |
| CDX-1140 | Celldex Therapeutics | I/II | Melanoma | Active | NCT04364230 | |
| APX005M | Apexigen, Inc. | II | Soft tissue sarcoma | Active | NCT03719430 | |
| ADC-1013 | Janssen Research & Development, LLC | I | Advanced solid neoplasms | Active | NCT02829099 | |
| ChiLob 7/4 | Cancer Research UK | I | B-cell lymphoma | Completed | NCT01561911 | |
| Selicrelumab | Hoffmann-La Roche | I/II | Pancreatic adenocarcinoma | Active | NCT03193190 | |
| FcγRIIb | BI-1206 | BioInvent International AB | I/II | Indolent B-cell non-Hodgkin lymphoma | Active | NCT03571568 |
| SIRPα | BI 765063 | OSE Immunotherapeutics | I | Solid tumor | Active | NCT03990233 |
| CC-95251 | Celgene | I | Neoplasms | Active | NCT03783403 | |
| GS-0189 | Gilead Sciences | I | Non-Hodgkin lymphoma | Active | NCT04502706 | |
| VISTA | CI-8993 | Curis, Inc. | I | Solid tumor | Active | NCT04475523 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, K.; Cragg, M.S.; Beers, S.A. Remodeling the Tumor Myeloid Landscape to Enhance Antitumor Antibody Immunotherapies. Cancers 2021, 13, 4904. https://doi.org/10.3390/cancers13194904
Hussain K, Cragg MS, Beers SA. Remodeling the Tumor Myeloid Landscape to Enhance Antitumor Antibody Immunotherapies. Cancers. 2021; 13(19):4904. https://doi.org/10.3390/cancers13194904
Chicago/Turabian StyleHussain, Khiyam, Mark S. Cragg, and Stephen A. Beers. 2021. "Remodeling the Tumor Myeloid Landscape to Enhance Antitumor Antibody Immunotherapies" Cancers 13, no. 19: 4904. https://doi.org/10.3390/cancers13194904
APA StyleHussain, K., Cragg, M. S., & Beers, S. A. (2021). Remodeling the Tumor Myeloid Landscape to Enhance Antitumor Antibody Immunotherapies. Cancers, 13(19), 4904. https://doi.org/10.3390/cancers13194904