Cancer and Stress: Does It Make a Difference to the Patient When These Two Challenges Collide?



Abstract
:Simple Summary
Abstract
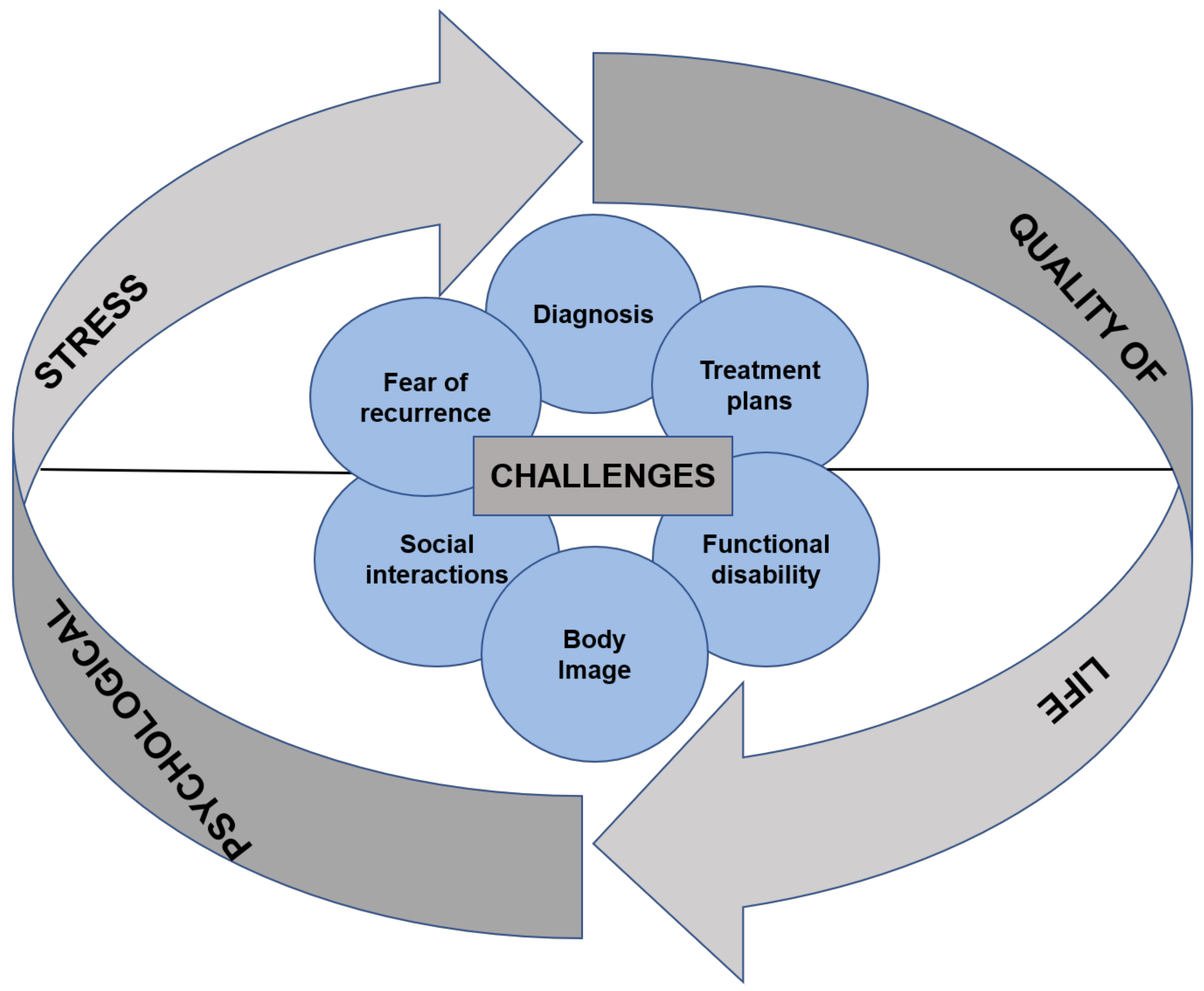
1. Introduction
2. Response to Stress
2.1. Adrenergic Signalling Pathway
2.2. Glucocorticoid Receptor Signalling
3. Stress Hormones (Glucocorticoids and Catecholamines) and Their Effect on the Biology of Cancer
3.1. Role of Stress Hormones in Cell Proliferation
3.2. Role of Stress Hormones in Angiogenesis
3.3. Role of Stress Hormones in Invasion and Migration
3.4. Role of Stress Hormones in Cell Survival
3.5. Role of Stress Hormones in DNA Damage
3.6. Role of Stress Hormones in Immunity
4. Stress Measurement
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gupta, B.; Johnson, N.W.; Kumar, N. Global epidemiology of head and neck cancers: A continuing challenge. Oncology 2016, 91, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Howren, M.B.; Christensen, A.J.; Karnell, L.H.; Funk, G.F. Psychological factors associated with head and neck cancer treatment and survivorship: Evidence and opportunities for behavioral medicine. J. Consult. Clin. Psychol. 2013, 81, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Vigneswaran, N.; Williams, M.D. Epidemiologic trends in head and neck cancer and aids in diagnosis. Oral Maxillofac. Surg. Clin. N. Am. 2014, 26, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Tshering Vogel, D.W.; Zbaeren, P.; Thoeny, H.C. Cancer of the oral cavity and oropharynx. Cancer Imaging 2010, 10, 62–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadakos, J.; McQuestion, M.; Gokhale, A.; Damji, A.; Trang, A.; Abdelmutti, N.; Ringash, J. Informational Needs of Head and Neck Cancer Patients. J. Cancer Educ. 2018, 33, 847–856. [Google Scholar] [CrossRef]
- Kar, A.; Asheem, M.R.; Bhaumik, U.; Rao, V.U.S. Psychological issues in head and neck cancer survivors: Need for addressal in rehabilitation. Oral Oncol. 2020, 110, 104859. [Google Scholar] [CrossRef]
- Ringash, J.; Bernstein, L.J.; Devins, G.; Dunphy, C.; Giuliani, M.; Martino, R.; McEwen, S. Head and Neck Cancer Survivorship: Learning the Needs, Meeting the Needs. Semin. Radiat. Oncol. 2018, 28, 64–74. [Google Scholar] [CrossRef]
- Humphris, G.M.; Ozakinci, G. Psychological responses and support needs of patients following head and neck cancer. Int. J. Surg. 2006, 4, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Bernabé, D.G.; Tamae, A.C.; Biasoli, É.R.; Oliveira, S.H.P. Stress hormones increase cell proliferation and regulates interleukin-6 secretion in human oral squamous cell carcinoma cells. Brain Behav. Immun. 2011, 25, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Li, C.; He, Y.; Griffin, R.; Ye, Q.; Li, L. Chronic stress promotes oral cancer growth and angiogenesis with increased circulating catecholamine and glucocorticoid levels in a mouse model. Oral Oncol. 2015, 51, 991–997. [Google Scholar] [CrossRef]
- Xie, H.; Li, B.; Li, L.; Zou, X.L.; Zhu, C.R.; Li, Y.; Gao, N.; Chen, Q.; Li, L. Association of increased circulating catecholamine and glucocorticoid levels with risk of psychological problems in oral neoplasm patients. PLoS ONE 2014, 9, e99179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo-Calderón, D.M.; Oliveira, D.T.; Marana, A.N.; Nonogaki, S.; Carvalho, A.L.; Kowalski, L.P. Prognostic significance of beta-2 adrenergic receptor in oral squamous cell carcinoma. Cancer Biomark. 2011, 10, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.J.; Liu, K.; Liang, D.F. Expression of β2-adrenergic receptor in oral squamous cell carcinoma. J. Oral Pathol. Med. 2009, 38, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Celentano, A.; McCullough, M.; Cirillo, N. Glucocorticoids reduce chemotherapeutic effectiveness on OSCC cells via glucose-dependent mechanisms. J. Cell. Physiol. 2019, 234, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- De Sena, L.S.B.; da Silveira, E.J.D.; Batista, A.C.; Mendonca, E.F.; Alves, P.M.; Nonaka, C.F.W. Immunoexpression of glucocorticoid receptor alpha (GRalpha) isoform and apoptotic proteins (Bcl-2 and Bax) in actinic cheilitis and lower lip squamous cell carcinoma. J. Oral Pathol. Med. 2018, 47, 788–795. [Google Scholar] [CrossRef]
- Bastos, D.B.; Sarafim-Silva, B.A.M.; Sundefeld, M.; Ribeiro, A.A.; Brandao, J.D.P.; Biasoli, E.R.; Miyahara, G.I.; Casarini, D.E.; Bernabe, D.G. Circulating catecholamines are associated with biobehavioral factors and anxiety symptoms in head and neck cancer patients. PLoS ONE 2018, 13, e0202515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirillo, N.; Hassona, Y.; Pignatelli, M.; Gasparoto, T.H.; Morgan, D.J.; Prime, S.S. Characterization of a novel oral glucocorticoid system and its possible role in disease. J. Dent. Res. 2012, 91, 97–103. [Google Scholar] [CrossRef]
- Bernabé, D.G.; Tamae, A.C.; Miyahara, G.I.; Sundefeld, M.L.M.; Oliveira, S.P.; Biasoli, É.R. Increased plasma and salivary cortisol levels in patients with oral cancer and their association with clinical stage. J. Clin. Pathol. 2012, 65, 934–939. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, C.; Chen, W.; Qiu, L.; Li, S.; Wang, T.; Xie, H.; Li, Y.; Li, C.; Li, L. The stress hormone norepinephrine promotes tumor progression through beta2-adrenoreceptors in oral cancer. Arch. Oral Biol. 2020, 113, 104712. [Google Scholar] [CrossRef]
- Valente, V.B.; Verza, F.A.; Lopes, F.Y.K.; Ferreira, J.Z.; Dos Santos, P.S.P.; Sundefeld, M.; Biasoli, E.R.; Miyahara, G.I.; Soubhia, A.M.P.; de Andrade, M.; et al. Stress hormones concentrations in the normal microenvironment predict risk for chemically induced cancer in rats. Psychoneuroendocrinology 2018, 89, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, C.; Xie, N.; Zhuang, Z.; Liu, X.; Hou, J.; Huang, H. Activation of adrenergic receptor β2 promotes tumor progression and epithelial mesenchymal transition in tongue squamous cell carcinoma. Int. J. Mol. Med. 2017, 41, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Shands, H.C.; Finesinger, J.E.; Cobb, S.; Abrams, R.D. Psychological mechanisms in patients with cancer. Cancer 1951, 4, 1159–1170. [Google Scholar] [CrossRef]
- Hinton, J. Bearing cancer. Br. J. Med Psychol. 1973, 46, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.; Lund, V.J.; Howard, D.J.; Greenberg, M.P.; McCarthy, M. Quality of life of patients treated surgically for head and neck cancer. J. Laryngol. Otol. 1992, 106, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxford Cancer Intelligence Unit. Major Surgical Resections in England: Head and Neck Cancers; Oxford Cancer Intelligence Unit: Oxford, UK, 2011. [Google Scholar]
- Katz, M.R.; Irish, J.C.; Devins, G.M.; Rodin, G.M.; Gullane, P.J. Reliability and validity of an observer-rated disfigurement scale for head and neck cancer patients. Head Neck 2000, 22, 132–141. [Google Scholar] [CrossRef]
- Rumsey, N.; Clarke, A.; White, P.; Wyn-Williams, M.; Garlick, W. Altered body image: Appearance-related concerns of people with visible disfigurement. J. Adv. Nurs. 2004, 48, 443–453. [Google Scholar] [CrossRef]
- Fingeret, M.C.; Yuan, Y.; Urbauer, D.; Weston, J.; Nipomnick, S.; Weber, R. The nature and extent of body image concerns among surgically treated patients with head and neck cancer. Psycho-Oncology 2012, 21, 836–844. [Google Scholar] [CrossRef] [Green Version]
- Dropkin, M.J. Coping with disfigurement and dysfunction after head and neck cancer surgery: A conceptual framework. Semin. Oncol. Nurs. 1989, 5, 213–219. [Google Scholar] [CrossRef]
- Suzuki, M.; Deno, M.; Myers, M.; Asakage, T.; Takahashi, K.; Saito, K.; Mori, Y.; Saito, H.; Ichikawa, Y.; Yamamoto-Mitani, N.; et al. Anxiety and depression in patients after surgery for head and neck cancer in Japan. Palliat. Support. Care 2016, 14, 269–277. [Google Scholar] [CrossRef]
- List, M.A.; Siston, A.; Haraf, D.; Schumm, P.; Kies, M.; Stenson, K.; Vokes, E.E. Quality of life and performance in advanced head and neck cancer patients on concomitant chemoradiotherapy: A prospective examination. J. Clin. Oncol. 1999, 17, 1020–1028. [Google Scholar] [CrossRef]
- Clarke, A. Psychosocial aspects of facial disfigurement: Problems, management and the role of a lay-led organization. Psychol. Health Med. 1999, 4, 127–142. [Google Scholar] [CrossRef]
- Doss, J.G.; Ghani, W.M.N.; Razak, I.A.; Yang, Y.H.; Rogers, S.N.; Zain, R.B. Changes in health-related quality of life of oral cancer patients treated with curative intent: Experience of a developing country. Int. J. Oral Maxillofac. Surg. 2017, 46, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Kent, G. Understanding the experiences of people with disfigurements: An integration of four models of social and psychological functioning. Psychol. Health Med. 2000, 5, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Kish, V.; Lansdown, R. Meeting the Psychosocial Impact of Facial Disfigurement: Developing a Clinical Service for Children and Families. Clin. Child Psychol. Psychiatry 2000, 5, 497–512. [Google Scholar] [CrossRef]
- Wu, Y.S.; Lin, P.Y.; Chien, C.Y.; Fang, F.M.; Chiu, N.M.; Hung, C.F.; Lee, Y.; Chong, M.Y. Anxiety and depression in patients with head and neck cancer: 6-month follow-up study. Neuropsychiatr. Dis. Treat. 2016, 12, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Costa, E.F.; Nogueira, T.E.; De Souza Lima, N.C.; Mendonça, E.F.; Leles, C.R. A qualitative study of the dimensions of patients’ perceptions of facial disfigurement after head and neck cancer surgery. Spec. Care Dent. 2014, 34, 114–121. [Google Scholar] [CrossRef]
- Henry, M.; Ho, A.; Lambert, S.D.; Carnevale, F.A.; Greenfield, B.; MacDonald, C.; Mlynarek, A.; Zeitouni, A.; Rosberger, Z.; Hier, M.; et al. Looking beyond disfigurement: The experience of patients with head and neck cancer. J. Palliat. Care 2014, 30, 5–15. [Google Scholar] [CrossRef]
- List, M.A.; D’Antonio, L.L.; Cella, D.F.; Siston, A.; Mumby, P.; Haraf, D.; Vokes, E. The Performance Status Scale for Head and Neck Cancer Patients and the Functional Assessment of Cancer Therapy-Head and Neck Scale. A study of utility and validity. Cancer 1996, 77, 2294–2301. [Google Scholar] [CrossRef]
- Ryan, S.; Oaten, M.; Stevenson, R.J.; Case, T.I. Facial disfigurement is treated like an infectious disease. Evol. Hum. Behav. 2012, 33, 639–646. [Google Scholar] [CrossRef]
- Murphy, B.A.; Ridner, S.; Wells, N.; Dietrich, M. Quality of life research in head and neck cancer: A review of the current state of the science. Crit. Rev. Oncol. Hematol. 2007, 62, 251–267. [Google Scholar] [CrossRef]
- Davidson, A.; Williams, J. Factors affecting quality of life in patients experiencing facial disfigurement due to surgery for head and neck cancer. Br. J. Nurs. 2019, 28, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Djan, R.; Penington, A. A systematic review of questionnaires to measure the impact of appearance on quality of life for head and neck cancer patients. J. Plast. Reconstr. Aesthet. Surg. 2013, 66, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.A.; Newell, R.; Thompson, A.; Harcourt, D.; Lindenmeyer, A. Appearance concerns and psychosocial adjustment following head and neck cancer: A cross-sectional study and nine-month follow-up. Psychol. Health Med. 2014, 19, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Flexen, J.; Ghazali, N.; Lowe, D.; Rogers, S.N. Identifying appearance-related concerns in routine follow-up clinics following treatment for oral and oropharyngeal cancer. Br. J. Oral Maxillofac. Surg. 2012, 50, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Lu, M.H.; Kuo, P.L.; Chen, Y.W.; Lee, S.C.; Liang, S.Y. Influences of facial disfigurement and social support for psychosocial adjustment among patients with oral cancer in Taiwan: A cross-sectional study. BMJ Open 2018, 8, 23670. [Google Scholar] [CrossRef]
- Nogueira, T.E.; Adorno, M.; Mendonça, E.; Leles, C. Factors associated with the quality of life of subjects with facial disfigurement due to surgical treatment of head and neck cancer. Med. Oral Patol. Oral Cir. Bucal 2018, 23, e132–e137. [Google Scholar] [CrossRef]
- Hagedoorn, M.; Molleman, E. Facial disfigurement in patients with head and neck cancer: The role of social self-efficacy. Health Psychol. 2006, 25, 643–647. [Google Scholar] [CrossRef] [Green Version]
- Lebel, S.; Ozakinci, G.; Humphris, G.; Mutsaers, B.; Thewes, B.; Prins, J.; Dinkel, A.; Butow, P. From normal response to clinical problem: Definition and clinical features of fear of cancer recurrence. Support. Care Cancer 2016, 24, 3265–3268. [Google Scholar] [CrossRef] [Green Version]
- Simard, S.; Savard, J.; Ivers, H. Fear of cancer recurrence: Specific profiles and nature of intrusive thoughts. J. Cancer Surviv. 2010, 4, 361–371. [Google Scholar] [CrossRef]
- Simard, S.; Thewes, B.; Humphris, G.; Dixon, M.; Hayden, C.; Mireskandari, S.; Ozakinci, G. Fear of cancer recurrence in adult cancer survivors: A systematic review of quantitative studies. J. Cancer Surviv. 2013, 7, 300–322. [Google Scholar] [CrossRef]
- O’Neill, M.P. Psychological aspects of cancer recovery. Cancer 1975, 36, 271–273. [Google Scholar] [CrossRef]
- Baker, F.; Denniston, M.; Smith, T.; West, M.M. Adult cancer survivors: How are they faring? Cancer 2005, 104, 2565–2576. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.N.; Scott, B.; Lowe, D.; Ozakinci, G.; Humphris, G.M. Fear of recurrence following head and neck cancer in the outpatient clinic. Eur. Arch. Oto-Rhino-Laryngol. 2010, 267, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- Hodges, L.J.; Humphris, G.M. Fear of recurrence and psychological distress in head and neck cancer patients and their carers. Psycho-Oncology 2009, 18, 841–848. [Google Scholar] [CrossRef]
- Simard, S.; Savard, J. Fear of Cancer Recurrence Inventory: Development and initial validation of a multidimensional measure of fear of cancer recurrence. Support. Care Cancer 2009, 17, 241–251. [Google Scholar] [CrossRef]
- Gill, K.M.; Mishel, M.; Belyea, M.; Germino, B.; Germino, L.S.; Porter, L.; LaNey, I.C.; Stewart, J. Triggers of uncertainty about recurrence and long-term treatment side effects in older African American and Caucasian breast cancer survivors. Oncol. Nurs. Forum 2004, 31, 633–639. [Google Scholar] [CrossRef]
- Hall, D.L.; Lennes, I.T.; Pirl, W.F.; Friedman, E.R.; Park, E.R. Fear of recurrence or progression as a link between somatic symptoms and perceived stress among cancer survivors. Support. Care Cancer 2017, 25, 1401–1407. [Google Scholar] [CrossRef] [Green Version]
- Tsay, S.L.; Wang, J.Y.; Lee, Y.H.; Chen, Y.J. Fear of recurrence: A mediator of the relationship between physical symptoms and quality of life in head and neck cancer patients. Eur. J. Cancer Care 2020, 29, e13243. [Google Scholar] [CrossRef]
- Llewellyn, C.D.; Weinman, J.; McGurk, M.; Humphris, G. Can we predict which head and neck cancer survivors develop fears of recurrence? J. Psychosom. Res. 2008, 65, 525–532. [Google Scholar] [CrossRef]
- Savard, J.; Ivers, H. The evolution of fear of cancer recurrence during the cancer care trajectory and its relationship with cancer characteristics. J. Psychosom. Res. 2013, 74, 354–360. [Google Scholar] [CrossRef]
- Mirosevic, S.; Thewes, B.; van Herpen, C.; Kaanders, J.; Merkx, T.; Humphris, G.; Baatenburg de Jong, R.J.; Langendijk, J.A.; Leemans, C.R.; Terhaard, C.H.J.; et al. Prevalence and clinical and psychological correlates of high fear of cancer recurrence in patients newly diagnosed with head and neck cancer. Head Neck 2019, 41, 3187–3200. [Google Scholar] [CrossRef] [PubMed]
- Semple, C.; Parahoo, K.; Norman, A.; McCaughan, E.; Humphris, G.; Mills, M. Psychosocial interventions for patients with head and neck cancer. Cochrane Database Syst. Rev. 2013, 2013, CD009441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaplin, J.M.; Morton, R.P. A prospective, longitudinal study of pain in head and neck cancer patients. Head Neck 1999, 21, 531–537. [Google Scholar] [CrossRef]
- Gellrich, N.C.; Schimming, R.; Schramm, A.; Schmalohr, D.; Bremerich, A.; Kugler, J. Pain, function, and psychologic outcome before, during, and after intraoral tumor resection. J. Oral Maxillofac. Surg. 2002, 60, 772–777. [Google Scholar] [CrossRef]
- Potter, J.; Higginson, I.J.; Scadding, J.W.; Quigley, C. Identifying neuropathic pain in patients with head and neck cancer: Use of the Leeds Assessment of Neuropathic Symptoms and Signs Scale. J. R. Soc. Med. 2003, 96, 379–383. [Google Scholar] [CrossRef]
- Ribeiro, K.d.C.B.; Kowalski, L.P.; Latorre, M.d.R.D.D.O. Perioperative complications, comorbidities, and survival in oral or oropharyngeal cancer. Arch. Otolaryngol.Head Neck Surg. 2003, 129, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Chua, K.S.G.; Reddy, S.K.; Lee, M.C.; Patt, R.B. Pain and loss of function in head and neck cancer survivors. J. Pain Symptom Manag. 1999, 18, 193–202. [Google Scholar] [CrossRef]
- Breivik, H.; Cherny, N.; Collett, B.; de Conno, F.; Filbet, M.; Foubert, A.J.; Cohen, R.; Dow, L. Cancer-related pain: A pan-European survey of prevalence, treatment, and patient attitudes. Ann. Oncol. 2009, 20, 1420–1433. [Google Scholar] [CrossRef]
- Gellrich, N.C.; Schramm, A.; Buckmann, R.; Kugler, J. Follow-up in patients with oral cancer. J. Oral Maxillofac. Surg. 2002, 60, 380–386. [Google Scholar] [CrossRef]
- Van den Beuken-van Everdingen, M.H.; de Rijke, J.M.; Kessels, A.G.; Schouten, H.C.; van Kleef, M.; Patijn, J. Prevalence of pain in patients with cancer: A systematic review of the past 40 years. Ann. Oncol. 2007, 18, 1437–1449. [Google Scholar] [CrossRef]
- Sato, J.; Yamazaki, Y.; Satoh, A.; Notani, K.i.; Kitagawa, Y. Pain is associated with an endophytic cancer growth pattern in patients with oral squamous cell carcinoma before treatment. Odontology 2010, 98, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Black, B.; Herr, K.; Fine, P.; Sanders, S.; Tang, X.; Bergen-Jackson, K.; Titler, M.; Forcucci, C. The Relationships among Pain, Nonpain Symptoms, and Quality of Life Measures in Older Adults with Cancer Receiving Hospice Care. Pain Med. 2011, 12, 880–889. [Google Scholar] [CrossRef]
- Cuffari, L.; Tesseroli de Siqueira, J.T.; Nemr, K.; Rapaport, A. Pain complaint as the first symptom of oral cancer: A descriptive study. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2006, 102, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, A.K.; Roy, S.; Thakar, A.; Sharma, A.; Mohanti, B.K. Symptom burden and quality of life in advanced head and neck cancer patients: AIIMS study of 100 patients. Indian J. Palliat. Care 2014, 20, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Infante-Cossio, P.; Torres-Carranza, E.; Cayuela, A.; Gutierrez-Perez, J.L.; Gili-Miner, M. Quality of life in patients with oral and oropharyngeal cancer. Int. J. Oral Maxillofac. Surg. 2009, 38, 250–255. [Google Scholar] [CrossRef]
- Weymuller, E.A.; Yueh, B.; Deleyiannis, F.W.; Kuntz, A.L.; Alsarraf, R.; Coltrera, M.D. Quality of life in patients with head and neck cancer: Lessons learned from 549 prospectively evaluated patients. Arch. Otolaryngol.-Head Neck Surg. 2000, 126, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Morris, N.; Moghaddam, N.; Tickle, A.; Biswas, S. The relationship between coping style and psychological distress in people with head and neck cancer: A systematic review. Psychooncology 2018, 27, 734–747. [Google Scholar] [CrossRef]
- Zimmaro, L.A.; Sephton, S.E.; Siwik, C.J.; Phillips, K.M.; Rebholz, W.N.; Kraemer, H.C.; Giese-Davis, J.; Wilson, L.; Bumpous, J.M.; Cash, E.D. Depressive symptoms predict head and neck cancer survival: Examining plausible behavioral and biological pathways. Cancer 2018, 124, 1053–1060. [Google Scholar] [CrossRef]
- Vichaya, E.G.; Vermeer, D.W.; Christian, D.L.; Molkentine, J.M.; Mason, K.A.; Lee, J.H.; Dantzer, R. Neuroimmune mechanisms of behavioral alterations in a syngeneic murine model of human papilloma virus-related head and neck cancer. Psychoneuroendocrinology 2017, 79, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Chida, Y.; Hamer, M.; Wardle, J.; Steptoe, A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat. Clin. Pract. Oncol. 2008, 5, 466–475. [Google Scholar] [CrossRef]
- Cohen, L.; Cole, S.W.; Sood, A.K.; Prinsloo, S.; Kirschbaum, C.; Arevalo, J.M.; Jennings, N.B.; Scott, S.; Vence, L.; Wei, Q.; et al. Depressive symptoms and cortisol rhythmicity predict survival in patients with renal cell carcinoma: Role of inflammatory signaling. PLoS ONE 2012, 7, e42324. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, E.S.; Sood, A.K.; Lutgendorf, S.K. Biobehavioral influences on cancer progression. Immunol. Allergy Clin. N. Am. 2011, 31, 109–132. [Google Scholar] [CrossRef] [Green Version]
- Schneiderman, N.; Ironson, G.; Siegel, S.D. Stress and health: Psychological, behavioral, and biological determinants. Annu. Rev. Clin. Psychol. 2005, 1, 607–628. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoni, M.H.; Lutgendorf, S.K.; Cole, S.W.; Dhabhar, F.S.; Sephton, S.E.; McDonald, P.G.; Stefanek, M.; Sood, A.K. The influence of bio-behavioural factors on tumour biology: Pathways and mechanisms. Nat. Rev. Cancer 2006, 6, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Colon-Echevarria, C.B.; Lamboy-Caraballo, R.; Aquino-Acevedo, A.N.; Armaiz-Pena, G.N. Neuroendocrine Regulation of Tumor-Associated Immune Cells. Front. Oncol. 2019, 9, 1077. [Google Scholar] [CrossRef]
- Reiche, E.M.; Nunes, S.O.; Morimoto, H.K. Stress, depression, the immune system, and cancer. Lancet Oncol. 2004, 5, 617–625. [Google Scholar] [CrossRef]
- Glaser, R.; Kiecolt-Glaser, J.K. Stress-induced immune dysfunction: Implications for health. Nat. Rev. Immunol. 2005, 5, 243–251. [Google Scholar] [CrossRef]
- Lutgendorf, S.K.; Costanzo, E.; Siegel, S. Psychosocial influences in oncology: An expanded model of biobehavioral mechanisms. In Psychoneuroimmunology, 4th ed.; Ader, R., Glaser, R., Cohen, N., Irwin, M., Eds.; Academic Press: New York, NY, USA, 2007; pp. 869–895. [Google Scholar]
- Schuller, H.M.; Al-Wadei, H.A. Beta-adrenergic signaling in the development and progression of pulmonary and pancreatic adenocarcinoma. Curr. Cancer Ther. Rev. 2012, 8, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Daly, C.J.; McGrath, J.C. Previously unsuspected widespread cellular and tissue distribution of beta-adrenoceptors and its relevance to drug action. Trends Pharmacol. Sci. 2011, 32, 219–226. [Google Scholar] [CrossRef]
- Eng, J.W.; Kokolus, K.M.; Reed, C.B.; Hylander, B.L.; Ma, W.W.; Repasky, E.A. A nervous tumor microenvironment: The impact of adrenergic stress on cancer cells, immunosuppression, and immunotherapeutic response. Cancer Immunol. Immunother. 2014, 63, 1115–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rooij, J.; Zwartkruis, F.J.T.; Verheijen, M.H.G.; Cool, R.H.; Nijman, S.M.B.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.B.; Le, X.; Heymach, J.V. β-Adrenergic Signaling in Lung Cancer: A Potential Role for Beta-Blockers. J. Neuroimmune Pharmacol. 2020, 15, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M.; Cole, B. Regulation of cell proliferation by beta-adrenergic receptors in a human lung adenocarcinoma cell line. Carcinogenesis 1989, 10, 1753–1755. [Google Scholar] [CrossRef]
- Zhang, X.; Odom, D.T.; Koo, S.H.; Conkright, M.D.; Canettieri, G.; Best, J.; Chen, H.; Jenner, R.; Herbolsheimer, E.; Jacobsen, E.; et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 4459–4464. [Google Scholar] [CrossRef] [Green Version]
- Luttrell, L.M.; Ferguson, S.S.G.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.T.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. β-arrestin-dependent formation of β2 adrenergic receptor-src protein kinase complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef]
- Li, Y.; Yang, S.; Sadaoui, N.C.; Hu, W.; Dasari, S.K.; Mangala, L.S.; Sun, Y.; Zhao, S.; Wang, L.; Liu, Y.; et al. Sustained Adrenergic Activation of YAP1 Induces Anoikis Resistance in Cervical Cancer Cells. iScience 2020, 23, 101289. [Google Scholar] [CrossRef]
- Vegiopoulos, A.; Herzig, S. Glucocorticoids, metabolism and metabolic diseases. Mol. Cell. Endocrinol. 2007, 275, 43–61. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Topete, D.; Cidlowski, J.A. One hormone, two actions: Anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation 2014, 22, 20–32. [Google Scholar] [CrossRef]
- Donatti, T.L.; Koch, V.H.; Takayama, L.; Pereira, R.M. Effects of glucocorticoids on growth and bone mineralization. J. Pediatr. 2011, 87, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Joëls, M. Impact of glucocorticoids on brain function: Relevance for mood disorders. Psychoneuroendocrinology 2011, 36, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Holsboer, F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 2000, 23, 477–501. [Google Scholar] [CrossRef] [Green Version]
- Selye, H. Pharmacological Classification of Steroid Hormones. Nature 1941, 148, 84–85. [Google Scholar] [CrossRef]
- Burns, C.M. The History of Cortisone Discovery and Development. Rheum. Dis. Clin. N. Am. 2016, 42, 1–14. [Google Scholar] [CrossRef]
- Whitfield, G.K.; Jurutka, P.W.; Haussler, C.A.; Haussler, M.R. Steroid hormone receptors: Evolution, ligands, and molecular basis of biologic function. J. Cell. Biochem. 1999, 75, 110–122. [Google Scholar] [CrossRef]
- Petta, I.; Dejager, L.; Ballegeer, M.; Lievens, S.; Tavernier, J.; De Bosscher, K.; Libert, C. The Interactome of the Glucocorticoid Receptor and Its Influence on the Actions of Glucocorticoids in Combatting Inflammatory and Infectious Diseases. Microbiol. Mol. Biol. Rev. 2016, 80, 495–522. [Google Scholar] [CrossRef] [Green Version]
- Scheschowitsch, K.; Leite, J.A.; Assreuy, J. New Insights in Glucocorticoid Receptor Signaling-More Than Just a Ligand-Binding Receptor. Front. Endocrinol. 2017, 8, 16. [Google Scholar] [CrossRef]
- Kino, T.; Su, Y.A.; Chrousos, G.P. Human glucocorticoid receptor isoform beta: Recent understanding of its potential implications in physiology and pathophysiology. Cell. Mol. Life Sci. 2009, 66, 3435–3448. [Google Scholar] [CrossRef] [Green Version]
- Chrousos, G.P.; Kino, T. Intracellular glucocorticoid signaling: A formerly simple system turns stochastic. Sci. STKE 2005, 2005, pe48. [Google Scholar] [CrossRef]
- Yudt, M.R.; Jewell, C.M.; Bienstock, R.J.; Cidlowski, J.A. Molecular origins for the dominant negative function of human glucocorticoid receptor beta. Mol. Cell. Biol. 2003, 23, 4319–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieken, E.S.; Miesfeld, R.L. Transcriptional transactivation functions localized to the glucocorticoid receptor N terminus are necessary for steroid induction of lymphocyte apoptosis. Mol. Cell. Biol. 1992, 12, 589–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almlöf, T.; Wallberg, A.E.; Gustafsson, J.Å.; Wright, A.P.H. Role of important hydrophobic amino acids in the interaction between the glucocorticoid receptor τ1-core activation domain and target factors. Biochemistry 1998, 37, 9586–9594. [Google Scholar] [CrossRef] [PubMed]
- Howard, K.J.; Holley, S.J.; Yamamoto, K.R.; Distelhorst, C.W. Mapping the HSP90 binding region of the glucocorticoid receptor. J. Biol. Chem. 1990, 265, 11928–11935. [Google Scholar] [PubMed]
- Schüle, R.; Rangarajan, P.; Kliewer, S.; Ransone, L.J.; Bolado, J.; Yang, N.; Verma, I.M.; Evans, R.M. Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell 1990, 62, 1217–1226. [Google Scholar] [CrossRef]
- Kumar, R.; Thompson, E.B. Gene regulation by the glucocorticoid receptor: Structure:function relationship. J. Steroid Biochem. Mol. Biol. 2005, 94, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Duma, D.; Jewell, C.M.; Cidlowski, J.A. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J. Steroid Biochem. Mol. Biol. 2006, 102, 11–21. [Google Scholar] [CrossRef]
- Vandevyver, S.; Dejager, L.; Libert, C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr. Rev. 2014, 35, 671–693. [Google Scholar] [CrossRef] [Green Version]
- Bledsoe, R.K.; Montana, V.G.; Stanley, T.B.; Delves, C.J.; Apolito, C.J.; McKee, D.D.; Consler, T.G.; Parks, D.J.; Stewart, E.L.; Willson, T.M.; et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 2002, 110, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Pratt, W.B.; Galigniana, M.D.; Morishima, Y.; Murphy, P.J. Role of molecular chaperones in steroid receptor action. Essays Biochem. 2004, 40, 41–58. [Google Scholar] [CrossRef]
- Pratt, W.B.; Silverstein, A.M.; Galigniana, M.D. A Model for the Cytoplasmic Trafficking of Signalling Proteins Involving the hsp90-Binding Immunophilins and p50cdc37. Cell. Signal. 1999, 11, 839–851. [Google Scholar] [CrossRef]
- Liberman, A.C.; Antunica-Noguerol, M.; Arzt, E. Modulation of the Glucocorticoid Receptor Activity by Post-Translational Modifications. Nucl. Recept. Res. 2014, 1, 1–15. [Google Scholar] [CrossRef]
- Shaheen, M.; Schindler, L.; Saar-Ashkenazy, R.; Bani Odeh, K.; Soreq, H.; Friedman, A.; Kirschbaum, C. Victims of war-Psychoendocrine evidence for the impact of traumatic stress on psychological well-being of adolescents growing up during the Israeli-Palestinian conflict. Psychophysiology 2020, 57, e13271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandevyver, S.; Dejager, L.; Libert, C. On the trail of the glucocorticoid receptor: Into the nucleus and back. Traffic 2012, 13, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Mitre-Aguilar, I.B.; Cabrera-Quintero, A.J.; Zentella-Dehesa, A. Genomic and non-genomic effects of glucocorticoids: Implications for breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 1–10. [Google Scholar]
- Vilasco, M.; Communal, L.; Mourra, N.; Courtin, A.; Forgez, P.; Gompel, A. Glucocorticoid receptor and breast cancer. Breast Cancer Res. Treat. 2011, 130, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Perez Kerkvliet, C.; Dwyer, A.R.; Diep, C.H.; Oakley, R.H.; Liddle, C.; Cidlowski, J.A.; Lange, C.A. Glucocorticoid receptors are required effectors of TGFbeta1-induced p38 MAPK signaling to advanced cancer phenotypes in triple-negative breast cancer. Breast Cancer Res. 2020, 22, 39. [Google Scholar] [CrossRef]
- Barnes, P.J. Anti-inflammatory actions of glucocorticoids: Molecular mechanisms. Clin. Sci. 1998, 94, 557–572. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Cidlowski, J.A. The human glucocorticoid receptor: One gene, multiple proteins and diverse responses. Steroids 2005, 70, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Stellato, C. Post-transcriptional and nongenomic effects of glucocorticoids. Proc. Am. Thorac. Soc. 2004, 1, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Losel, R.M.; Falkenstein, E.; Feuring, M.; Schultz, A.; Tillmann, H.C.; Rossol-Haseroth, K.; Wehling, M. Nongenomic steroid action: Controversies, questions, and answers. Physiol. Rev. 2003, 83, 965–1016. [Google Scholar] [CrossRef] [PubMed]
- Ordonez-Moran, P.; Munoz, A. Nuclear receptors: Genomic and non-genomic effects converge. Cell Cycle 2009, 8, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Leis, H.; Page, A.; Ramírez, A.; Bravo, A.; Segrelles, C.; Paramio, J.; Barettino, D.; Jorcano, J.L.; Pérez, P. Glucocorticoid Receptor Counteracts Tumorigenic Activity of Akt in Skin through Interference with the Phosphatidylinositol 3-Kinase Signaling Pathway. Mol. Endocrinol. 2004, 18, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafezi-Moghadam, A.; Simoncini, T.; Yang, Z.; Limbourg, F.P.; Plumier, J.C.; Rebsamen, M.C.; Hsieh, C.M.; Chui, D.S.; Thomas, K.L.; Prorock, A.J.; et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat. Med. 2002, 8, 473–479. [Google Scholar] [CrossRef]
- Molinolo, A.A.; Amornphimoltham, P.; Squarize, C.H.; Castilho, R.M.; Patel, V.; Gutkind, J.S. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009, 45, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical update on head and neck cancer: Molecular biology and ongoing challenges. Cell Death Dis. 2019, 10, 540. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Ortega, S.; Malumbres, M.; Barbacid, M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta 2002, 1602, 73–87. [Google Scholar] [CrossRef]
- Moreno-Smith, M.; Lutgendorf, S.K.; Sood, A.K. Impact of stress on cancer metastasis. Future Oncol. 2010, 6, 1863–1881. [Google Scholar] [CrossRef] [Green Version]
- Heffner, K.L.; Loving, T.J.; Robles, T.F.; Kiecolt-Glaser, J.K. Examining psychosocial factors related to cancer incidence and progression: In search of the silver lining. Brain Behav. Immun. 2003, 17 (Suppl. 1), S109–S111. [Google Scholar] [CrossRef]
- Gao, Y.; Yuan, L.; Pan, B.; Wang, L. Resilience and associated factors among Chinese patients diagnosed with oral cancer. BMC Cancer 2019, 19, 447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutgendorf, S.K.; DeGeest, K.; Dahmoush, L.; Farley, D.; Penedo, F.; Bender, D.; Goodheart, M.; Buekers, T.E.; Mendez, L.; Krueger, G.; et al. Social isolation is associated with elevated tumor norepinephrine in ovarian carcinoma patients. Brain Behav. Immun. 2011, 25, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaker, P.H.; Han, L.Y.; Kamat, A.A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; Jennings, N.B.; Armaiz-Pena, G.; Bankson, J.A.; Ravoori, M.; et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 2006, 12, 939–944. [Google Scholar] [CrossRef]
- Shin, V.Y.; Wu, W.K.; Chu, K.M.; Koo, M.W.; Wong, H.P.; Lam, E.K.; Tai, E.K.; Cho, C.H. Functional role of beta-adrenergic receptors in the mitogenic action of nicotine on gastric cancer cells. Toxicol. Sci. 2007, 96, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.P.; Yu, L.; Lam, E.K.; Tai, E.K.; Wu, W.K.; Cho, C.H. Nicotine promotes colon tumor growth and angiogenesis through beta-adrenergic activation. Toxicol. Sci. 2007, 97, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Re, G.; Badino, P.; Novelli, A.; Girardi, C.; Di Carlo, F. Evidence for functional β-adrenoceptor subtypes in CG-5 breast cancer cells. Pharmacol. Res. 1996, 33, 255–260. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; He, Z.; Yin, K.; Li, B.; Zhang, L.; Xu, Z. Chronic stress promotes gastric cancer progression and metastasis: An essential role for ADRB2. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Obeid, E.I.; Conzen, S.D. The role of adrenergic signaling in breast cancer biology. Cancer Biomark. 2013, 13, 161–169. [Google Scholar] [CrossRef]
- Montoya, A.; Amaya, C.N.; Belmont, A.; Diab, N.; Trevino, R.; Villanueva, G.; Rains, S.; Sanchez, L.A.; Badri, N.; Otoukesh, S.; et al. Use of non-selective β-blockers is associated with decreased tumor proliferative indices in early stage breast cancer. Oncotarget 2017, 8, 6446–6460. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.P.S.; Ho, J.W.C.; Koo, M.W.L.; Yu, L.; Wu, W.K.K.; Lam, E.K.Y.; Tai, E.K.K.; Ko, J.K.S.; Shin, V.Y.; Chu, K.M.; et al. Effects of adrenaline in human colon adenocarcinoma HT-29 cells. Life Sci. 2011, 88, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Wang, F.; Yang, R.; Zheng, X.; Gao, H.; Zhang, P. Effect of chronic restraint stress on human colorectal carcinoma growth in mice. PLoS ONE 2013, 8, e61435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wu, W.K.K.; Yu, L.; Sung, J.J.Y.; Srivastava, G.; Zhang, S.T.; Cho, C.H. Epinephrine stimulates esophageal squamous-cell carcinoma cell proliferation via β-adrenoceptor-dependent transactivation of extracellular signal-regulated kinase/cyclooxygenase-2 pathway. J. Cell. Biochem. 2008, 105, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Dal Monte, M.; Fornaciari, I.; Nicchia, G.P.; Svelto, M.; Casini, G.; Bagnoli, P. β3-adrenergic receptor activity modulates melanoma cell proliferation and survival through nitric oxide signaling. Naunyn Schmiedebergs Arch. Pharmacol. 2014, 387, 533–543. [Google Scholar] [CrossRef]
- Carie, A.E.; Sebti, S.M. A chemical biology approach identifies a beta-2 adrenergic receptor agonist that causes human tumor regression by blocking the Raf-1/Mek-1/Erk1/2 pathway. Oncogene 2007, 26, 3777–3788. [Google Scholar] [CrossRef] [Green Version]
- Yap, A.; Lopez-Olivo, M.A.; Dubowitz, J.; Pratt, G.; Hiller, J.; Gottumukkala, V.; Sloan, E.; Riedel, B.; Schier, R. Effect of beta-blockers on cancer recurrence and survival: A meta-analysis of epidemiological and perioperative studies. Br. J. Anaesth. 2018, 121, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Thaker, P.H.; Sood, A.K. Neuroendocrine influences on cancer biology. Semin. Cancer Biol. 2008, 18, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Simon, W.E.; Albrecht, M.; Trams, G.; Dietel, M.; Hölzel, F. In vitro growth promotion of human mammary carcinoma cells by steroid hormones, tamoxifen, and prolactin. J. Natl. Cancer Inst. 1984, 73, 313–321. [Google Scholar] [CrossRef]
- Buxant, F.; Kindt, N.; Laurent, G.; Noël, J.C.; Saussez, S. Antiproliferative effect of dexamethasone in the MCF-7 breast cancer cell line. Mol. Med. Rep. 2015, 12, 4051–4054. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Song, L.; Wang, Z. Effects of Dexamethasone on glucocorticoid receptor expression in a human ovarian carcinoma cell line 3AO. Chin. Med. J. 2003, 116, 392–395. [Google Scholar]
- Gundisch, S.; Boeckeler, E.; Behrends, U.; Amtmann, E.; Ehrhardt, H.; Jeremias, I. Glucocorticoids augment survival and proliferation of tumor cells. Anticancer Res. 2012, 32, 4251–4261. [Google Scholar] [PubMed]
- Mikosz, C.A.; Brickley, D.R.; Sharkey, M.S.; Moran, T.W.; Conzen, S.D. Glucocorticoid Receptor-mediated Protection from Apoptosis Is Associated with Induction of the Serine/Threonine Survival Kinase Gene, sgk-1. J. Biol. Chem. 2001, 276, 16649–16654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruhn, M.A.; Pearson, R.B.; Hannan, R.D.; Sheppard, K.E. Second AKT: The rise of SGK in cancer signalling. Growth Factors 2010, 28, 394–408. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, D.; Phung, S.; Masri, S.; Smith, D.; Chen, S. SGK3 is an estrogen-inducible kinase promoting estrogen-mediated survival of breast cancer cells. Mol. Endocrinol. 2011, 25, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slagsvold, T.; Marchese, A.; Brech, A.; Stenmark, H. CISK attenuates degradation of the chemokine receptor CXCR4 via the ubiquitin ligase AIP4. EMBO J. 2006, 25, 3738–3749. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, H.L.; Li, B.B.; Li, W.; Ma, D.; Li, Y.H.; Liu, T. Serum- and glucocorticoid-inducible kinase 3 is a potential oncogene in nasopharyngeal carcinoma. Braz. J. Otorhinolaryngol. 2019, 85, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Liu, L.; Zhang, C.; Zheng, T.; Wang, J.; Lin, M.; Zhao, Y.; Wang, X.; Levine, A.J.; Hu, W. Chronic restraint stress attenuates p53 function and promotes tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 7013–7018. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Huang, W.; Zeng, G.; Ma, X.; Zhou, X.; Wang, Y.; Ouyang, C.; Cheng, A. Expression of the Annexin A1 gene is associated with suppression of growth, invasion and metastasis of nasopharyngeal carcinoma. Mol. Med. Rep. 2014, 10, 3059–3067. [Google Scholar] [CrossRef] [Green Version]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef]
- Sakurai, T.; Kudo, M. Signaling pathways governing tumor angiogenesis. Oncology 2011, 81 (Suppl. 1), 24–29. [Google Scholar] [CrossRef]
- Lee, J.W.; Shahzad, M.M.K.; Lin, Y.G.; Armaiz-Pena, G.; Mangala, L.S.; Han, H.D.; Kim, H.S.; Nam, E.J.; Jennings, N.B.; Halder, J.; et al. Surgical stress promotes tumor growth in ovarian carcinoma. Clin. Cancer Res. 2009, 15, 2695–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutgendorf, S.K.; Johnsen, E.L.; Cooper, B.; Anderson, B.; Sorosky, J.I.; Buller, R.E.; Sood, A.K. Vascular endothelial growth factor and social support in patients with ovarian carcinoma. Cancer 2002, 95, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, E.S.; Lutgendorf, S.K.; Sood, A.K.; Andersen, B.; Sorosky, J.; Lubaroff, D.M. Psychosocial factors and interleukin-6 among women with advanced ovarian cancer. Cancer 2005, 104, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Tasken, K.A. beta-Adrenergic Receptor Signaling in Prostate Cancer. Front. Oncol. 2014, 4, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, S.; Karpova, Y.; Baiz, D.; Yancey, D.; Pullikuth, A.; Flores, A.; Register, T.; Cline, J.M.; D’Agostino, R.; Danial, N.; et al. Behavioral stress accelerates prostate cancer development in mice. J. Clin. Investig. 2013, 123, 874–886. [Google Scholar] [CrossRef] [Green Version]
- Hulsurkar, M.; Li, Z.; Zhang, Y.; Li, X.; Zheng, D.; Li, W. Beta-adrenergic signaling promotes tumor angiogenesis and prostate cancer progression through HDAC2-mediated suppression of thrombospondin-1. Oncogene 2017, 36, 1525–1536. [Google Scholar] [CrossRef]
- Wu, X.; Liu, B.J.; Ji, S.; Wu, J.F.; Xu, C.Q.; Du, Y.J.; You, X.F.; Li, B.; Le, J.J.; Xu, H.L.; et al. Social defeat stress promotes tumor growth and angiogenesis by upregulating vascular endothelial growth factor/extracellular signal-regulated kinase/matrix metalloproteinase signaling in a mouse model of lung carcinoma. Mol. Med. Rep. 2015, 12, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- Garg, J.; Feng, Y.X.; Jansen, S.R.; Friedrich, J.; Lezoualc’h, F.; Schmidt, M.; Wieland, T. Catecholamines facilitate VEGF-dependent angiogenesis via β2-adrenoceptor-induced Epac1 and PKA activation. Oncotarget 2017, 8, 44732–44748. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Wei, Y.; Li, Z.Y.; Cai, X.Y.; Zhang, L.L.; Dong, X.R.; Zhang, S.; Zhang, R.G.; Meng, R.; Zhu, F.; et al. Catecholamines contribute to the neovascularization of lung cancer via tumor-associated macrophages. Brain Behav. Immun. 2019, 81, 111–121. [Google Scholar] [CrossRef]
- Calvani, M.; Pelon, F.; Comito, G.; Taddei, M.L.; Moretti, S.; Innocenti, S.; Nassini, R.; Gerlini, G.; Borgognoni, L.; Bambi, F.; et al. Norepinephrine promotes tumor microenvironment reactivity through β3-adrenoreceptors during melanoma progression. Oncotarget 2015, 6, 4615–4632. [Google Scholar] [CrossRef] [Green Version]
- Bruno, G.; Cencetti, F.; Pini, A.; Tondo, A.; Cuzzubbo, D.; Fontani, F.; Strinna, V.; Buccoliero, A.M.; Casazza, G.; Donati, C.; et al. β3-adrenoreceptor blockade reduces tumor growth and increases neuronal differentiation in neuroblastoma via SK2/S1P2 modulation. Oncogene 2020, 39, 368–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, E.V.; Sood, A.K.; Chen, M.; Li, Y.; Eubank, T.D.; Marsh, C.B.; Jewell, S.; Flavahan, N.A.; Morrison, C.; Yeh, P.E.; et al. Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 2006, 66, 10357–10364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, E.V.; Kim, S.j.; Donovan, E.L.; Chen, M.; Gross, A.C.; Webster Marketon, J.I.; Barsky, S.H.; Glaser, R. Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: Implications for stress-related enhancement of tumor progression. Brain Behav. Immun. 2009, 23, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landen, C.N.; Lin, Y.G.; Pena, G.N.A.; Das, P.D.; Arevalo, J.M.; Kamat, A.A.; Han, L.Y.; Jennings, N.B.; Spannuth, W.A.; Thaker, P.H.; et al. Neuroendocrine modulation of signal transducer and activator of transcription-3 in ovarian cancer. Cancer Res. 2007, 67, 10389–10396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Sinnott, J.A.; Valdimarsdottir, U.; Fang, F.; Gerke, T.; Tyekucheva, S.; Fiorentino, M.; Lambe, M.; Sesso, H.D.; Sweeney, C.J.; et al. Stress-Related signaling pathways in lethal and nonlethal prostate cancer. Clin. Cancer Res. 2016, 22, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Kassi, E.; Moutsatsou, P. Glucocorticoid receptor signaling and prostate cancer. Cancer Lett. 2011, 302, 1–10. [Google Scholar] [CrossRef]
- Flaherty, R.L.; Intabli, H.; Falcinelli, M.; Bucca, G.; Hesketh, A.; Patel, B.A.; Allen, M.C.; Smith, C.P.; Flint, M.S. Stress hormone-mediated acceleration of breast cancer metastasis is halted by inhibition of nitric oxide synthase. Cancer Lett. 2019, 459, 59–71. [Google Scholar] [CrossRef]
- Ishiguro, H.; Kawahara, T.; Zheng, Y.; Kashiwagi, E.; Li, Y.; Miyamoto, H. Differential regulation of bladder cancer growth by various glucocorticoids: Corticosterone and prednisone inhibit cell invasion without promoting cell proliferation or reducing cisplatin cytotoxicity. Cancer Chemother. Pharmacol. 2014, 74, 249–255. [Google Scholar] [CrossRef]
- Llaguno-Munive, M.; Medina, L.A.; Jurado, R.; Romero-Piña, M.; Garcia-Lopez, P. Mifepristone improves chemo-radiation response in glioblastoma xenografts. Cancer Cell Int. 2013, 13, 29. [Google Scholar] [CrossRef] [Green Version]
- Rathore, B.; Chandra Sekhar Jaggarapu, M.M.; Ganguly, A.; Reddy Rachamalla, H.K.; Banerjee, R. Cationic lipid-conjugated hydrocortisone as selective antitumor agent. Eur. J. Med. Chem. 2016, 108, 309–321. [Google Scholar] [CrossRef]
- Geng, Y.; Wang, J.; Jing, H.; Wang, H.W.; Bao, Y.X. Inhibitory effect of dexamethasone on Lewis mice lung cancer cells. Genet. Mol. Res. 2014, 13, 6827–6836. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Ji, H.; Wang, W.; Zhu, Q.; Cao, M.; Zang, Q. Inhibitory effect of dexamethasone on residual Lewis lung cancer cells in mice following palliative surgery. Oncol. Lett. 2017, 13, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, M.; Itoh, M.; Tsuchida, A.; Ooishi, H.; Kanada, K.; Kajiyama, G. Dexamethasone inhibits invasiveness of a human pancreatic cancer cell line. Int. J. Oncol. 1996, 8, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Aleksandrowicz, E.; Schonsiegel, F.; Groner, D.; Bauer, N.; Nwaeburu, C.C.; Zhao, Z.; Gladkich, J.; Hoppe-Tichy, T.; Yefenof, E.; et al. Dexamethasone mediates pancreatic cancer progression by glucocorticoid receptor, TGFbeta and JNK/AP-1. Cell Death Dis. 2017, 8, e3064. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.X.; Wang, Y.; Su, J.; Zhou, P.; Li, B.; Yin, L.J.; Lu, J. Up-regulation of Rho-associated kinase 1/2 by glucocorticoids promotes migration, invasion and metastasis of melanoma. Cancer Lett. 2017, 410, 1–11. [Google Scholar] [CrossRef]
- Entschladen, F.; Drell, T.L.t.; Lang, K.; Joseph, J.; Zaenker, K.S. Neurotransmitters and chemokines regulate tumor cell migration: Potential for a new pharmacological approach to inhibit invasion and metastasis development. Curr. Pharm. Des. 2005, 11, 403–411. [Google Scholar] [CrossRef]
- Armaiz-Pena, G.N.; Allen, J.K.; Cruz, A.; Stone, R.L.; Nick, A.M.; Lin, Y.G.; Han, L.Y.; Mangala, L.S.; Villares, G.J.; Vivas-Mejia, P.; et al. Src activation by beta-adrenoreceptors is a key switch for tumour metastasis. Nat. Commun. 2013, 4, 1403. [Google Scholar] [CrossRef] [Green Version]
- Guo, K.; Ma, Q.; Wang, L.; Hu, H.; Li, J.; Zhang, D.; Zhang, M. Norepinephrine-induced invasion by pancreatic cancer cells is inhibited by propranolol. Oncol. Rep. 2009, 22, 825–830. [Google Scholar] [CrossRef]
- Lang, K.; Drell, T.L.; Lindecke, A.; Niggemann, B.; Kaltschmidt, C.; Zaenker, K.S.; Entschladen, F. Induction of a metastatogenic tumor cell type by neurotransmitters and its pharmacological inhibition by established drugs. Int. J. Cancer 2004, 112, 231–238. [Google Scholar] [CrossRef]
- Le, C.P.; Nowell, C.J.; Kim-Fuchs, C.; Botteri, E.; Hiller, J.G.; Ismail, H.; Pimentel, M.A.; Chai, M.G.; Karnezis, T.; Rotmensz, N.; et al. Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination. Nat. Commun. 2016, 7, 10634. [Google Scholar] [CrossRef] [Green Version]
- Sloan, E.K.; Priceman, S.J.; Cox, B.F.; Yu, S.; Pimentel, M.A.; Tangkanangnukul, V.; Arevalo, J.M.G.; Morizono, K.; Karanikolas, B.D.W.; Wu, L.; et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010, 70, 7042–7052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sood, A.K.; Bhatty, R.; Kamat, A.A.; Landen, C.N.; Han, L.; Thaker, P.H.; Li, Y.; Gershenson, D.M.; Lutgendorf, S.; Cole, S.W. Stress hormone-mediated invasion of ovarian cancer cells. Clin. Cancer Res. 2006, 12, 369–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.P.; Karolak, M.R.; Ma, Y.; Perrien, D.S.; Masood-Campbell, S.K.; Penner, N.L.; Munoz, S.A.; Zijlstra, A.; Yang, X.; Sterling, J.A.; et al. Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PLoS Biol. 2012, 10, e1001363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creed, S.J.; Le, C.P.; Hassan, M.; Pon, C.K.; Albold, S.; Chan, K.T.; Berginski, M.E.; Huang, Z.; Bear, J.E.; Lane, J.R.; et al. β2-adrenoceptor signaling regulates invadopodia formation to enhance tumor cell invasion. Breast Cancer Res. 2015, 17, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruet, M.; Cotton, D.; Coveney, C.; Boocock, D.J.; Wagner, S.; Komorowski, L.; Rees, R.C.; Pockley, A.G.; Garner, A.C.; Wallis, J.D.; et al. beta2-Adrenergic Signalling Promotes Cell Migration by Upregulating Expression of the Metastasis-Associated Molecule LYPD3. Biology 2020, 9, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.H.; Gill, N.K.; Nyberg, K.D.; Nguyen, A.V.; Hohlbauch, S.V.; Geisse, N.A.; Nowell, C.J.; Sloan, E.K.; Rowat, A.C. Cancer cells become less deformable and more invasive with activation of β-adrenergic signaling. J. Cell Sci. 2016, 129, 4563–4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Deng, Y.T.; Liu, J.; Wang, Y.Q.; Yi, T.W.; Huang, B.Y.; He, S.S.; Zheng, B.; Jiang, Y. Norepinephrine induced epithelial-mesenchymal transition in HT-29 and A549 cells in vitro. J. Cancer Res. Clin. Oncol. 2016, 142, 423–435. [Google Scholar] [CrossRef]
- Choi, M.J.; Cho, K.H.; Lee, S.; Bae, Y.J.; Jeong, K.J.; Rha, S.Y.; Choi, E.J.; Park, J.H.; Kim, J.M.; Lee, J.S.; et al. HTERT mediates norepinephrine-induced Slug expression and ovarian cancer aggressiveness. Oncogene 2015, 34, 3402–3412. [Google Scholar] [CrossRef]
- Barbieri, A.; Bimonte, S.; Palma, G.; Luciano, A.; Rea, D.; Giudice, A.; Scognamiglio, G.; La Mantia, E.; Franco, R.; Perdonà, S.; et al. The stress hormone norepinephrine increases migration of prostate cancer cells in vitro and in vivo. Int. J. Oncol. 2015, 47, 527–534. [Google Scholar] [CrossRef] [Green Version]
- Shan, T.; Cui, X.; Li, W.; Lin, W.; Li, Y.; Chen, X.; Wu, T. Novel regulatory program for norepinephrine-induced epithelial-mesenchymal transition in gastric adenocarcinoma cell lines. Cancer Sci. 2014, 105, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Sood, A.K.; Armaiz-Pena, G.N.; Halder, J.; Nick, A.M.; Stone, R.L.; Hu, W.; Carroll, A.R.; Spannuth, W.A.; Deavers, M.T.; Allen, J.K.; et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J. Clin. Investig. 2010, 120, 1515–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, K.S.R.; Karpova, Y.; Prokopovich, S.; Smith, A.J.; Essau, B.; Gersappe, A.; Carson, J.P.; Weber, M.J.; Register, T.C.; Chen, Y.Q.; et al. Epinephrine protects cancer cells from apoptosis via activation of cAMP-dependent protein kinase and BAD phosphorylation. J. Biol. Chem. 2007, 282, 14094–14100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.Y.; Malloy, P.J.; Krishnan, A.V.; Swami, S.; Navone, N.M.; Peehl, D.M.; Feldman, D. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat. Med. 2000, 6, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Tian, M.; Han, G.; Li, J.L. Increased glucocorticoid receptor activity and proliferation in metastatic colon cancer. Sci. Rep. 2019, 9, 11257. [Google Scholar] [CrossRef] [PubMed]
- Runnebaum, I.B.; Brüning, A. Glucocorticoids inhibit cell death in ovarian cancer and up-regulate caspase inhibitor cIAP2. Clin. Cancer Res. 2005, 11, 6325–6332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, J.C.; Huber, R.M.; Hanson, R.L.; Collier, P.M.; Haws, T.F.; Mills, J.K.; Burn, T.C.; Allegretto, E.A. Dexamethasone and tumor necrosis factor-α act together to induce the cellular inhibitor of apoptosis-2 gene and prevent apoptosis in a variety of cell types. Endocrinology 2002, 143, 3866–3874. [Google Scholar] [CrossRef] [Green Version]
- Gascoyne, D.M.; Kypta, R.M.; Vivanco, M. Glucocorticoids inhibit apoptosis during fibrosarcoma development by transcriptionally activating Bcl-xL. J. Biol. Chem. 2003, 278, 18022–18029. [Google Scholar] [CrossRef] [Green Version]
- Gorman, A.M.; Hirt, U.A.; Orrenius, S.; Ceccatelli, S. Dexamethasone pre-treatment interferes with apoptotic death in glioma cells. Neuroscience 2000, 96, 417–425. [Google Scholar] [CrossRef]
- Moran, T.J.; Gray, S.; Mikosz, C.A.; Conzen, S.D. The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Res. 2000, 60, 867–872. [Google Scholar]
- Herr, I.; Buchler, M.W.; Mattern, J. Glucocorticoid-mediated apoptosis resistance of solid tumors. Results Probl. Cell Differ. 2009, 49, 191–218. [Google Scholar] [CrossRef]
- Zhang, C.; Kolb, A.; Mattern, J.; Gassler, N.; Wenger, T.; Herzer, K.; Debatin, K.M.; Büchler, M.; Friess, H.; Rittgen, W.; et al. Dexamethasone desensitizes hepatocellular and colorectal tumours toward cytotoxic therapy. Cancer Lett. 2006, 242, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Beckermann, B.; Kallifatidis, G.; Liu, Z.; Rittgen, W.; Edler, L.; Büchler, P.; Debatin, K.M.; Büchler, M.W.; Friess, H.; et al. Corticosteroids induce chemotherapy resistance in the majority of tumour cells from bone, brain, breast, cervix, melanoma and neuroblastoma. Int. J. Oncol. 2006, 29, 1295–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Yuan, L.; Sun, Y.; Wang, P.; Zhang, H.; Feng, X.; Wang, Z.; Zhang, W.; Yang, C.; Zeng, Y.A.; et al. Glucocorticoid receptor signaling activates TEAD4 to promote breast cancer progression. Cancer Res. 2019, 79, 4399–4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrella, A.; Ercolino, S.F.; Festa, M.; Gentilella, A.; Tosco, A.; Conzen, S.D.; Parente, L. Dexamethasone inhibits TRAIL-induced apoptosis of thyroid cancer cells via Bcl-xL induction. Eur. J. Cancer 2006, 42, 3287–3293. [Google Scholar] [CrossRef]
- Kim, K.M.; Jung, B.H.; Lho, D.S.; Chung, W.Y.; Paeng, K.J.; Chung, B.C. Alteration of urinary profiles of endogenous steroids and polyunsaturated fatty acids in thyroid cancer. Cancer Lett. 2003, 202, 173–179. [Google Scholar] [CrossRef]
- Flint, M.S.; Baum, A.; Chambers, W.H.; Jenkins, F.J. Induction of DNA damage, alteration of DNA repair and transcriptional activation by stress hormones. Psychoneuroendocrinology 2007, 32, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Gidron, Y.; Russ, K.; Tissarchondou, H.; Warner, J. The relation between psychological factors and DNA-damage: A critical review. Biol. Psychol. 2006, 72, 291–304. [Google Scholar] [CrossRef]
- Flaherty, R.L.; Owen, M.; Fagan-Murphy, A.; Intabli, H.; Healy, D.; Patel, A.; Allen, M.C.; Patel, B.A.; Flint, M.S. Glucocorticoids induce production of reactive oxygen species/reactive nitrogen species and DNA damage through an iNOS mediated pathway in breast cancer. Breast Cancer Res 2017, 19, 35. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, L.; Mims, J.; Punska, E.C.; Williams, K.E.; Zhao, W.; Arcaro, K.F.; Tsang, A.W.; Zhou, X.; Furdui, C.M. Analysis of DNA methylation and gene expression in radiation-resistant head and neck tumors. Epigenetics 2015, 10, 545–561. [Google Scholar] [CrossRef] [Green Version]
- Alyusuf, R.; Wazir, J.F.; Brahmi, U.P.; Fakhro, A.R.; Bakhiet, M. The Immunoexpression of Glucocorticoid Receptors in Breast Carcinomas, Lactational Change, and Normal Breast Epithelium and Its Possible Role in Mammary Carcinogenesis. Int. J. Breast Cancer 2017, 2017, 1403054. [Google Scholar] [CrossRef] [Green Version]
- Pan, D.; Kocherginsky, M.; Conzen, S.D. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res. 2011, 71, 6360–6370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Pew, T.; Zou, M.; Pang, D.; Conzen, S.D. Glucocorticoid receptor-induced MAPK phosphatase-1 (MPK-1) expression inhibits paclitaxel-associated MAPK activation and contributes to breast cancer cell survival. J. Biol. Chem. 2005, 280, 4117–4124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, D.C.; Kocherginsky, M.; Tonsing-Carter, E.Y.; Dolcen, D.N.; Hosfield, D.J.; Lastra, R.R.; Sinnwell, J.P.; Thompson, K.J.; Bowie, K.R.; Harkless, R.V.; et al. Discovery of a glucocorticoid receptor (gr) activity signature using selective gr antagonis in er-negative breast cancer. Clin. Cancer Res. 2018, 24, 3433–3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catteau, X.; Simon, P.; Buxant, F.; Noël, J.-C. Expression of the glucocorticoid receptor in breast cancer-associated fibroblasts. Mol. Clin. Oncol. 2016, 5, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Veneris, J.T.; Darcy, K.M.; Mhawech-Fauceglia, P.; Tian, C.; Lengyel, E.; Lastra, R.R.; Pejovic, T.; Conzen, S.D.; Fleming, G.F. High glucocorticoid receptor expression predicts short progression-free survival in ovarian cancer. Gynecol. Oncol. 2017, 146, 153–160. [Google Scholar] [CrossRef]
- Veneris, J.T.; Huang, L.; Churpek, J.E.; Conzen, S.D.; Fleming, G.F. Glucocorticoid receptor expression is associated with inferior overall survival independent of BRCA mutation status in ovarian cancer. Int. J. Gynecol. Cancer 2019, 29, 357–364. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Ploner, C.; Handle, F.; Schaefer, G.; Kroon, J.; Leo, A.; Heidegger, I.; Eder, I.; et al. The glucocorticoid receptor is a key player for prostate cancer cell survival and a target for improved antiandrogen therapy. Clin. Cancer Res. 2018, 24, 927–938. [Google Scholar] [CrossRef] [Green Version]
- Abduljabbar, R.; Negm, O.H.; Lai, C.F.; Jerjees, D.A.; Al-Kaabi, M.; Hamed, M.R.; Tighe, P.J.; Buluwela, L.; Mukherjee, A.; Green, A.R.; et al. Clinical and biological significance of glucocorticoid receptor (GR) expression in breast cancer. Breast Cancer Res. Treat. 2015, 150, 335–346. [Google Scholar] [CrossRef]
- Yemelyanov, A.; Czwornog, J.; Chebotaev, D.; Karseladze, A.; Kulevitch, E.; Yang, X.; Budunova, I. Tumor suppressor activity of glucocorticoid receptor in the prostate. Oncogene 2007, 26, 1885–1896. [Google Scholar] [CrossRef] [Green Version]
- Mitani, Y.; Lin, S.H.; Pytynia, K.B.; Ferrarotto, R.; El-Naggar, A.K. Reciprocal and autonomous glucocorticoid and androgen receptor activation in salivary duct carcinoma. Clin. Cancer Res. 2020, 26, 1175–1184. [Google Scholar] [CrossRef]
- Chebotaev, D.; Yemelyanov, A.; Zhu, L.; Lavker, R.M.; Budunova, I. The tumor suppressor effect of the glucocorticoid receptor in skin is mediated via its effect on follicular epithelial stem cells. Oncogene 2007, 26, 3060–3068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chebotaev, D.; Yemelyanov, A.; Budunova, I. The mechanisms of tumor suppressor effect of glucocorticoid receptor in skin. Mol. Carcinog. 2007, 46, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Budunova, I.V.; Kowalczyk, D.; Pérez, P.; Yao, Y.J.; Jorcano, J.L.; Slaga, T.J. Glucocorticoid receptor functions as a potent suppressor of mouse skin carcinogenesis. Oncogene 2003, 22, 3279–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancha-Ramirez, A.M.; Yang, X.; Liang, H.; Junco, J.; Lee, K.P.; Bovio, S.F.; Espinoza, M.; Wool, J.; Slaga, A.; Glade, D.C.; et al. Harnessing the gatekeepers of glucocorticoids for chemoprevention of non-melanoma skin cancer. Mol. Carcinog. 2019, 58, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Ancoli-Israel, S.; Bower, J.E.; Capuron, L.; Irwin, M.R. Neuroendocrine-immune mechanisms of behavioral comorbidities in patients with cancer. J. Clin. Oncol. 2008, 26, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Foss, F.M. Immunologic mechanisms of antitumor activity. Semin. Oncol. 2002, 29, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S. CD4+ T lymphocytes: A critical component of antitumor immunity. Cancer Investig. 2005, 23, 413–419. [Google Scholar]
- Yuen, G.J.; Demissie, E.; Pillai, S. B lymphocytes and cancer: A love-hate relationship. Trends Cancer 2016, 2, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Dhabhar, F.S.; McEwen, B.S. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: A potential role for leukocyte trafficking. Brain Behav. Immun. 1997, 11, 286–306. [Google Scholar] [CrossRef] [Green Version]
- Elenkov, I.J. Systemic stress-induced Th2 shift and its clinical implications. Int. Rev. Neurobiol. 2002, 52, 163–186. [Google Scholar] [CrossRef]
- Glaser, R.; MacCallum, R.C.; Laskowski, B.F.; Malarkey, W.B.; Sheridan, J.F.; Kiecolt-Glaser, J.K. Evidence for a shift in the Th-1 to Th-2 cytokine response associated with chronic stress and aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2001, 56, M477–M482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, B.F.; Fauci, A.S. The differential effect of in vivo hydrocortisone on the kinetics of subpopulations of human peripheral blood thymus-derived lymphocytes. J. Clin. Investig. 1978, 61, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Klein, A. Does endogenous cortisol play a role in the development of cancer? Med. Hypotheses 1982, 8, 163–171. [Google Scholar] [CrossRef]
- Thomson, S.P.; McMahon, L.J.; Nugent, C.A. Endogenous cortisol: A regulator of the number of lymphocytes in peripheral blood. Clin. Immunol. Immunopathol. 1980, 17, 506–514. [Google Scholar] [CrossRef]
- Penn, I. Tumor incidence in human allograft recipients. Transplant. Proc. 1979, 11, 1047–1051. [Google Scholar]
- Baserga, R.; Shubik, P.; Baum, J. Action of cortisone on disseminated tumor cells after removal of the primary growth. Science 1955, 121, 100–101. [Google Scholar] [CrossRef]
- Frick, L.R.; Barreiro Arcos, M.L.; Rapanelli, M.; Zappia, M.P.; Brocco, M.; Mongini, C.; Genaro, A.M.; Cremaschi, G.A. Chronic restraint stress impairs T-cell immunity and promotes tumor progression in mice. Stress 2009, 12, 134–143. [Google Scholar] [CrossRef]
- Schmidt, D.; Peterlik, D.; Reber, S.O.; Lechner, A.; Mannel, D.N. Induction of Suppressor Cells and Increased Tumor Growth following Chronic Psychosocial Stress in Male Mice. PLoS ONE 2016, 11, e0159059. [Google Scholar] [CrossRef] [Green Version]
- Nissen, M.D.; Sloan, E.K.; Mattarollo, S.R. β-Adrenergic signaling impairs antitumor CD8+ T-cell responses to B-cell lymphoma immunotherapy. Cancer Immunol. Res. 2018, 6, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Eldridge, R.C.; Beitler, J.J.; Higgins, K.A.; Chico, C.E.; Felger, J.C.; Wommack, E.C.; Knobf, T.; Saba, N.F.; Shin, D.M.; et al. Association Among Glucocorticoid Receptor Sensitivity, Fatigue, and Inflammation in Patients with Head and Neck Cancer. Psychosom. Med. 2020, 82, 508–516. [Google Scholar] [CrossRef]
- Li, L.; Wang, B.Q.; Gao, T.H.; Tian, J. Assessment of psychological status of inpatients with head and neck cancer before surgery. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi Chin. J. Otorhinolaryngol. Head Neck Surg. 2018, 53, 21–26. [Google Scholar] [CrossRef]
- Sah, G.S. Psychosocial and Functional Distress of Cancer Patients in A Tertiary Care Hospital: A Descriptive Cross-sectional Study. JNMA J. Nepal Med. Assoc. 2019, 57, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Kamarck, T.; Mermelstein, R. A global measure of perceived stress. J. Health Soc. Behav. 1983, 24, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Lovibond, P.F.; Lovibond, S.H. The structure of negative emotional states: Comparison of the Depression Anxiety Stress Scales (DASS) with the Beck Depression and Anxiety Inventories. Behav. Res. Ther. 1995, 33, 335–343. [Google Scholar] [CrossRef]
- Ryff, C.D. Happiness is everything, or is it? Explorations on the meaning of psychological well-being. J. Personal. Soc. Psychol. 1989, 57, 1069–1081. [Google Scholar] [CrossRef]
- Diener, E.; Emmons, R.A.; Larsem, R.J.; Griffin, S. The satisfaction with life scale. J. Personal. Assess. 1985, 49, 71–75. [Google Scholar] [CrossRef]
- Hellhammer, J.; Fries, E.; Schweisthal, O.W.; Schlotz, W.; Stone, A.A.; Hagemann, D. Several daily measurements are necessary to reliably assess the cortisol rise after awakening: State- and trait components. Psychoneuroendocrinology 2007, 32, 80–86. [Google Scholar] [CrossRef]
- Almeida, D.M.; Piazza, J.R.; Stawski, R.S. Interindividual differences and intraindividual variability in the cortisol awakening response: An examination of age and gender. Psychol. Aging 2009, 24, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Stalder, T.; Steudte, S.; Alexander, N.; Miller, R.; Gao, W.; Dettenborn, L.; Kirschbaum, C. Cortisol in hair, body mass index and stress-related measures. Biol. Psychol. 2012, 90, 218–223. [Google Scholar] [CrossRef]
- Dettenborn, L.; Tietze, A.; Bruckner, F.; Kirschbaum, C. Higher cortisol content in hair among long-term unemployed individuals compared to controls. Psychoneuroendocrinology 2010, 35, 1404–1409. [Google Scholar] [CrossRef]
- Kirschbaum, C.; Tietze, A.; Skoluda, N.; Dettenborn, L. Hair as a retrospective calendar of cortisol production-Increased cortisol incorporation into hair in the third trimester of pregnancy. Psychoneuroendocrinology 2009, 34, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Yamada, J.; Stevens, B.; de Silva, N.; Gibbins, S.; Beyene, J.; Taddio, A.; Newman, C.; Koren, G. Hair cortisol as a potential biologic marker of chronic stress in hospitalized neonates. Neonatology 2007, 92, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Karlén, J.; Ludvigsson, J.; Frostell, A.; Theodorsson, E.; Faresjö, T. Cortisol in hair measured in young adults-a biomarker of major life stressors? BMC Clin. Pathol. 2011, 11, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.E.; Rasmussen, A. War exposure, daily stressors, and mental health in conflict and post-conflict settings: Bridging the divide between trauma-focused and psychosocial frameworks. Soc. Sci. Med. 2010, 70, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Löwenberg, M.; Verhaar, A.P.; van den Brink, G.R.; Hommes, D.W. Glucocorticoid signaling: A nongenomic mechanism for T-cell immunosuppression. Trends Mol. Med. 2007, 13, 158–163. [Google Scholar] [CrossRef]
- Massie, M.J. Prevalence of depression in patients with cancer. J. Natl. Cancer Inst. Monogr. 2004, 2004, 57–71. [Google Scholar] [CrossRef]
- Carlson, L.E.; Angen, M.; Cullum, J.; Goodey, E.; Koopmans, J.; Lamont, L.; MacRae, J.H.; Martin, M.; Pelletier, G.; Robinson, J.; et al. High levels of untreated distress and fatigue in cancer patients. Br. J. Cancer 2004, 90, 2297–2304. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Performed on | Techniques Used | Findings |
|---|---|---|---|
| 1. Stress hormones increase cell proliferation and regulates interleukin-6 secretion in human oral squamous cell carcinoma cells [9]. | Oral squamous cell carcinoma (OSCC) cell lines (SCC9, SCC15, SCC25), 20 OSCC biopsies, 17 leukoplakia biopsies, 15 Normal oral mucosae. | Polymerase Chain Reaction (PCR), to determine Interleukin-6 (IL-6) gene expression in cell lines and tissues, (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT) to determine cell proliferation, enzyme-linked immunosorbent assay (ELISA) to determine the IL-6 protein levels. | Norepinephrine (NE) increased IL-6 expression and cell proliferation in OSCC cell lines. Pharmacological dose of cortisol decreased VEGF and IL-6 whereas, stress dose increased VEGF and IL-6 expressions. Mean expression of β1 mRNA in OSCC was higher compared to normal mucosa (p < 0.05). |
| 2. Chronic stress promotes oral cancer growth and angiogenesis with increased circulating catecholamine and glucocorticoid levels in a mouse model [10]. | Oral Cancer Cell line CAL 27 implanted into mice. | Catecholamine levels were determined by High Performance Liquid Chromatography with Mass spectrometry (HPLC-MS/MS). Expression of VEGF and MMP by was observed with immunohistochemistry (IHC). Physical restraint system was used to induce characteristic chronic stress. | Chronic stress increased tumour size, matrix metalloproteinases (MMP), VEGF expression, level of plasma catecholamines, cortisone and caused more invasive growth of oral carcinoma cells in a mice model. |
| 3. Association of Increased Circulating Catecholamine and Glucocorticoid Levels with Risk of Psychological Problems in Oral Neoplasm Patients [11]. | 75 patients (49 men and 26 women) with oral tumours were included. | Checklist 90-revised Inventory (SCL90-R) which is a self-assessment survey of 90 questions, as well as a demographic questionnaire were used to assess the psychosocial status of patients. Blood samples were taken two hours before surgery, between approximately 9:00 and 11:00 a.m. Catecholamine levels were determined by High Performance Liquid Chromatography with Mass spectrometry (HPLC-MS-MS). | Significant difference in the scores of SCL90-R between the benign tumour and cancer patients’ groups was only seen in the dimensions of depression (p = 0.0201) and obsessive-compulsion (p = 0.0093) Peripheral blood mean concentrations of catecholamines and glucocorticoids in the oral cancer group were higher than in the benign tumour group (p < 0.01) (p < 0.001), respectively. Stage I and II cancer showed comparatively low concentrations of epinephrine and Stage III and IV cancer showed substantially greater concentrations of epinephrine, norepinephrine, cortisone, hydrocortisone. |
| 4. Prognostic significance of beta-2 adrenergic receptor in oral squamous cell carcinoma [12]. | Clinicopathological data, treatment, tumour outcome, prognosis and expression of β2-adrenergic receptor was examined for 106 OSCC patients. | Immunohistochemistry was used to analyse the expression of β2-adrenergic receptors and its relation to clinicopathological variables. | Strong cytoplasmic and membranous β2-adrenergic receptor expression was found in malignant OSCC (72.6%). Significant association between β2-adrenergic receptor expression and alcohol (p = 0.021), simultaneous use of alcohol and tobacco (p = 0.014) and T stage (p = 0.07), was observed. |
| 5. Expression of β2-adrenergic receptor in oral squamous cell carcinoma [13]. | 65 OSCC patients with pathologically confirmed diagnosis of OSCC. Ten cases of adjacent normal mucosa as controls. TCa8113—cell line from OSCC of tongue. ACC—cell line from Salivary Adenoid Cystic Carcinoma. | Immunohistochemistry (IHC) Western blot RT PCR Migration assay Proliferation assay. | β2 expression significantly correlated with cervical lymph node metastasis (p = 0.001), age (0.003), tumour size (0.001), clinical stage (0.001). |
| 6. Glucocorticoids reduce chemotherapeutic effectiveness on OSCC cells via glucose-dependent mechanisms [14]. | Oral malignant keratinocytes: H314, H357, H400, BICR16, BICR56. | Annexin V-FITC assay to study apoptosis. Enzyme-linked immunosorbent assay (ELISA)—to measure the concentration of cortisol after adrenocorticotropic hormone (ACTH) stimulation. | Glucocorticoids had an antiapoptotic and protective effect on OSCC against chemotherapy in a glucose dependent manner. |
| 7. Immunoexpression of glucocorticoid receptor alpha (GRα) isoform and apoptotic proteins (Bcl-2 and Bax) in actinic cheilitis and lower lip squamous cell carcinoma [15]. | 22 cases of actinic cheilitis (AC), 44 cases of lower lip squamous cell carcinoma (LLSCC) (22 with normal mucosa, 22 without normal mucosa) The percentages of nuclear (GRα) and cytoplasmic (GRα, Bcl-2, and Bax) staining in epithelial cells were correlated with clinical (tumour size/extent and clinical stage) and histopathological parameters (risk of malignant transformation for AC and histopathological grade of malignancy for LLSCCs). | Immunohistochemistry (IHC). | A relatively high median percentages of GRα positive staining was observed in all cases. A lower nuclear expression and higher cytoplasmic expression of GRα was observed in LLSCC specimens compared to actinic cheilitis (p < 0.05). A higher GRα expression was observed in high grade tumours compared to low grade tumours (p = 0.036). |
| 8. Circulating catecholamines are associated with biobehavioural factors and anxiety syptoms in head and neck cancer patients [16]. | Plasma epinephrine and norepinephrine were measured. Psychological anxiety levels in 93 patients with HNSCC and 32 patients with oral leukoplakia. | Plasma epinephrine and norepinephrine were measured by High Performance Liquid Chromatography-Electrochemical Detection (HPLC-ED). Psychological anxiety levels measured by Beck Anxiety Inventory (BAI). | Significantly higher levels of plasma epinephrine and norepinephrine were observed in OSCC patients than in oropharyngeal and oral leukoplakia patients. The total BAI mean scores did not show a significant difference among the three groups. |
| 9. Characterisation of a Novel Oral Glucocorticoid System and Its Possible Role in Disease [17]. | Normal oral keratinocytes (NOK), normal oral fibroblasts (NOF), normal oral mucosa (NOM) and malignant tissue. | Western Blot, Immunohistochemistry, ELISA. | NOK and NOF synthesise cortisol in the presence of 10 nM ACTH. NOK expressed 11-β hydroxysteroid dehydrogenase type 1 (11-β HSD1), 11-β hydroxysteroid dehydrogenase type 2 (11-β HSD 2), Glucocorticoid Receptor and Mineralocorticoid Receptor. NOK lacked 11-β-HSD 2 showing their inability to degrade cortisol. 11-β-HSD expression was not detected in OSCC. |
| 10. Increased plasma and salivary cortisol levels in patients with oral cancer and their association with clinical stage [18]. | 34 oral squamous cell carcinoma (OSCC) patients, 17 oropharyngeal SCC patients, 17 oral leukoplakia patients, 27 smokers and/or drinkers and 25 healthy volunteers. | The plasma and salivary cortisol levels of patients with OSCC were compared with other groups by enzyme immunoassay with a commercial kit. | OSCC patients showed significantly higher levels of plasma (p < 0.05) and salivary (p < 0.01) cortisol compared to all the other groups. Patients at advanced stage of OSCC showed significantly higher cortisol levels than those at initial stage. |
| 11. The stress hormone Norepinephrine promotes tumour progression through β2-adrenoreceptors in oral cancer [19]. | 40 OSCC samples from patients and 20 para cancer Normal Oral Mucosa. SCC25 and CAL 27 cell lines. | RT-PCR Immunohistochemistry Cell proliferation assay CCK-8 Matrigel coated transwell assay Colony forming assay Sphere forming assay. | OSCC showed significantly higher β2 adrenergic receptor expression than normal para cancer tissue. Norepinephrine promoted proliferation, invasion and stem cell characteristics of OSCC cell lines. |
| 12. Stress hormones concentrations in the normal microenvironment predict risk for chemically induced cancer in rats [20]. | Male Wistar rats | Measurement of stress hormone levels, norepinephrine, corticosterone, ACTH and brain-derived neurotropic factor (BDNF) in the tongue microenvironment prior to carcinogen induction was done by ELISA and Milliplex Multi-Analyte Profiling method. To induce tumours, mice were treated with 4-nitroquinoline-1-oxide. The tongues with carcinogen induced lesions were used to perform histochemical analysis and RT PCR. | Increased concentrations of norepinephrine and BDNF positively correlated to OSCC occurrence whereas decreased basal corticosterone levels were predictive for OSCC occurrence. |
| 13. Activation of adrenergic receptor β2 promotes tumour progression and epithelial mesenchymal transition in tongue squamous cell carcinoma (2018) [21]. | Tongue squamous cell carcinoma (TSCC) specimens (n = 70) and adjacent non-cancerous tissue samples (n = 20). CAL 27 and SCC 15 cell lines. | Immunohistochemistry Cell migration and invasion assay Immunofluorescence. | Increased expression of β-adrenergic receptor was observed in TSCC and was associated with lymph node metastasis and reduced overall survival. Treatment of cells with isoproterenol induced epithelial–mesenchymal transition (EMT) by activating IL-6/STAT-3 SNAIL 1 pathway. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iftikhar, A.; Islam, M.; Shepherd, S.; Jones, S.; Ellis, I. Cancer and Stress: Does It Make a Difference to the Patient When These Two Challenges Collide? Cancers 2021, 13, 163. https://doi.org/10.3390/cancers13020163
Iftikhar A, Islam M, Shepherd S, Jones S, Ellis I. Cancer and Stress: Does It Make a Difference to the Patient When These Two Challenges Collide? Cancers. 2021; 13(2):163. https://doi.org/10.3390/cancers13020163
Chicago/Turabian StyleIftikhar, Anem, Mohammad Islam, Simon Shepherd, Sarah Jones, and Ian Ellis. 2021. "Cancer and Stress: Does It Make a Difference to the Patient When These Two Challenges Collide?" Cancers 13, no. 2: 163. https://doi.org/10.3390/cancers13020163
APA StyleIftikhar, A., Islam, M., Shepherd, S., Jones, S., & Ellis, I. (2021). Cancer and Stress: Does It Make a Difference to the Patient When These Two Challenges Collide? Cancers, 13(2), 163. https://doi.org/10.3390/cancers13020163