
The Cellular Prion Protein and the Hallmarks of Cancer


Abstract
:Simple Summary
Abstract
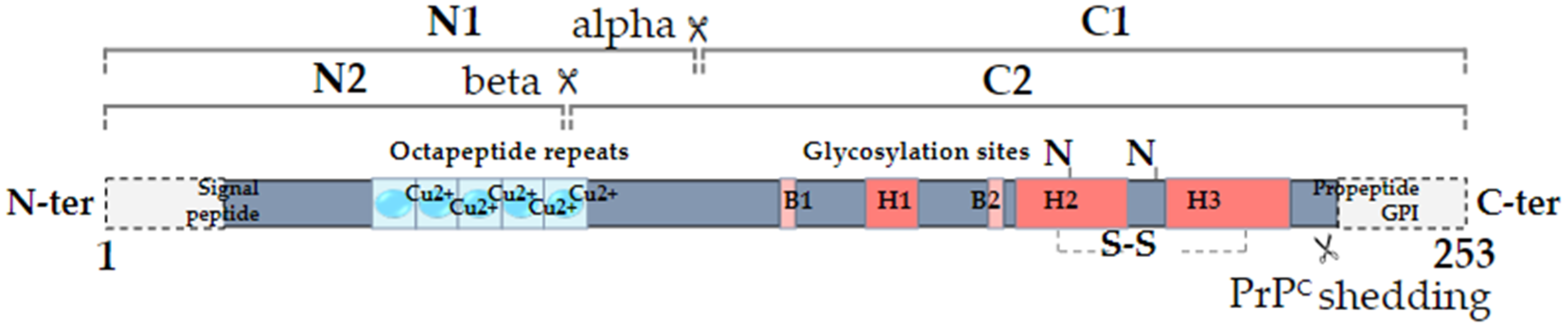
1. Introduction
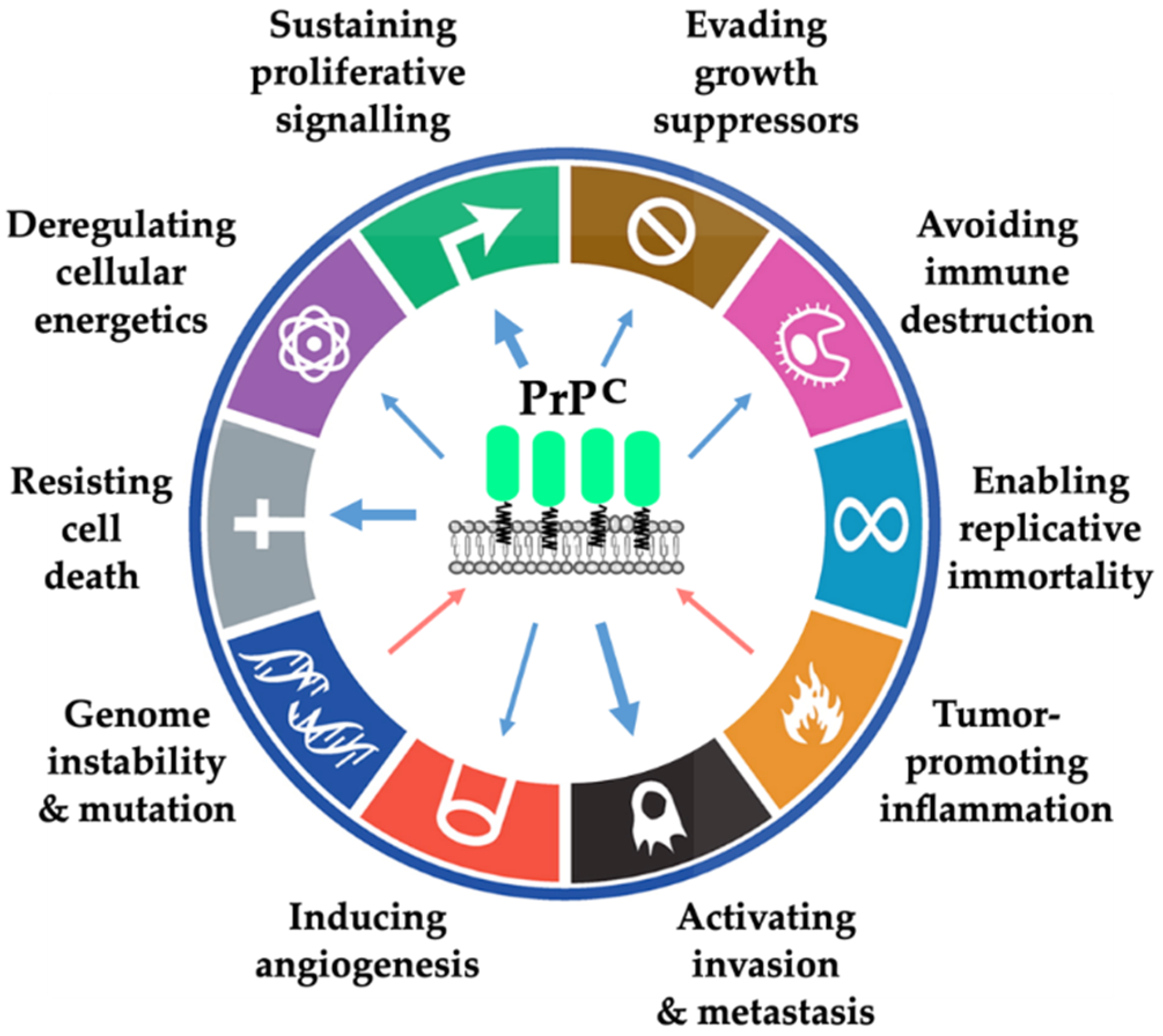
2. Sustaining Proliferative Signalling
3. Evading Growth Suppressors
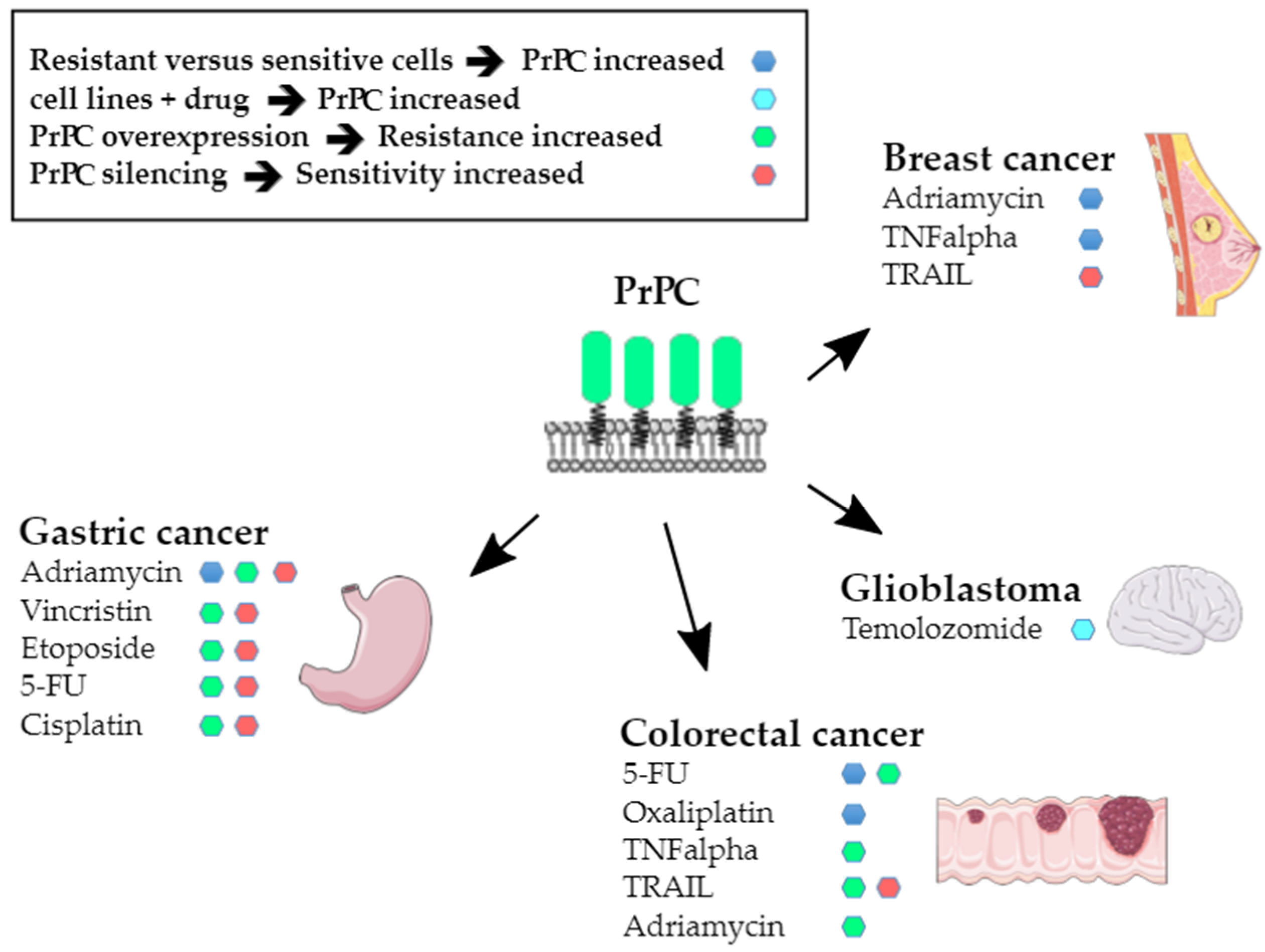
4. Resisting Cell Death
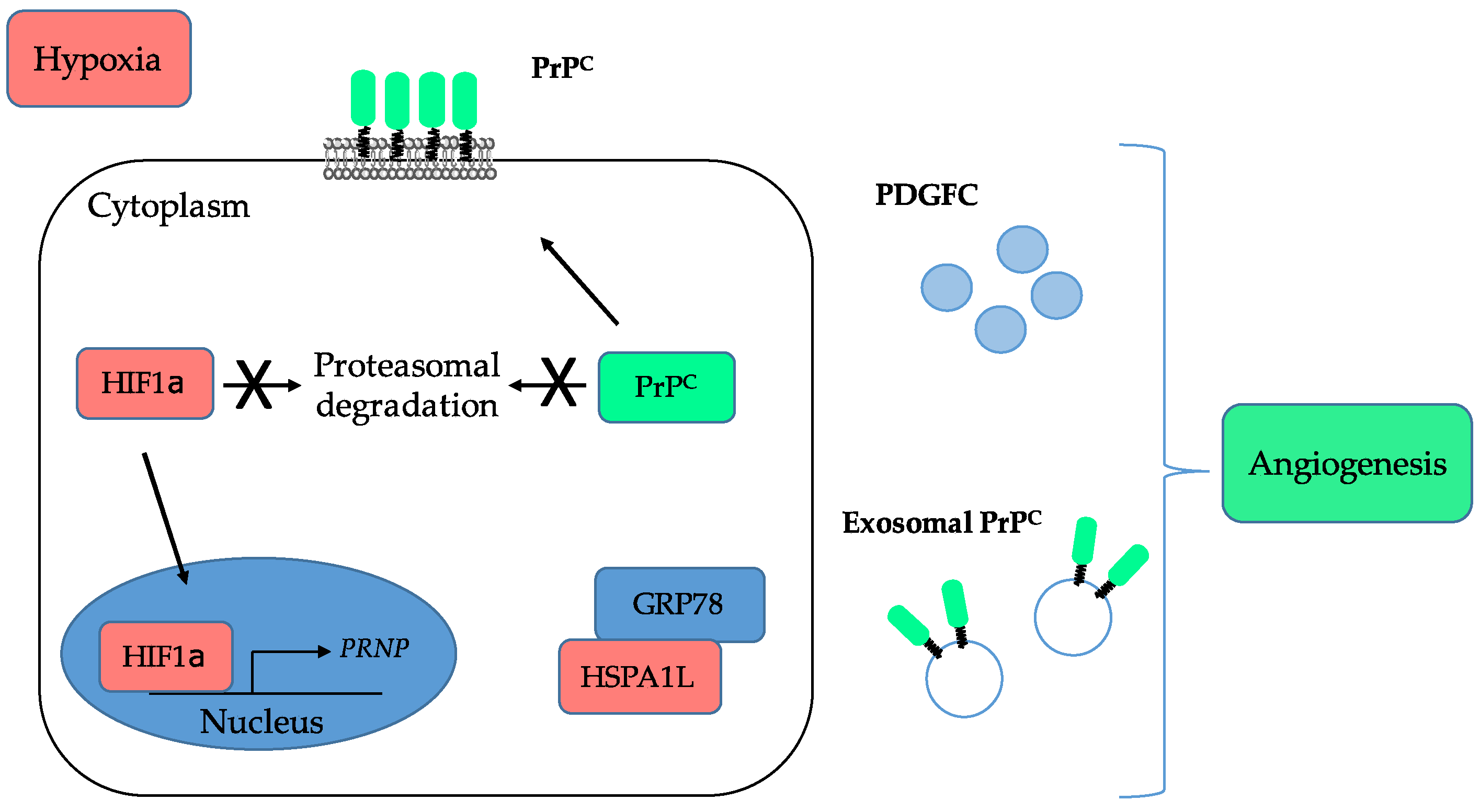
5. Inducing Angiogenesis
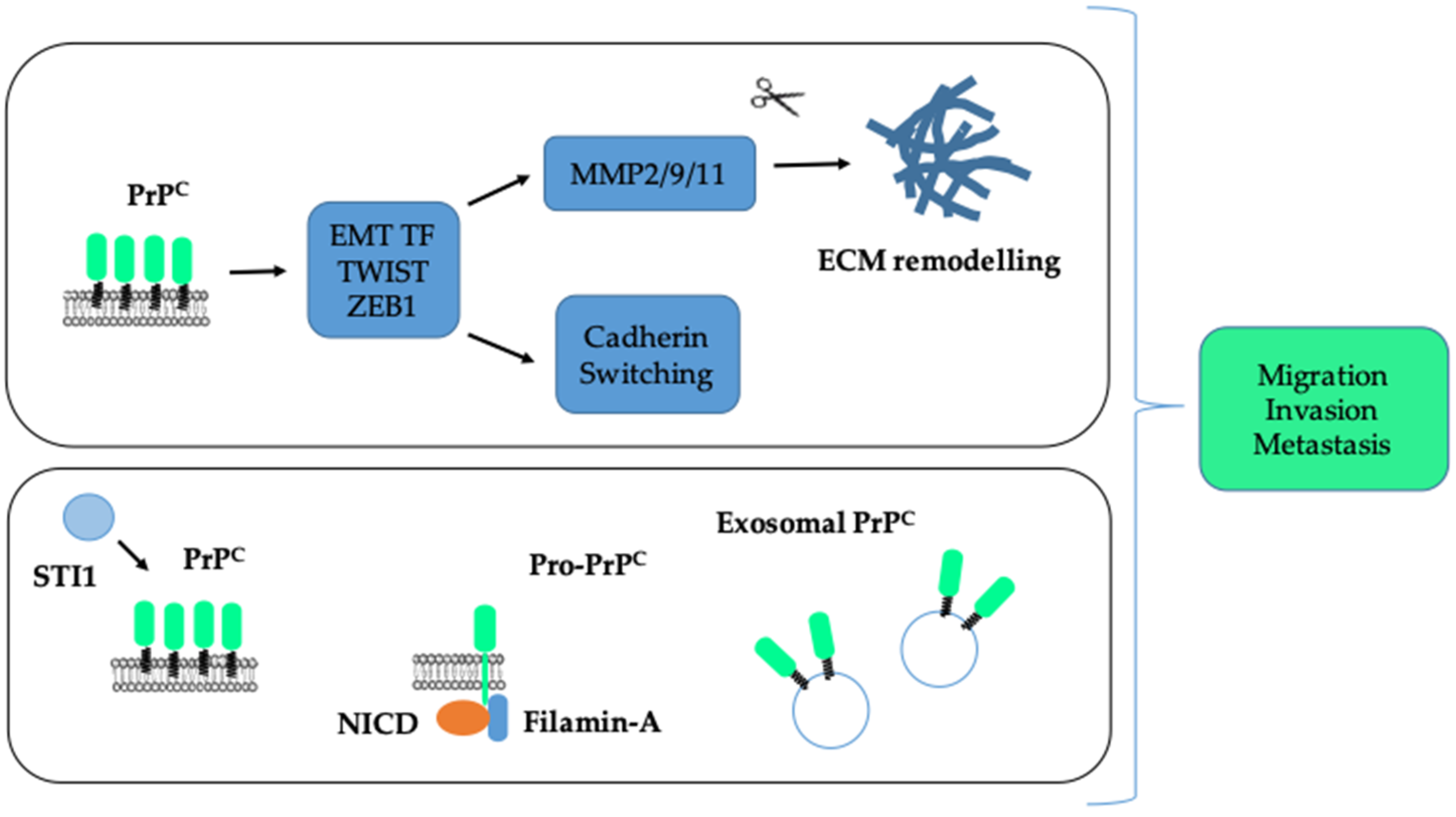
6. Activating Invasion and Metastasis
7. Reprogramming of Energy Metabolism (Emerging Hallmark)
8. Evading Immune Destruction (Emerging Hallmark)
9. Genome Instability and Mutation
10. Tumour-Promoting Inflammation
11. Conclusions/Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluorouracil |
| APE1 | apurinic/apyrimidinic endonuclease 1 |
| ATM | ataxia telangiectasia mutated |
| EMT | epithelial to mesenchymal transition |
| EGFR | epidermal growth factor receptor |
| GPI | glycosyl-phosphatidylinositol |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| HSPA1L | heat shock protein 70 member 1-like |
| HUVEC | human umbilical vein endothelial cell |
| ILK | integrin linked kinase |
| NF2 | neurofibromatosis type 2 |
| PDGFC | platelet-derived growth factor C |
| PrPC | cellular prion protein |
| PrPSc | scrapie prion protein |
| SNU-C5/FUR | 5-FU resistant SNU-C5 cells |
| TGFβ | transforming growth factor β |
| TNFα | tumour necrosis factor-α |
References
- Oesch, B.; Westaway, D.; Walchli, M.; McKinley, M.P.; Kent, S.B.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A Cellular Gene Encodes Scrapie PrP 27-30 Protein. Cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Aguzzi, A.; Baumann, F.; Bremer, J. The Prion’s Elusive Reason for Being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D.; Pepe, A.; Zurzolo, C. Chapter Three—Cell Biology of Prion Protein. In Progress in Molecular Biology and Translational Science; Legname, G., Vanni, S., Eds.; Prion Protein; Academic Press: Cambridge, MA, USA, 2017; Volume 150, pp. 57–82. [Google Scholar]
- Evans, E.G.B.; Millhauser, G.L. Copper- and Zinc-Promoted Interdomain Structure in the Prion Protein: A Mechanism for Autoinhibition of the Neurotoxic N-Terminus. Prog. Mol. Biol. Transl. Sci. 2017, 150, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Linsenmeier, L.; Altmeppen, H.C.; Wetzel, S.; Mohammadi, B.; Saftig, P.; Glatzel, M. Diverse Functions of the Prion Protein—Does Proteolytic Processing Hold the Key? Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2128–2137. [Google Scholar] [CrossRef]
- Hirsch, T.Z.; Martin-Lannerée, S.; Mouillet-Richard, S. Functions of the Prion Protein. Prog. Mol. Biol. Transl. Sci. 2017, 150, 1–34. [Google Scholar] [CrossRef]
- Hirsch, T.Z.; Hernandez-Rapp, J.; Martin-Lanneree, S.; Launay, J.M.; Mouillet-Richard, S. PrPC Signalling in Neurons: From Basics to Clinical Challenges. Biochimie 2014, 104C, 2–11. [Google Scholar] [CrossRef]
- Manni, G.; Lewis, V.; Senesi, M.; Spagnolli, G.; Fallarino, F.; Collins, S.J.; Mouillet-Richard, S.; Biasini, E. The Cellular Prion Protein beyond Prion Diseases. Swiss Med. Wkly. 2020, 150, w20222. [Google Scholar] [CrossRef] [PubMed]
- Corsaro, A.; Bajetto, A.; Thellung, S.; Begani, G.; Villa, V.; Nizzari, M.; Pattarozzi, A.; Solari, A.; Gatti, M.; Pagano, A.; et al. Cellular Prion Protein Controls Stem Cell-like Properties of Human Glioblastoma Tumor-Initiating Cells. Oncotarget 2016, 7, 38638. [Google Scholar] [CrossRef]
- Go, G.; Lee, S.H. The Cellular Prion Protein: A Promising Therapeutic Target for Cancer. Int. J. Mol. Sci. 2020, 21, 9208. [Google Scholar] [CrossRef]
- Martin-Lannerée, S.; Hirsch, T.Z.; Hernandez-Rapp, J.; Halliez, S.; Vilotte, J.L.; Launay, J.M.; Mouillet-Richard, S. PrPC from Stem Cells to Cancer. Front. Cell Dev. Biol. 2014, 2, 55. [Google Scholar] [PubMed] [Green Version]
- Ryskalin, L.; Biagioni, F.; Busceti, C.L.; Giambelluca, M.A.; Morelli, L.; Frati, A.; Fornai, F. The Role of Cellular Prion Protein in Promoting Stemness and Differentiation in Cancer. Cancers 2021, 13, 170. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Pan, Y.; Zhang, D.; Guo, C.; Shi, Y.; Wang, J.; Chen, Y.; Wang, X.; Liu, J.; Guo, X.; et al. Cellular Prion Protein Promotes Proliferation and G1/S Transition of Human Gastric Cancer Cells SGC7901 and AGS. FASEB J. 2007, 21, 2247–2256. [Google Scholar] [CrossRef]
- Vassallo, N.; Herms, J.; Behrens, C.; Krebs, B.; Saeki, K.; Onodera, T.; Windl, O.; Kretzschmar, H.A. Activation of Phosphatidylinositol 3-Kinase by Cellular Prion Protein and Its Role in Cell Survival. Biochem. Biophys. Res. Commun. 2005, 332, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yu, S.; Nakamura, F.; Yin, S.; Xu, J.; Petrolla, A.A.; Singh, N.; Tartakoff, A.; Abbott, D.W.; Xin, W.; et al. Binding of Pro-Prion to Filamin a Disrupts Cytoskeleton and Correlates with Poor Prognosis in Pancreatic Cancer. J. Clin. Invest. 2009, 119, 2725–2736. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, S.; Huang, D.; Cui, M.; Hu, H.; Zhang, L.; Wang, W.; Parameswaran, N.; Jackson, M.; Osborne, B.; et al. Cellular Prion Protein Mediates Pancreatic Cancer Cell Survival and Invasion through Association with and Enhanced Signaling of Notch1. Am. J. Pathol. 2016, 186, 2945–2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.T.; Jiang, G.; Chen, Q.; Zheng, J.N. Ki67 Is a Promising Molecular Target in the Diagnosis of Cancer (Review). Mol. Med. Rep. 2015, 11, 1566–1572. [Google Scholar] [CrossRef] [Green Version]
- Martin-Lannerée, S.; Halliez, S.; Hirsch, T.Z.; Hernandez-Rapp, J.; Passet, B.; Tomkiewicz, C.; Villa-Diaz, A.; Torres, J.-M.; Launay, J.-M.; Béringue, V.; et al. The Cellular Prion Protein Controls Notch Signaling in Neural Stem/Progenitor Cells. Stem Cells 2017, 35, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.H.; Santos, T.G.; Rodrigues, B.R.; Queiroz-Hazarbassanov, N.; Cunha, I.W.; Wasilewska-Sampaio, A.P.; Costa-Silva, B.; Marchi, F.A.; Bleggi-Torres, L.F.; Sanematsu, P.I.; et al. Disruption of Prion Protein-HOP Engagement Impairs Glioblastoma Growth and Cognitive Decline and Improves Overall Survival. Oncogene 2014, 34, 3305–3314. [Google Scholar] [CrossRef]
- Iglesia, R.P.; Prado, M.B.; Cruz, L.; Martins, V.R.; Santos, T.G.; Lopes, M.H. Engagement of Cellular Prion Protein with the Co-Chaperone Hsp70/90 Organizing Protein Regulates the Proliferation of Glioblastoma Stem-like Cells. Stem Cell Res. Ther. 2017, 8, 76. [Google Scholar] [CrossRef]
- Zhuang, D.; Liu, Y.; Mao, Y.; Gao, L.; Zhang, H.; Luan, S.; Huang, F.; Li, Q. TMZ-Induced PrPc/Par-4 Interaction Promotes the Survival of Human Glioma Cells. Int. J. Cancer 2012, 130, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, L.; Ryan, Y.; Hilton, D.A.; Lyons-Rimmer, J.; Dave, F.; Maze, E.A.; Adams, C.L.; Rigby-Jones, R.; Ammoun, S.; Hanemann, C.O. Cellular Prion Protein (PrPC) in the Development of Merlin-Deficient Tumours. Oncogene 2017, 36, 6132–6142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chieng, C.K.-L.; Say, Y.-H. Cellular Prion Protein Contributes to LS 174T Colon Cancer Cell Carcinogenesis by Increasing Invasiveness and Resistance against Doxorubicin-Induced Apoptosis. Tumour Biol. 2015, 36, 8107–8120. [Google Scholar] [CrossRef] [PubMed]
- Ghazi, A.; Le Corre, D.; Pilati, C.; Taieb, J.; Aparicio, T.; Didelot, A.; Dedhar, S.; Mulot, C.; Le Malicot, K.; Djouadi, F.; et al. Prognostic Value of the PrPC-ILK-IDO1 Axis in the Mesenchymal Colorectal Cancer Subtype. Oncoimmunology 2021, 10, 1940674. [Google Scholar] [CrossRef]
- Go, G.; Yun, C.W.; Yoon, Y.M.; Lim, J.H.; Lee, J.H.; Lee, S.H. Role of PrPC in Cancer Stem Cell Characteristics and Drug Resistance in Colon Cancer Cells. Anticancer Res. 2020, 40, 5611–5620. [Google Scholar] [CrossRef]
- Le Corre, D.; Ghazi, A.; Balogoun, R.; Pilati, C.; Aparicio, T.; Martin-Lannerée, S.; Marisa, L.; Djouadi, F.; Poindessous, V.; Crozet, C.; et al. The Cellular Prion Protein Controls the Mesenchymal-like Molecular Subtype and Predicts Disease Outcome in Colorectal Cancer. EBioMedicine 2019, 46, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.Q.; Sun, Y.P.; Ruan, C.P.; Xu, X.Y.; Ge, J.H.; He, J.; Xu, Z.D.; Wang, Q.; Gao, W.C. Cellular Prion Protein Promotes Glucose Uptake through the Fyn-HIF-2α-Glut1 Pathway to Support Colorectal Cancer Cell Survival. Cancer Sci. 2011, 102, 400–406. [Google Scholar] [CrossRef]
- Yap, Y.H.-Y.; Say, Y.-H. Resistance against Tumour Necrosis Factor α Apoptosis by the Cellular Prion Protein Is Cell-Specific for Oral, Colon and Kidney Cancer Cell Lines. Cell Biol. Int. 2012, 36, 273–277. [Google Scholar] [CrossRef]
- Yun, C.-W.; Lee, J.-H.; Go, G.; Jeon, J.; Yoon, S.; Lee, S.-H. Prion Protein of Extracellular Vesicle Regulates the Progression of Colorectal Cancer. Cancers 2021, 13, 2144. [Google Scholar] [CrossRef]
- Dias, M.V.S.; Teixeira, B.L.; Rodrigues, B.R.; Sinigaglia-Coimbra, R.; Porto-Carreiro, I.; Roffé, M.; Hajj, G.N.M.; Martins, V.R. PRNP/Prion Protein Regulates the Secretion of Exosomes Modulating CAV1/Caveolin-1-Suppressed Autophagy. Autophagy 2016, 12, 2113–2128. [Google Scholar] [CrossRef] [Green Version]
- Llorens, F.; Carulla, P.; Villa, A.; Torres, J.M.; Fortes, P.; Ferrer, I.; del Rio, J.A. PrP(C) Regulates Epidermal Growth Factor Receptor Function and Cell Shape Dynamics in Neuro2a Cells. J. Neurochem. 2013, 127, 124–138. [Google Scholar] [CrossRef]
- Atkinson, C.J.; Kawamata, F.; Liu, C.; Ham, S.; Győrffy, B.; Munn, A.L.; Wei, M.Q.; Möller, A.; Whitehall, V.; Wiegmans, A.P. EGFR and Prion Protein Promote Signaling via FOXO3a-KLF5 Resulting in Clinical Resistance to Platinum Agents in Colorectal Cancer. Mol. Oncol. 2019, 13, 725–737. [Google Scholar] [CrossRef] [Green Version]
- Checler, F.; Alves da Costa, C. P53 in Neurodegenerative Diseases and Brain Cancers. Pharmacol. Ther. 2014, 142, 99–113. [Google Scholar] [CrossRef]
- Paitel, E.; Fahraeus, R.; Checler, F. Cellular Prion Protein Sensitizes Neurons to Apoptotic Stimuli through Mdm2-Regulated and P53-Dependent Caspase 3-like Activation. J. Biol. Chem. 2003, 278, 10061–10066. [Google Scholar] [CrossRef] [Green Version]
- Sunyach, C.; Cisse, M.A.; da Costa, C.A.; Vincent, B.; Checler, F. The C-Terminal Products of Cellular Prion Protein Processing, C1 and C2, Exert Distinct Influence on P53-Dependent Staurosporine-Induced Caspase-3 Activation. J. Biol. Chem. 2007, 282, 1956–1963. [Google Scholar] [CrossRef] [Green Version]
- Guillot-Sestier, M.V.; Sunyach, C.; Druon, C.; Scarzello, S.; Checler, F. The α-Secretase-Derived N-Terminal Product of Cellular Prion, N1, Displays Neuroprotective Function In Vitro and In Vivo. J. Biol. Chem. 2009, 284, 35973–35986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altmeppen, H.C.; Prox, J.; Puig, B.; Kluth, M.A.; Bernreuther, C.; Thurm, D.; Jorissen, E.; Petrowitz, B.; Bartsch, U.; De Strooper, B.; et al. Lack of A-Disintegrin-and-Metalloproteinase ADAM10 Leads to Intracellular Accumulation and Loss of Shedding of the Cellular Prion Protein In Vivo. Mol. Neurodegener. 2011, 6, 36. [Google Scholar] [CrossRef] [Green Version]
- Weiss, E.; Ramljak, S.; Asif, A.R.; Ciesielczyk, B.; Schmitz, M.; Gawinecka, J.; Schulz-Schaeffer, W.; Behrens, C.; Zerr, I. Cellular Prion Protein Overexpression Disturbs Cellular Homeostasis in SH-SY5Y Neuroblastoma Cells but Does Not Alter P53 Expression: A Proteomic Study. Neuroscience 2010, 169, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Parchaliuk, D.; Medina, S.; Sorensen, G.; Landry, L.; Huang, S.; Wang, M.; Kong, Q.; Booth, S.A. Activation of P53-Regulated pro-Apoptotic Signaling Pathways in PrP-Mediated Myopathy. BMC Genomics 2009, 10, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Pan, Y.L.; Ning, X.X.; Sun, L.J.; Lan, M.; Hong, L.; Du, J.P.; Liu, N.; Liu, C.J.; Qiao, T.D.; et al. Overexpression of PrPC and Its Antiapoptosis Function in Gastric Cancer. Tumour Biol. 2006, 27, 84–91. [Google Scholar] [CrossRef]
- Curto, M.; McClatchey, A.I. Nf2/Merlin: A Coordinator of Receptor Signalling and Intercellular Contact. Br. J. Cancer 2008, 98, 256–262. [Google Scholar] [CrossRef] [Green Version]
- Serrano, I.; McDonald, P.C.; Lock, F.; Muller, W.J.; Dedhar, S. Inactivation of the Hippo Tumour Suppressor Pathway by Integrin-Linked Kinase. Nat. Commun. 2013, 4, 2976. [Google Scholar] [CrossRef]
- Mouillet-Richard, S.; Laurent-Puig, P. YAP/TAZ Signalling in Colorectal Cancer: Lessons from Consensus Molecular Subtypes. Cancers 2020, 12, 3160. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Tumaneng, K.; Guan, K.-L. The Hippo Pathway in Organ Size Control, Tissue Regeneration and Stem Cell Self-Renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Batlle, E. Targeting the Microenvironment in Advanced Colorectal Cancer. Trends Cancer 2016, 2, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; You, H.; Liu, F.; An, H.; Shi, Y.; Yu, Q.; Fan, D. Differentially Expressed Gene Profiles between Multidrug Resistant Gastric Adenocarcinoma Cells and Their Parental Cells. Cancer Lett. 2002, 185, 211–218. [Google Scholar] [CrossRef]
- Diarra-Mehrpour, M.; Arrabal, S.; Jalil, A.; Pinson, X.; Gaudin, C.; Pietu, G.; Pitaval, A.; Ripoche, H.; Eloit, M.; Dormont, D.; et al. Prion Protein Prevents Human Breast Carcinoma Cell Line from Tumor Necrosis Factor α-Induced Cell Death. Cancer Res. 2004, 64, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Meslin, F.; Hamai, A.; Gao, P.; Jalil, A.; Cahuzac, N.; Chouaib, S.; Mehrpour, M. Silencing of Prion Protein Sensitizes Breast Adriamycin-Resistant Carcinoma Cells to TRAIL-Mediated Cell Death. Cancer Res. 2007, 67, 10910–10919. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Tao, L.; Xu, J.; Li, Q.; Yu, J.; Jin, Y.; Chen, Q.; Xu, Z.; Zou, Q.; Liu, X. CD44/Cellular Prion Protein Interact in Multidrug Resistant Breast Cancer Cells and Correlate with Responses to Neoadjuvant Chemotherapy in Breast Cancer Patients. Mol. Carcinog. 2013, 53, 686–697. [Google Scholar] [CrossRef]
- Lee, J.H.; Yun, C.W.; Lee, S.H. Cellular Prion Protein Enhances Drug Resistance of Colorectal Cancer Cells via Regulation of a Survival Signal Pathway. Biomol. Ther. 2018, 26, 313–321. [Google Scholar] [CrossRef]
- Du, J.; Pan, Y.; Shi, Y.; Guo, C.; Jin, X.; Sun, L.; Liu, N.; Qiao, T.; Fan, D. Overexpression and Significance of Prion Protein in Gastric Cancer and Multidrug-Resistant Gastric Carcinoma Cell Line SGC7901/ADR. Int. J. Cancer 2005, 113, 213–220. [Google Scholar] [CrossRef]
- Liang, J.; Bai, F.; Luo, G.; Wang, J.; Liu, J.; Ge, F.; Pan, Y.; Yao, L.; Du, R.; Li, X.; et al. Hypoxia Induced Overexpression of PrP(C) in Gastric Cancer Cell Lines. Cancer Biol. Ther. 2007, 6, 769–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Han, Y.-S.; Yoon, Y.M.; Yun, C.W.; Yun, S.P.; Kim, S.M.; Kwon, H.Y.; Jeong, D.; Baek, M.J.; Lee, H.J.; et al. Role of HSPA1L as a Cellular Prion Protein Stabilizer in Tumor Progression via HIF-1α/GP78 Axis. Oncogene 2017, 36, 6555–6567. [Google Scholar] [CrossRef]
- Park, J.-Y.; Jeong, J.-K.; Lee, J.-H.; Moon, J.-H.; Kim, S.-W.; Lee, Y.-J.; Park, S.-Y. Induction of Cellular Prion Protein (PrPc) under Hypoxia Inhibits Apoptosis Caused by TRAIL Treatment. Oncotarget 2015, 6, 5342–5353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-H.; Du, J.-P.; Zhang, Y.-H.; Zhao, X.-J.; Fan, R.-Y.; Wang, Z.-H.; Wu, Z.-T.; Han, Y. Dynamic Changes and Surveillance Function of Prion Protein Expression in Gastric Cancer Drug Resistance. World J. Gastroenterol. 2011, 17, 3986–3993. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Ge, F.; Guo, C.; Luo, G.; Wang, X.; Han, G.; Zhang, D.; Wang, J.; Li, K.; Pan, Y.; et al. Inhibition of PI3K/Akt Partially Leads to the Inhibition of PrP(C)-Induced Drug Resistance in Gastric Cancer Cells. FEBS J. 2009, 276, 685–694. [Google Scholar] [CrossRef]
- Wang, J.H.; Du, J.P.; Li, S.J.; Zhai, L.P.; Yang, X.Y.; Wang, Z.H.; Wu, Z.T.; Han, Y. Octarepeat Peptides of Prion Are Essential for Multidrug Resistance in Gastric Cancer Cells. J. Dig. Dis. 2012, 13, 143–152. [Google Scholar] [CrossRef]
- Liang, J.; Wang, J.; Luo, G.; Pan, Y.; Wang, X.; Guo, C.; Zhang, D.; Yin, F.; Zhang, X.; Liu, J.; et al. Function of PrPC (1-OPRD) in Biological Activities of Gastric Cancer Cell Lines. J. Cell. Mol. Med. 2009, 13, 4453–4464. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Wang, W.; Wu, Q.; Lu, Y.; Su, T.; Gu, N.; Li, K.; Wang, J.; Du, R.; Zhao, X.; et al. MGr1-Antigen/37 KDa Laminin Receptor Precursor Promotes Cellular Prion Protein Induced Multi-Drug-Resistance of Gastric Cancer. Oncotarget 2017, 8, 71630–71641. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, G.; Palumbo, S.; Gabrusiewicz, K.; Azzalin, A.; Marchesi, N.; Spedito, A.; Biggiogera, M.; Sbalchiero, E.; Mazzini, G.; Miracco, C.; et al. Silencing of Cellular Prion Protein (PrPC) Expression by DNA-Antisense Oligonucleotides Induces Autophagy-Dependent Cell Death in Glioma Cells. Autophagy 2011, 7, 840–853. [Google Scholar] [CrossRef] [Green Version]
- Roucou, X.; Gains, M.; LeBlanc, A.C. Neuroprotective Functions of Prion Protein. J. Neurosci. Res. 2004, 75, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.S.; Zafar, S.; Latif, U.; Llorens, F.; Sabine, M.; Kumar, P.; Tahir, W.; Thüne, K.; Shafiq, M.; Schmitz, M.; et al. The Role of Cellular Prion Protein in Lipid Metabolism in the Liver. Prion 2020, 14, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Wiegmans, A.P.; Saunus, J.M.; Ham, S.; Lobb, R.; Kutasovic, J.R.; Dalley, A.J.; Miranda, M.; Atkinson, C.; Foliaki, S.T.; Ferguson, K.; et al. Secreted Cellular Prion Protein Binds Doxorubicin and Correlates with Anthracycline Resistance in Breast Cancer. JCI Insight 2019, 5, e124092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardino-Sgherri, J.; Siberchicot, C.; Auvré, F.; Busso, D.; Brocas, C.; El Masri, G.; Lioutsko, A.; Ferri, F.; Radicella, J.P.; Romeo, P.-H.; et al. Tumor Resistance to Radiotherapy Is Triggered by an ATM/TAK1-Dependent-Increased Expression of the Cellular Prion Protein. Oncogene 2021, 40, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Siberchicot, C.; Gault, N.; Déchamps, N.; Barroca, V.; Aguzzi, A.; Roméo, P.-H.; Radicella, J.P.; Bravard, A.; Bernardino-Sgherri, J. Prion Protein Deficiency Impairs Hematopoietic Stem Cell Determination and Sensitizes Myeloid Progenitors to Irradiation. Haematologica 2020, 105, 1216–1222. [Google Scholar] [CrossRef] [Green Version]
- Bravard, A.; Auvré, F.; Fantini, D.; Bernardino-Sgherri, J.; Sissoëff, L.; Daynac, M.; Xu, Z.; Etienne, O.; Dehen, C.; Comoy, E.; et al. The Prion Protein Is Critical for DNA Repair and Cell Survival after Genotoxic Stress. Nucleic Acids Res. 2015, 43, 904–916. [Google Scholar] [CrossRef] [Green Version]
- Turu, M.; Slevin, M.; Ethirajan, P.; Luque, A.; Elasbali, A.; Font, A.; Gaffney, J.; Cairols, M.; Kumar, P.; Kumar, S.; et al. The Normal Cellular Prion Protein and Its Possible Role in Angiogenesis. Front. Biosci. J. Virtual Libr. 2008, 13, 6491–6500. [Google Scholar] [CrossRef] [Green Version]
- Doeppner, T.R.; Kaltwasser, B.; Schlechter, J.; Jaschke, J.; Kilic, E.; Bähr, M.; Hermann, D.M.; Weise, J. Cellular Prion Protein Promotes Post-Ischemic Neuronal Survival, Angioneurogenesis and Enhances Neural Progenitor Cell Homing via Proteasome Inhibition. Cell Death Dis. 2015, 6, e2024. [Google Scholar] [CrossRef] [Green Version]
- Alfaidy, N.; Chauvet, S.; Andrei, S.; Salomon, A.; Saoudi, Y.; Richaud, P.; Aude-Garcia, C.; Hoffmann, P.; Andrieux, A.; Moulis, J.M.; et al. Prion Protein Expression and Functional Importance in Developmental Angiogenesis: Role in Oxidative Stress and Copper Homeostasis. Antioxid. Redox Signal. 2012, 18, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramljak, S.; Herlyn, H.; Zerr, I. Cellular Prion Protein (PrPc) and Hypoxia: True to Each Other in Good Times and in Bad, in Sickness, and in Health. Front. Cell. Neurosci. 2016, 10, 292. [Google Scholar] [CrossRef] [Green Version]
- Singleton, D.C.; Macann, A.; Wilson, W.R. Therapeutic Targeting of the Hypoxic Tumour Microenvironment. Nat. Rev. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Möller, A.; Lobb, R.J. The Evolving Translational Potential of Small Extracellular Vesicles in Cancer. Nat. Rev. Cancer 2020, 20, 697–709. [Google Scholar] [CrossRef]
- Cao, R.; Bråkenhielm, E.; Li, X.; Pietras, K.; Widenfalk, J.; Ostman, A.; Eriksson, U.; Cao, Y. Angiogenesis Stimulated by PDGF-CC, a Novel Member in the PDGF Family, Involves Activation of PDGFR-αα and-αβ Receptors. FASEB J. 2002, 16, 1575–1583. [Google Scholar] [CrossRef]
- Azad, T.; Ghahremani, M.; Yang, X. The Role of YAP and TAZ in Angiogenesis and Vascular Mimicry. Cells 2019, 8, 407. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Morales, J.E.; Avci, N.; Guerrero, P.A.; Rao, G.; Seo, J.H.; McCarty, J.H. The Vascular Endothelial Cell-Expressed Prion Protein Doppel Promotes Angiogenesis and Blood-Brain Barrier Development. Development 2020, 147, dev193094. [Google Scholar] [CrossRef]
- Al-Hilal, T.A.; Chung, S.W.; Choi, J.U.; Alam, F.; Park, J.; Kim, S.W.; Kim, S.Y.; Ahsan, F.; Kim, I.-S.; Byun, Y. Targeting Prion-like Protein Doppel Selectively Suppresses Tumor Angiogenesis. J. Clin. Invest. 2016, 126, 1251–1266. [Google Scholar] [CrossRef]
- Ciric, D.; Rezaei, H. Biochemical Insight into the Prion Protein Family. Front. Cell Dev. Biol. 2015, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.; Kim, Y.K.; Kim, K.-E.; Kim, W.; Park, C.-S.; Lee, K.J. Cellular Prion Protein Regulates Invasion and Migration of Breast Cancer Cells through MMP-9 Activity. Biochem. Biophys. Res. Commun. 2016, 470, 213–219. [Google Scholar] [CrossRef]
- Pan, Y.; Zhao, L.; Liang, J.; Liu, J.; Shi, Y.; Liu, N.; Zhang, G.; Jin, H.; Gao, J.; Xie, H.; et al. Cellular Prion Protein Promotes Invasion and Metastasis of Gastric Cancer. FASEB J. 2006, 20, 1886–1888. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-C.; Lin, C.-H.; Shih, N.-C.; Liu, H.-L.; Wang, W.-C.; Lin, K.-Y.; Liu, Z.-Y.; Tseng, Y.-J.; Chang, H.-K.; Lin, Y.-C.; et al. Cellular Prion Protein Transcriptionally Regulated by NFIL3 Enhances Lung Cancer Cell Lamellipodium Formation and Migration through JNK Signaling. Oncogene 2020, 39, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qian, J.; Wang, F.; Ma, Z. Cellular Prion Protein Accelerates Colorectal Cancer Metastasis via the Fyn-SP1-SATB1 Axis. Oncol. Rep. 2012, 28, 2029–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Rao, G.; Wang, H.; Li, B.; Tian, W.; Cui, J.; He, L.; Laffin, B.; Tian, X.; Hao, C.; et al. CD44-Positive Cancer Stem Cells Expressing Cellular Prion Protein Contribute to Metastatic Capacity in Colorectal Cancer. Cancer Res. 2013, 73, 2682–2694. [Google Scholar] [CrossRef] [Green Version]
- De Lacerda, T.C.S.; Costa-Silva, B.; Giudice, F.S.; Dias, M.V.S.; de Oliveira, G.P.; Teixeira, B.L.; Dos Santos, T.G.; Martins, V.R. Prion Protein Binding to HOP Modulates the Migration and Invasion of Colorectal Cancer Cells. Clin. Exp. Metastasis 2016, 33, 441–451. [Google Scholar] [CrossRef]
- Li, C.; Yu, S.; Nakamura, F.; Pentikäinen, O.T.; Singh, N.; Yin, S.; Xin, W.; Sy, M.-S. Pro-Prion Binds Filamin A, Facilitating Its Interaction with Integrin β1, and Contributes to Melanomagenesis. J. Biol. Chem. 2010, 285, 30328–30339. [Google Scholar] [CrossRef] [Green Version]
- Dery, M.A.; Jodoin, J.; Ursini-Siegel, J.; Aleynikova, O.; Ferrario, C.; Hassan, S.; Basik, M.; Leblanc, A.C. Endoplasmic Reticulum Stress Induces PRNP Prion Protein Gene Expression in Breast Cancer. Breast Cancer Res. 2013, 15, R22. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Shang, Y.; Liu, C.; Li, J.; Hu, H.; Liang, C.; Han, Y.; Zhang, W.; Liang, J.; Wu, K. Overexpression of PrPc, Combined with MGr1-Ag/37LRP, Is Predictive of Poor Prognosis in Gastric Cancer. Int. J. Cancer 2014, 135, 2329–2337. [Google Scholar] [CrossRef]
- Coebergh van den Braak, R.R.J.; Ten Hoorn, S.; Sieuwerts, A.M.; Tuynman, J.B.; Smid, M.; Wilting, S.M.; Martens, J.W.M.; Punt, C.J.A.; Foekens, J.A.; Medema, J.P.; et al. Interconnectivity between Molecular Subtypes and Tumor Stage in Colorectal Cancer. BMC Cancer 2020, 20, 850. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- De Wit, M.; Jimenez, C.R.; Carvalho, B.; Belien, J.A.M.; Delis-van Diemen, P.M.; Mongera, S.; Piersma, S.R.; Vikas, M.; Navani, S.; Pontén, F.; et al. Cell Surface Proteomics Identifies Glucose Transporter Type 1 and Prion Protein as Candidate Biomarkers for Colorectal Adenoma-to-Carcinoma Progression. Gut 2012, 61, 855–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashok, A.; Singh, N. Prion Protein Modulates Glucose Homeostasis by Altering Intracellular Iron. Sci. Rep. 2018, 8, 6556. [Google Scholar] [CrossRef] [PubMed]
- Ramljak, S.; Schmitz, M.; Repond, C.; Zerr, I.; Pellerin, L. Altered MRNA and Protein Expression of Monocarboxylate Transporter MCT1 in the Cerebral Cortex and Cerebellum of Prion Protein Knockout Mice. Int. J. Mol. Sci. 2021, 22, 1566. [Google Scholar] [CrossRef] [PubMed]
- Kleene, R.; Loers, G.; Langer, J.; Frobert, Y.; Buck, F.; Schachner, M. Prion Protein Regulates Glutamate-Dependent Lactate Transport of Astrocytes. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 12331–12340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutishauser, D.; Mertz, K.D.; Moos, R.; Brunner, E.; Rulicke, T.; Calella, A.M.; Aguzzi, A. The Comprehensive Native Interactome of a Fully Functional Tagged Prion Protein. PLoS ONE 2009, 4, e4446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.C.; Huo, H.; Bai, Y.; Ehsani, S.; Jeon, A.H.; Shi, T.; Daude, N.; Lau, A.; Young, R.; Xu, L.; et al. Interactome Analyses Identify Ties of PrP and Its Mammalian Paralogs to Oligomannosidic N-Glycans and Endoplasmic Reticulum-Derived Chaperones. PLoS Pathog. 2009, 5, e1000608. [Google Scholar] [CrossRef]
- Zafar, S.; von Ahsen, N.; Oellerich, M.; Zerr, I.; Schulz-Schaeffer, W.J.; Armstrong, V.W.; Asif, A.R. Proteomics Approach to Identify the Interacting Partners of Cellular Prion Protein and Characterization of Rab7a Interaction in Neuronal Cells. J. Proteome Res. 2011, 10, 3123–3135. [Google Scholar] [CrossRef] [PubMed]
- Ramljak, S.; Schmitz, M.; Zafar, S.; Wrede, A.; Schenkel, S.; Asif, A.R.; Carimalo, J.; Doeppner, T.R.; Schulz-Schaeffer, W.J.; Weise, J.; et al. Cellular Prion Protein Directly Interacts with and Enhances Lactate Dehydrogenase Expression under Hypoxic Conditions. Exp. Neurol. 2015, 271, 155–167. [Google Scholar] [CrossRef]
- Strom, A.; Diecke, S.; Hunsmann, G.; Stuke, A.W. Identification of Prion Protein Binding Proteins by Combined Use of Far-Western Immunoblotting, Two Dimensional Gel Electrophoresis and Mass Spectrometry. Proteomics 2006, 6, 26–34. [Google Scholar] [CrossRef]
- Zafar, S.; Asif, A.R.; Ramljak, S.; Tahir, W.; Schmitz, M.; Zerr, I. Anchorless 23-230 PrPC Interactomics for Elucidation of PrPC Protective Role. Mol. Neurobiol. 2014, 49, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
- Faris, R.; Moore, R.A.; Ward, A.; Race, B.; Dorward, D.W.; Hollister, J.R.; Fischer, E.R.; Priola, S.A. Cellular Prion Protein Is Present in Mitochondria of Healthy Mice. Sci. Rep. 2017, 7, 41556. [Google Scholar] [CrossRef] [PubMed]
- Miele, G.; Jeffrey, M.; Turnbull, D.; Manson, J.; Clinton, M. Ablation of Cellular Prion Protein Expression Affects Mitochondrial Numbers and Morphology. Biochem. Biophys. Res. Commun. 2002, 291, 372–377. [Google Scholar] [CrossRef]
- Paterson, A.W.J.; Curtis, J.C.; Macleod, N.K. Complex I Specific Increase in Superoxide Formation and Respiration Rate by PrP-Null Mouse Brain Mitochondria. J. Neurochem. 2008, 105, 177–191. [Google Scholar] [CrossRef]
- Stella, R.; Cifani, P.; Peggion, C.; Hansson, K.; Lazzari, C.; Bendz, M.; Levander, F.; Sorgato, M.C.; Bertoli, A.; James, P. Relative Quantification of Membrane Proteins in Wild-Type and Prion Protein (PrP)-Knockout Cerebellar Granule Neurons. J. Proteome Res. 2012, 11, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; de Reyniès, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautès-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [Green Version]
- Bakkebø, M.K.; Mouillet-Richard, S.; Espenes, A.; Goldmann, W.; Tatzelt, J.; Tranulis, M.A. The Cellular Prion Protein: A Player in Immunological Quiescence. Front. Immunol. 2015, 6, 450. [Google Scholar] [CrossRef] [Green Version]
- Angioni, R.; Sánchez-Rodríguez, R.; Viola, A.; Molon, B. TGF-β in Cancer: Metabolic Driver of the Tolerogenic Crosstalk in the Tumor Microenvironment. Cancers 2021, 13, 401. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Domingues, P.H.; Nanduri, L.S.Y.; Seget, K.; Venkateswaran, S.V.; Agorku, D.; Viganó, C.; von Schubert, C.; Nigg, E.A.; Swanton, C.; Sotillo, R.; et al. Cellular Prion Protein PrPC and Ecto-5′-Nucleotidase Are Markers of the Cellular Stress Response to Aneuploidy. Cancer Res. 2017, 77, 2914–2926. [Google Scholar] [CrossRef] [Green Version]
- Qin, K.; Zhao, L.; Ash, R.D.; McDonough, W.F.; Zhao, R.Y. ATM-Mediated Transcriptional Elevation of Prion in Response to Copper-Induced Oxidative Stress. J. Biol. Chem. 2009, 284, 4582–4593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boddy, A.M.; Huang, W.; Aktipis, A. Life History Trade-Offs in Tumors. Curr. Pathobiol. Rep. 2018, 6, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-ΚB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.-R.; Mu, T.-C.; Gao, Z.-X.; Wang, J.; Sy, M.-S.; Li, C.-Y. Prion Protein Is Required for Tumor Necrosis Factor α (TNFα)-Triggered Nuclear Factor ΚB (NF-ΚB) Signaling and Cytokine Production. J. Biol. Chem. 2017, 292, 18747–18759. [Google Scholar] [CrossRef] [Green Version]
- Assi, K.; Mills, J.; Owen, D.; Ong, C.; St Arnaud, R.; Dedhar, S.; Salh, B. Integrin-Linked Kinase Regulates Cell Proliferation and Tumour Growth in Murine Colitis-Associated Carcinogenesis. Gut 2008, 57, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Pammer, J.; Cross, H.S.; Frobert, Y.; Tschachler, E.; Oberhuber, G. The Pattern of Prion-Related Protein Expression in the Gastrointestinal Tract. Virchows Arch. Int. J. Pathol. 2000, 436, 466–472. [Google Scholar] [CrossRef]
- Petit, C.S.V.; Barreau, F.; Besnier, L.; Gandille, P.; Riveau, B.; Chateau, D.; Roy, M.; Berrebi, D.; Svrcek, M.; Cardot, P.; et al. Requirement of Cellular Prion Protein for Intestinal Barrier Function and Mislocalization in Patients with Inflammatory Bowel Disease. Gastroenterology 2012, 143, 122–132.e15. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, N.; Zheng, Y.; de Wilde, R.F.; Maitra, A.; Pan, D. The Hippo Signaling Pathway Restricts the Oncogenic Potential of an Intestinal Regeneration Program. Genes Dev. 2010, 24, 2383–2388. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.; Halder, G. The Two Faces of Hippo: Targeting the Hippo Pathway for Regenerative Medicine and Cancer Treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hallmark | Partner | Cancer Type | Reference |
|---|---|---|---|
| Sustaining proliferative signalling | NOTCH1 | Pancreatic | [18] |
| Sustaining proliferative signalling | STI1 | Glioblastoma | [21] |
| Resisting cell death | 37LRP | Gastric | [62] |
| Activating invasion and metastasis | STI1 | Colorectal | [87] |
| Activating invasion and metastasis | Filamin-A | Melanoma | [88] |
| Hallmark | Partner | Cancer Type | Reference |
|---|---|---|---|
| Sustaining proliferative signalling | PI3K/AKT- CyclinD1 | Gastric | [15] |
| Sustaining proliferative signalling | NOTCH1 | Pancreatic | [18] |
| Sustaining proliferative signalling | ILK | Colorectal | [26] |
| Evading growth suppressors | NF2 | Schwannoma | [24] |
| Evading growth suppressors | TGFβ | Colorectal | [26] |
| Resisting cell death | MDR1 | Gastric | [54] |
| Resisting cell death | PI3K/AKT | Gastric | [59] |
| Resisting cell death | BCL2 | Gastric | [42,54,58] |
| Resisting cell death | BCL2 | Breast | [51] |
| Resisting cell death | BCL2 | Glioma | [63] |
| Resisting cell death | Survivin/cIAP-1/XIAP | Colorectal | [25] |
| Resisting cell death | Par4 | Glioblastoma | [23] |
| Resisting cell death | Soluble PrPC | Breast | [66] |
| Inducing angiogenesis | Hypoxia | Colorectal | [31] |
| Activating invasion and metastasis | MMPs | Breast | [52,82] |
| Activating invasion and metastasis | MMPs | Gastric | [83] |
| Activating invasion and metastasis | NOTCH1 | Pancreatic | [18] |
| Activating invasion and metastasis | JNK | Lung | [84] |
| Reprogramming of energy metabolism | GLUT1 | Colorectal | [29] |
| Evading immune surveillance | TGFβ | Colorectal | [26] |
| Evading immune surveillance | IDO | Colorectal | [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mouillet-Richard, S.; Ghazi, A.; Laurent-Puig, P. The Cellular Prion Protein and the Hallmarks of Cancer. Cancers 2021, 13, 5032. https://doi.org/10.3390/cancers13195032
Mouillet-Richard S, Ghazi A, Laurent-Puig P. The Cellular Prion Protein and the Hallmarks of Cancer. Cancers. 2021; 13(19):5032. https://doi.org/10.3390/cancers13195032
Chicago/Turabian StyleMouillet-Richard, Sophie, Alexandre Ghazi, and Pierre Laurent-Puig. 2021. "The Cellular Prion Protein and the Hallmarks of Cancer" Cancers 13, no. 19: 5032. https://doi.org/10.3390/cancers13195032
APA StyleMouillet-Richard, S., Ghazi, A., & Laurent-Puig, P. (2021). The Cellular Prion Protein and the Hallmarks of Cancer. Cancers, 13(19), 5032. https://doi.org/10.3390/cancers13195032