Dormant Tumor Cell Vaccination: A Mathematical Model of Immunological Dormancy in Triple-Negative Breast Cancer

 , , and
, , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
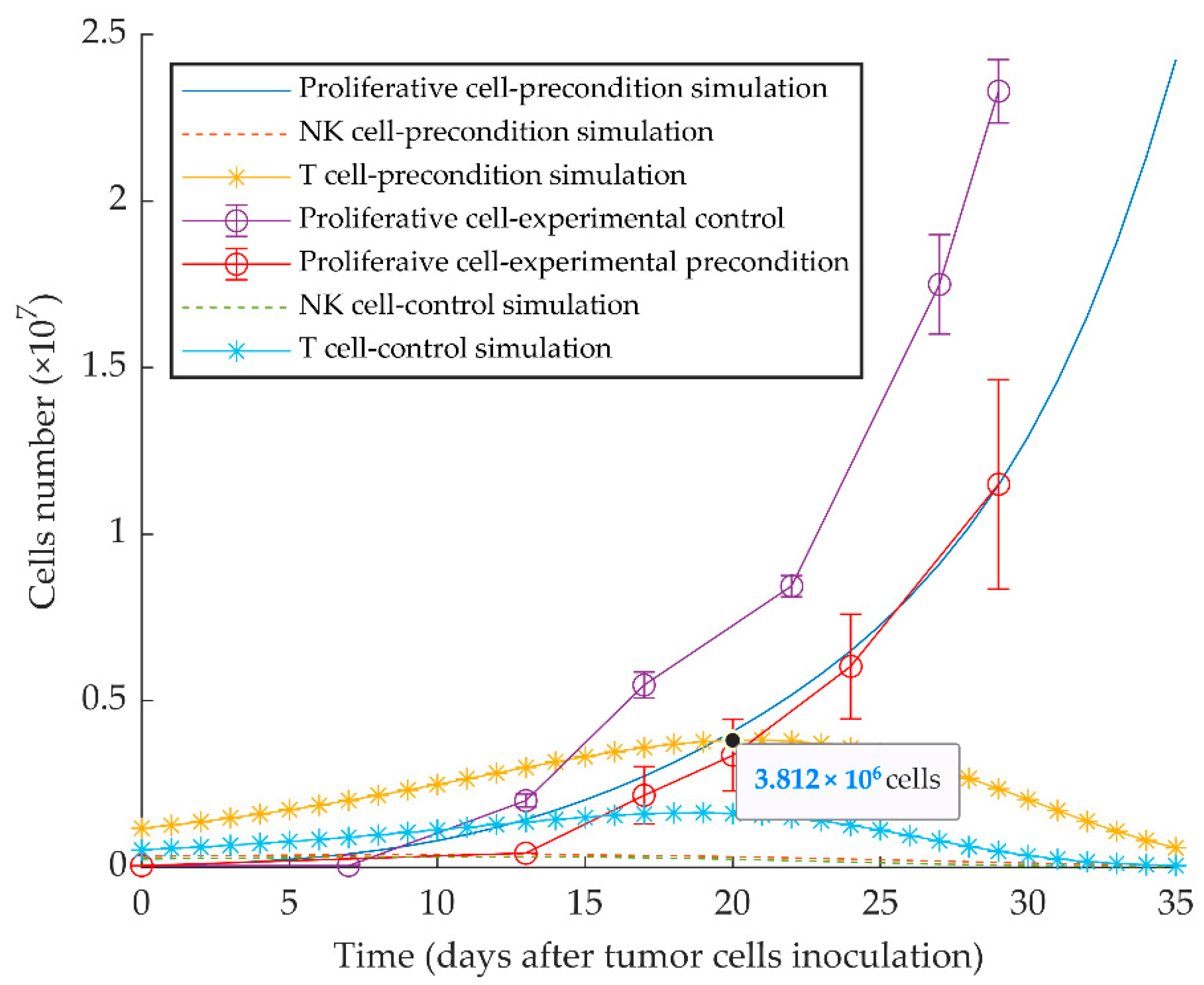
2.1. Previously Published Experimental Results
2.2. Assumptions
- Under the circumstances of insufficient nutrient supply and cell-contact inhibition, a self-limited growth model best explains the biological facts behind tumor growth. The Gompertzian growth model fits best with the published experimental results and leads to the theoretical maximal tumor size [67,68].
- The tumor growth rate represents the proliferation rate minus the natural death rate.
- NK cells, as a part of the innate immune system, are always present and active in tissues, even in the absence of tumor cells; there is a constant source of cytotoxic NK cells.
- Both NK and CD8+ T cells can be inactivated by tumor cells.
- As the number of dormant cells will remain at about 2000 cells, the dormant tumor might not have enough time to find necessary mutations to escape. For simplicity, the model assumes intra-tumoral heterogeneity as a biological element behind the eventual relapse of the MR20 tumor. In this case, we consider two different phenotypes: a quiescent (MR20) and a proliferative population (4T1).
- The model does not consider the spatiotemporal heterogeneity of tumor and immune cells in TME.
2.3. Mathematical Model
2.4. Initial Conditions and Parameters
2.5. Simulations
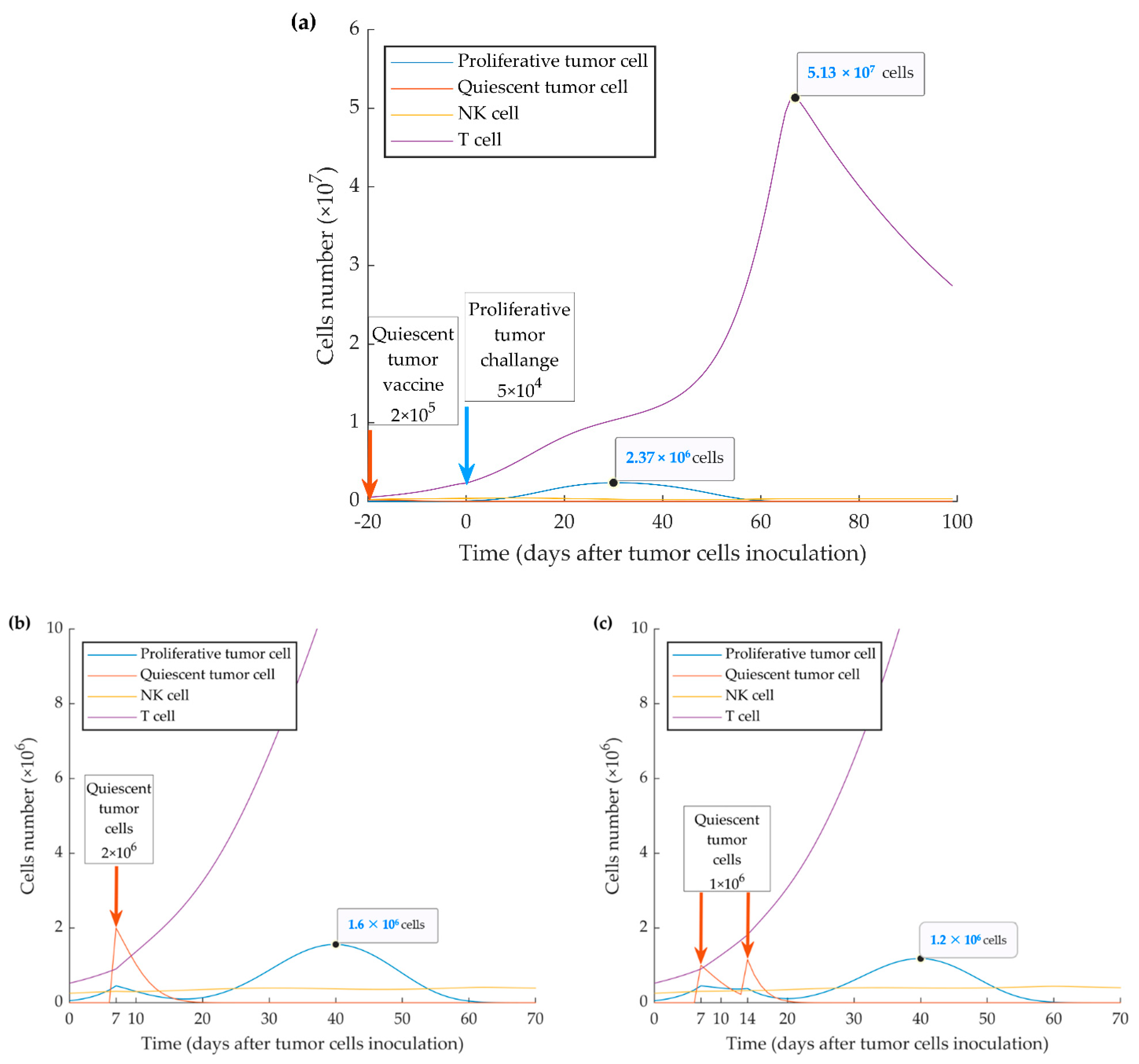
3. Results
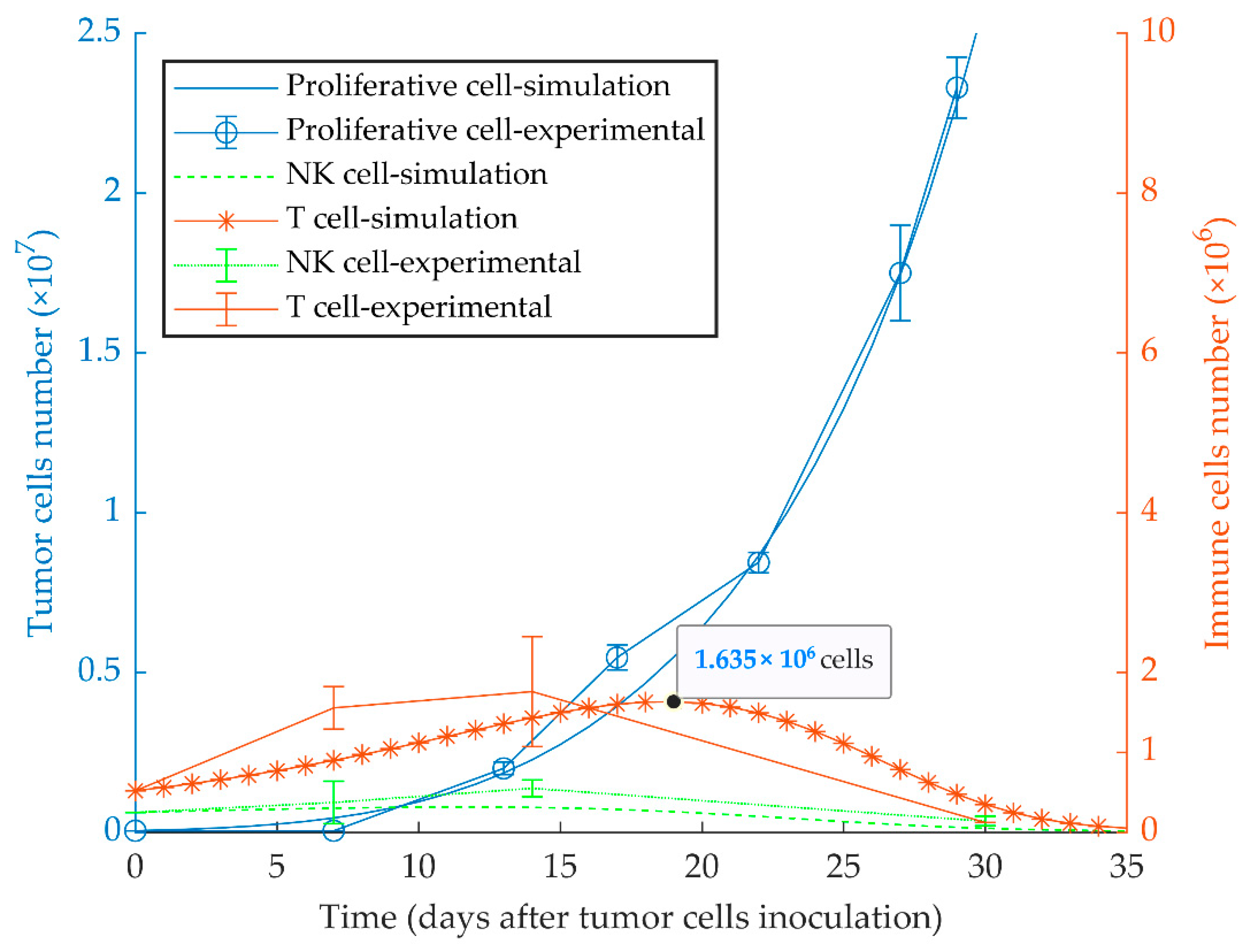
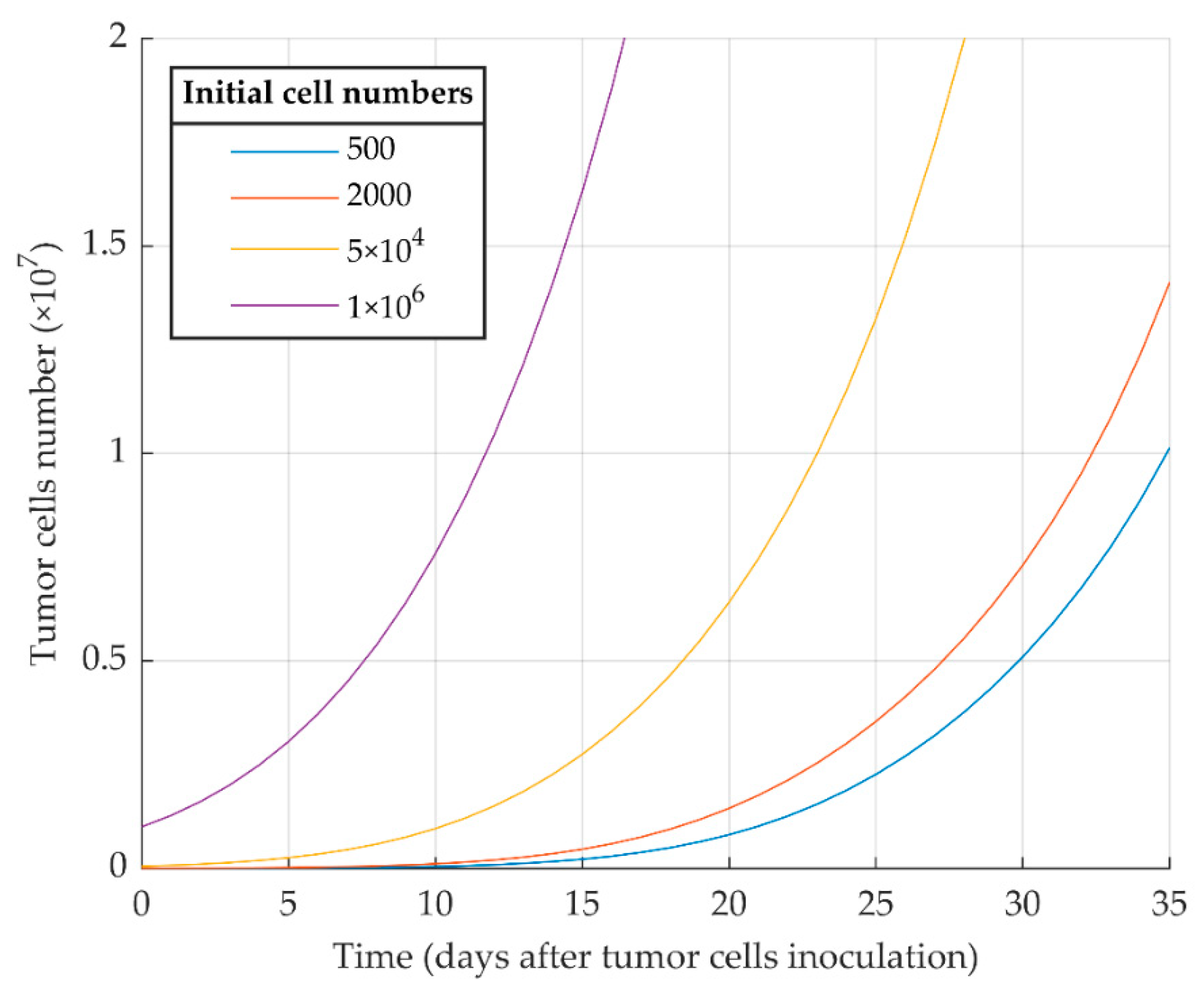
3.1. Simulations of Proliferative Tumor Growth (Series 1)
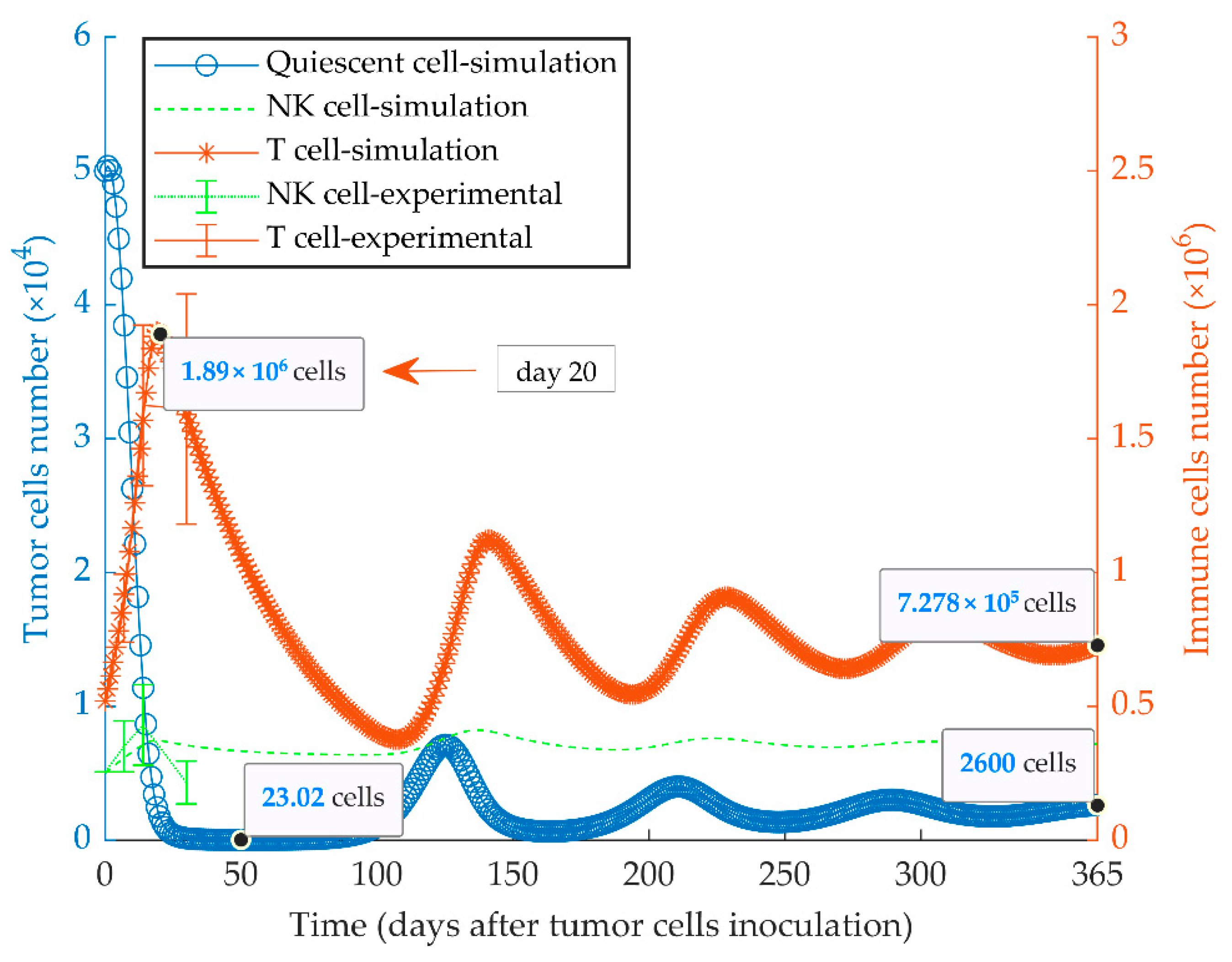
3.2. Simulations of Quiescent Tumor Growth (Series 2)
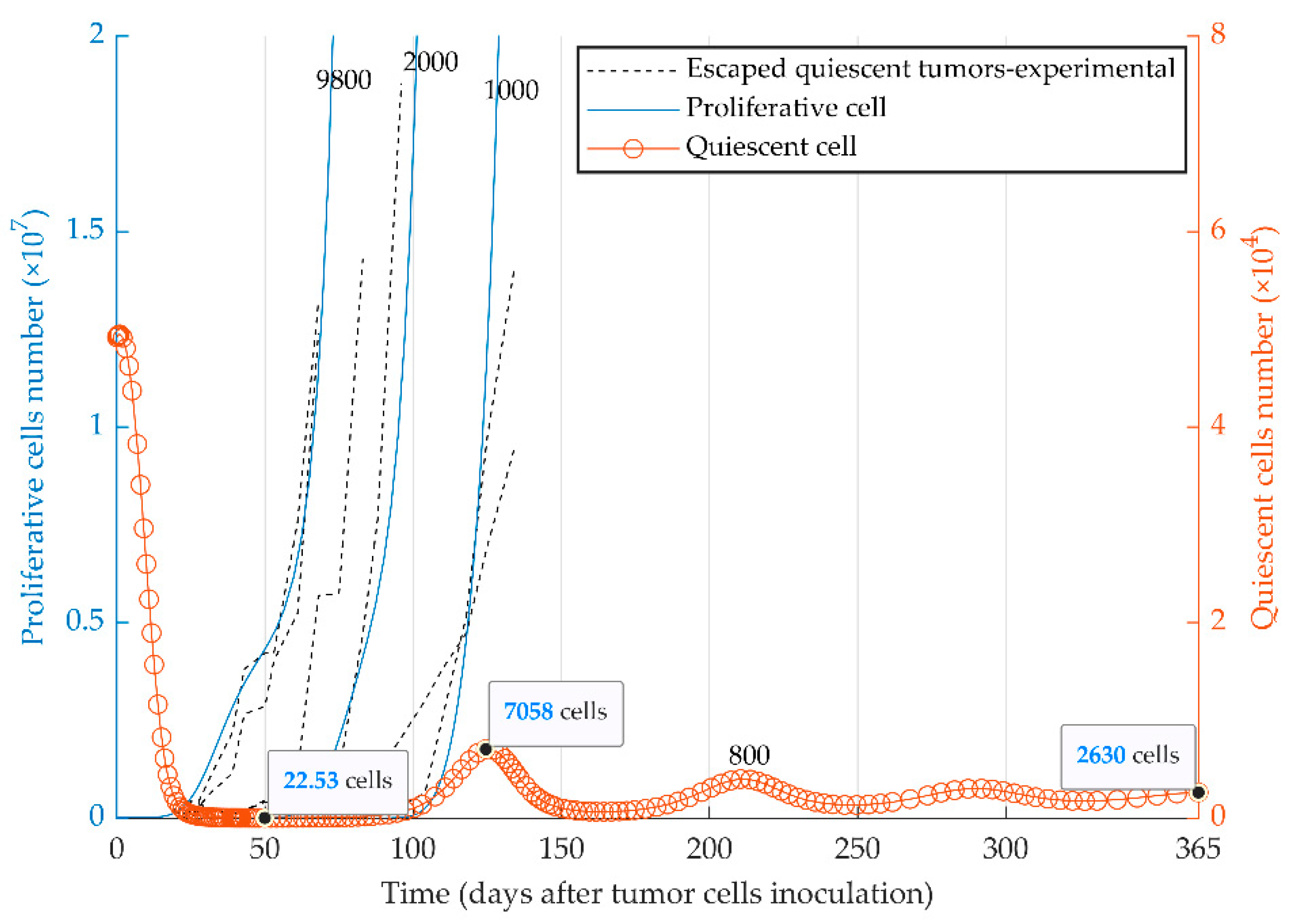
3.3. Simulations of Heterogenenous Tumor Growth (Series 3)
3.4. Simulations of Vaccination Scenarios (Series 4)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluz, O.; Liedtke, C.; Gottschalk, N.; Pusztai, L.; Nitz, U.; Harbeck, N. Triple-negative breast cancer—Current status and future directions. Ann. Oncol. 2009, 20, 1913–1927. [Google Scholar] [CrossRef]
- Vagia, E.; Mahalingam, D.; Cristofanilli, M. The Landscape of Targeted Therapies in TNBC. Cancers 2020, 12, 916. [Google Scholar] [CrossRef] [Green Version]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Colleoni, M.; Cole, B.F.; Viale, G.; Regan, M.M.; Price, K.N.; Maiorano, E.; Mastropasqua, M.G.; Crivellari, D.; Gelber, R.D.; Goldhirsch, A.; et al. Classical cyclophosphamide, methotrexate, and fluorouracil chemotherapy is more effective in triple-negative, node-negative breast cancer: Results from two randomized trials of adjuvant chemoendocrine therapy for node-negative breast cancer. J. Clin. Oncol. 2010, 28, 2966–2973. [Google Scholar] [CrossRef] [Green Version]
- Joensuu, H.; Gligorov, J. Adjuvant treatments for triple-negative breast cancers. Ann. Oncol. 2012, 23 (Suppl. 60), vi40–vi45. [Google Scholar] [CrossRef]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [CrossRef]
- Andreopoulou, E.; Schweber, S.J.; Sparano, J.A.; McDaid, H.M. Therapies for triple negative breast cancer. Expert Opin. Pharm. 2015, 16, 983–998. [Google Scholar] [CrossRef]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell 2018, 173, 879–893 e813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of chemotherapy resistance in triple-negative breast cancer-how we can rise to the challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre, F.; Zielinski, C.C. Optimal strategies for the treatment of metastatic triple-negative breast cancer with currently approved agents. Ann. Oncol. 2012, 23 (Suppl. 6), vi46–vi51. [Google Scholar] [CrossRef]
- Mehdizadeh, R.; Najafi, S.; Jahanzad, I. Evaluation of EGFR, VEGFR2, IGF-1R and HIF-1a Expression and Their Prognostic Value in Iranian Triple-negative Breast Cancer Patients. Eur. J. Cancer 2012, 48. [Google Scholar] [CrossRef]
- Emens, L.A.; Middleton, G. The interplay of immunotherapy and chemotherapy: Harnessing potential synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Kalimutho, M.; Parsons, K.; Mittal, D.; Lopez, J.A.; Srihari, S.; Khanna, K.K. Targeted therapies for triple-negative breast cancer: Combating a stubborn disease. Trends Pharm. Sci. 2015, 36, 822–846. [Google Scholar] [CrossRef]
- Katz, H.; Alsharedi, M. Immunotherapy in triple-negative breast cancer. Med. Oncol. 2017, 35, 13. [Google Scholar] [CrossRef]
- Gerlinger, M.; Swanton, C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br. J. Cancer 2010, 103, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Bentires-Alj, M. Breast tumor heterogeneity: Source of fitness, hurdle for therapy. Mol. Cell 2015, 60, 537–546. [Google Scholar] [CrossRef]
- Gatenby, R.; Brown, J. The evolution and ecology of resistance in cancer therapy. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, F.; De Giovanni, C.; Nanni, P.; Forni, G.; Lollini, P.L. 2011: The immune hallmarks of cancer. Cancer Immunol. Immunother. 2011, 60, 319–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzel, M.; Bovier, A.; Tuting, T. Plasticity of tumour and immune cells: A source of heterogeneity and a cause for therapy resistance? Nat. Rev. Cancer 2013, 13, 365–376. [Google Scholar] [CrossRef]
- Maley, C.C.; Aktipis, A.; Graham, T.A.; Sottoriva, A.; Boddy, A.M.; Janiszewska, M.; Silva, A.S.; Gerlinger, M.; Yuan, Y.; Pienta, K.J.; et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer 2017, 17, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Ruiz-Cabello, F.; Aptsiauri, N. Rejection versus escape: The tumor MHC dilemma. Cancer Immunol. Immunother. 2017, 66, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Caswell, D.R.; Swanton, C. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med. 2017, 15, 133. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [Green Version]
- Senovilla, L.; Vacchelli, E.; Galon, J.; Adjemian, S.; Eggermont, A.; Fridman, W.H.; Sautes-Fridman, C.; Ma, Y.; Tartour, E.; Zitvogel, L.; et al. Trial watch: Prognostic and predictive value of the immune infiltrate in cancer. Oncoimmunology 2012, 1, 1323–1343. [Google Scholar] [CrossRef] [Green Version]
- Dieci, M.V.; Mathieu, M.C.; Guarneri, V.; Conte, P.; Delaloge, S.; Andre, F.; Goubar, A. Prognostic and predictive value of tumor-infiltrating lymphocytes in two phase III randomized adjuvant breast cancer trials. Ann. Oncol. 2015, 26, 1698–1704. [Google Scholar] [CrossRef]
- Peyvandi, S.; Lan, Q.; Lorusso, G.; Rüegg, C. Chemotherapy-induced immunological breast cancer dormancy: A new function for old drugs? J. Cancer Metastasis Treat. 2019, 5, 44. [Google Scholar] [CrossRef] [Green Version]
- Pruneri, G.; Vingiani, A.; Bagnardi, V.; Rotmensz, N.; De Rose, A.; Palazzo, A.; Colleoni, A.M.; Goldhirsch, A.; Viale, G. Clinical validity of tumor-infiltrating lymphocytes analysis in patients with triple-negative breast cancer. Ann. Oncol. 2016, 27, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Burnet, M. Cancer: A biological approach. III. Viruses associated with neoplastic conditions. IV. Practical applications. Br. Med. J. 1957, 1, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L. Delayed hypersensitivity in health and disease. In Cellular and Humoral Aspects of the Hypersensitive States; PB Hoeber: New York, NY, USA, 1959. [Google Scholar]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Brackstone, M.; Townson, J.L.; Chambers, A.F. Tumour dormancy in breast cancer: An update. Breast Cancer Res. 2007, 9. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Senovilla, L.; Galluzzi, L.; André, F.; Zitvogel, L. Natural and therapy-induced immunosurveillance in breast cancer. Nat. Med. 2015, 21, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxevanis, C.N.; Perez, S.A. Cancer Dormancy: A Regulatory Role for Endogenous Immunity in Establishing and Maintaining the Tumor Dormant State. Vaccines 2015, 3, 597–619. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.F.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M.; et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005, 6, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidwell, B.N.; Slaney, C.Y.; Withana, N.P.; Forster, S.; Cao, Y.; Loi, S.; Andrews, D.; Mikeska, T.; Mangan, N.E.; Samarajiwa, S.A.; et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 2012, 18, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remedios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- Legrier, M.E.; Bieche, I.; Gaston, J.; Beurdeley, A.; Yvonnet, V.; Deas, O.; Thuleau, A.; Chateau-Joubert, S.; Servely, J.L.; Vacher, S.; et al. Activation of IFN/STAT1 signalling predicts response to chemotherapy in oestrogen receptor-negative breast cancer. Br. J. Cancer 2016, 114, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Budhwani, M.; Mazzieri, R.; Dolcetti, R. Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-tumor Immunity to Resistance. Front. Oncol. 2018, 8, 322. [Google Scholar] [CrossRef] [Green Version]
- Lan, Q.; Peyvandi, S.; Duffey, N.; Huang, Y.T.; Barras, D.; Held, W.; Richard, F.; Delorenzi, M.; Sotiriou, C.; Desmedt, C.; et al. Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene 2019, 38, 2814–2829. [Google Scholar] [CrossRef]
- Kirschner, D.; Panetta, J.C. Modeling immunotherapy of the tumor-immune interaction. J. Math. Biol. 1998, 37, 235–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Pillis, L.G.; Gu, W.; Radunskaya, A.E. Mixed immunotherapy and chemotherapy of tumors: Modeling, applications and biological interpretations. J. Biol. 2006, 238, 841–862. [Google Scholar] [CrossRef] [PubMed]
- Kareva, I.; Berezovskaya, F. Cancer immunoediting: A process driven by metabolic competition as a predator-prey-shared resource type model. J. Biol. 2015, 380, 463–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arabameri, A.; Asemani, D.; Hadjati, J. A structural methodology for modeling immune-tumor interactions including pro- and anti-tumor factors for clinical applications. Math. Biosci. 2018, 304, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Eftimie, R.; Bramson, J.L.; Earn, D.J.D. Interactions Between the immune system and cancer: A brief review of non-spatial mathematical models. Bull. Math. Biol. 2010, 73, 2–32. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, K.P. A review of mathematical models of cancer-immune interactions in the context of tumor dormancy. In Systems Biology of Tumor Dormancy. Advances in Experimental Medicine and Biology; Enderling, H.A.N., Hlatky, L., Eds.; Springer: New York, NY, USA, 2013; Volume 734, pp. 201–234. [Google Scholar]
- Wilkie, K.P.; Hahnfeldt, P. Mathematical models of immune-induced cancer dormancy and the emergence of immune evasion. Interface Focus 2013, 3, 20130010. [Google Scholar] [CrossRef] [Green Version]
- Wilkie, K.P.; Hahnfeldt, P. Tumor-Immune Dynamics Regulated in the Microenvironment Inform the Transient Nature of Immune-Induced Tumor Dormancy. Cancer Res. 2013, 73, 3534–3544. [Google Scholar] [CrossRef] [Green Version]
- Page, K.; Uhr, J.W. Mathematical models of cancer dormancy. Leuk Lymphoma 2005, 46, 313–327. [Google Scholar] [CrossRef]
- De Pillis, L.G.; Radunskaya, A. A mathematical model of immune response to tumor invasion. In Computational Fluid and Solid Mechanics 2003; Elsevier Science Ltd.: Cambridge, MA, USA, 2003; pp. 1661–1668. [Google Scholar]
- De Pillis, L.G.; Radunskaya, A.E.; Wiseman, C.L. A Validated Mathematical Model of Cell-Mediated Immune Response to Tumor Growth. Cancer Res. 2005, 65, 7950–7958. [Google Scholar] [CrossRef] [Green Version]
- Pulaski, B.A.; Ostrand-Rosenberg, S. Mouse 4T1 breast tumor model. Curr. Protoc. Immunol. 2001. [Google Scholar] [CrossRef]
- Tao, K.; Fang, M.; Alroy, J.; Sahagian, G.G. Imagable 4T1 model for the study of late stage breast cancer. BMC Cancer 2008, 8, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, V.; Makalkin, I.; Taylor, M.; Perelson, A. Nonlinear dynamics of immunogenic tumors: Parameter estimation and global bifurcation analysis. Bull. Math. Biol. 1994, 56, 295–321. [Google Scholar] [CrossRef]
- Shariatpanahi, S.P.; Shariatpanahi, S.P.; Madjidzadeh, K.; Hassan, M.; Abedi-Valugerdi, M. Mathematical modeling of tumor-induced immunosuppression by myeloid-derived suppressor cells: Implications for therapeutic targeting strategies. J. Theor. Biol. 2018, 442, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Benzekry, S.; Lamont, C.; Beheshti, A.; Tracz, A.; Ebos, J.M.; Hlatky, L.; Hahnfeldt, P. Classical mathematical models for description and prediction of experimental tumor growth. PLoS Comput. Biol. 2014, 10, e1003800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregorio, A.C.; Fonseca, N.A.; Moura, V.; Lacerda, M.; Figueiredo, P.; Simoes, S.; Dias, S.; Moreira, J.N. Inoculated Cell Density as a Determinant Factor of the Growth Dynamics and Metastatic Efficiency of a Breast Cancer Murine Model. PLoS ONE 2016, 11, e0165817. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, A.; Cerwenka, A. Natural killer cells and solid tumors. J. Innate Immun. 2011, 3, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Palucka, A.K.; Coussens, L.M. The Basis of Oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [Green Version]
- Rivera, A.; Fu, X.; Tao, L.; Zhang, X. Modification of a popular syngeneic murine mammary tumor model for immunotherapy studies. ISRN Immunol. 2011, 2011, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, A.; Brooks, M.W.; Houshyar, S.; Reinhardt, F.; Ardolino, M.; Fessler, E.; Chen, M.B.; Krall, J.A.; DeCock, J.; Zervantonakis, I.K.; et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discov. 2016, 6, 630–649. [Google Scholar] [CrossRef] [Green Version]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front. Immunol. 2019, 10, 3038. [Google Scholar] [CrossRef]
- Jamieson, A.M.; Isnard, P.; Dorfman, J.R.; Coles, M.C.; Raulet, D.H. Turnover and proliferation of NK cells in steady state and lymphopenic conditions. J. Immunol. 2004, 172, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Cha, E.; Graham, L.; Manjili, M.H.; Bear, H.D. IL-7 + IL-15 are superior to IL-2 for the ex vivo expansion of 4T1 mammary carcinoma-specific T cells with greater efficacy against tumors in vivo. Breast Cancer Res. Treat. 2010, 122, 359–369. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Spector, N.L.; Blackwell, K.L. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2-positive breast cancer. J. Clin. Oncol. 2009, 27, 5838–5847. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J. Treatment of HER2-overexpressing breast cancer. Ann. Oncol. 2010, 21 (Suppl. 7), vii36–vii40. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Hasmann, M.; Leyland-Jones, B. Molecular determinants of trastuzumab efficacy: What is their clinical relevance? Cancer Treat. Rev. 2013, 39, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Cabo, M.; Servitja, S.; Tusquets, I.; Martinez-Garcia, M.; Rovira, A.; Rojo, F.; Albanell, J.; Lopez-Botet, M. Interplay between Natural Killer Cells and Anti-HER2 Antibodies: Perspectives for Breast Cancer Immunotherapy. Front. Immunol. 2017, 8, 1544. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Lao, X.; Pan, Q.; Ning, N.; Yet, J.; Xu, Y.; Li, S.; Chang, A.E. Adoptive transfer of tumor reactive B cells confers host T-cell immunity and tumor regression. Clin. Cancer Res. 2011, 17, 4987–4995. [Google Scholar] [CrossRef] [Green Version]
- Rogers, D.; Vila-Leahey, A.; Pessoa, A.C.; Oldford, S.; Marignani, P.A.; Marshall, J.S. Ranitidine Inhibition of Breast Tumor Growth Is B Cell Dependent and Associated with an Enhanced Antitumor Antibody Response. Front. Immunol. 2018, 9, 1894. [Google Scholar] [CrossRef] [Green Version]
- Ragonnaud, E.; Moritoh, K.; Bodogai, M.; Gusev, F.; Garaud, S.; Chen, C.; Wang, X.; Baljinnyam, T.; Becker, K.G.; Maul, R.W.; et al. Tumor-Derived Thymic Stromal Lymphopoietin Expands Bone Marrow B-cell Precursors in Circulation to Support Metastasis. Cancer Res. 2019, 79, 5826–5838. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Lu, L.; Xia, Y.; Dai, F.; Wang, Y.; Bao, Y.; Lundy, S.K.; Ito, F.; Pan, Q.; Zhang, X.; et al. Antitumor effector B cells directly kill tumor cells via the Fas/FasL pathway and are regulated by IL-10. Eur. J. Immunol. 2015, 45, 999–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berryman, A.A. The orgins and evolution of predator-prey theory. Ecology 1992, 73, 1530–1535. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Chen, L. Modeling and analysis of a predator–prey model with disease in the prey. Math. Biosci. 2001, 171, 59–82. [Google Scholar] [CrossRef]
- d’Onofrio, A. A general framework for modeling tumor-immune system competition and immunotherapy: Mathematical analysis and biomedical inferences. Phys. D Nonlinear Phenom. 2005, 208, 220–235. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Ravindranathan, S.; Nguyen, K.G.; Kurtz, S.L.; Frazier, H.N.; Smith, S.G.; Koppolu, B.P.; Rajaram, N.; Zaharoff, D.A. Tumor-derived granulocyte colony-stimulating factor diminishes efficacy of breast tumor cell vaccines. Breast Cancer Res. 2018, 20, 126. [Google Scholar] [CrossRef]
- Tsai, S.C.; Gansbacher, B.; Tait, L.; Miller, F.R.; Heppner, G.H. Induction of antitumor immunity by interleukin-2 gene-transduced mouse mammary tumor cells versus transduced mammary stromal fibroblasts. J. Natl. Cancer Inst. 1993, 85, 546–553. [Google Scholar] [CrossRef]
- Majumdar, A.S.; Zolotorev, A.; Samuel, S.; Tran, K.; Vertin, B.; Hall-Meier, M.; Antoni, B.A.; Adeline, E.; Philip, M.; Philip, R. Efficacy of herpes simplex virus thymidine kinase in combination with cytokine gene therapy in an experimental metastatic breast cancer model. Cancer Gene 2000, 7, 1086–1099. [Google Scholar] [CrossRef] [Green Version]
- Brockstedt, D.G.; Diagana, M.; Zhang, Y.; Tran, K.; Belmar, N.; Meier, M.; Yang, A.; Boissiere, F.; Lin, A.; Chiang, Y. Development of Anti-tumor Immunity against a Non-immunogenic Mammary Carcinoma through in Vivo Somatic GM-CSF, IL-2, and HSVtk Combination Gene Therapy. Mol. Ther. 2002, 6, 627–636. [Google Scholar] [CrossRef]
- De Giovanni, C.; Nicoletti, G.; Landuzzi, L.; Astolfi, A.; Croci, S.; Comes, A.; Ferrini, S.; Meazza, R.; Iezzi, M.; Di Carlo, E.; et al. Immunoprevention of HER-2/neu transgenic mammary carcinoma through an interleukin 12-engineered allogeneic cell vaccine. Cancer Res. 2004, 64, 4001–4009. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, C.L.; Kharazi, A. Objective clinical regression of metastatic breast cancer in disparate sites after use of whole-cell vaccine genetically modified to release sargramostim. Breast J. 2006, 12, 475–480. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Units | Estimated Value | Description | Source |
|---|---|---|---|---|
| Day−1 | 0.38 × 10−1 | Proliferative (WT 4T1) tumor growth rate | [71] | |
| Day−1 | 0.174 × 10−1 | Quiescent (MR20) tumor growth rate | estimated from [51] | |
| Cell | 109 | Maximum tumor size | estimated | |
| Dimensionless | 0.3 | Proportion coefficient of NK cell cytotoxicity on tumor cells | estimated | |
| Cell−1 Day−1 | = 1.56 × 10−8 | Proliferative tumor cells kill rate by NK cells | estimated from [51] | |
| Cell−1 Day−1 | 5.2 × 10−8 | Quiescent tumor cells kill rate by NK cells | estimated from [51] | |
| Cell−1 Day−1 | 0.21 × 10−7 | Proliferative tumor cells kill rate by CD8+ T cells | estimated from [51] | |
| Cell−1 Day−1 | 2.8 × 10−7 | Quiescent tumor cells kill rate by CD8+ T cells | estimated from [51] | |
| Cell Day−1 | 1.3 × 104 | Constant source of NK cells. | [62,65] | |
| Cell−1 Day−1 | = 0.36 × 10−7 | CD8+ T cells stimulation coefficient due to proliferative tumor-NK cells interaction | estimated | |
| Cell−1 Day−1 | 1.2 × 10−7 | CD8+ T cells stimulation coefficient due to quiescent tumor-NK cells interaction | [62,66] estimated | |
| Day−1 | 2.5 × 10−2 | Maximum NK cell recruitment rate by tumor cells | [62,66] | |
| Cell2 | 2.02 × 107 | Steepness coefficient of the NK cell recruitment curve | [62,66] | |
| Day−1 | 4.12 × 10−2 | Death rate of NK cells | [62,66,74] | |
| Cell−1 Day−1 | 1.8 × 10−8 | NK cell inactivation rate by tumor cells | estimated from [51] | |
| Day−1 | 10 × 10−2 | Maximum CD8+ T-cell recruitment rate by tumor cells | [66] | |
| Cell2 | 2.02 × 107 | Steepness coefficient of the CD8+ T-cell recruitment curve | [62,65,66] | |
| Day−1 | 2 × 10−2 | Death rate of CD8+ T cells | [62,66] | |
| Cell−1 Day−1 | 2.1 × 10−8 | CD8+ T-cell inactivation rate by proliferative tumor cells | estimated from [51] | |
| Cell−1 Day−1 | 1.7 × 10−12 | CD8+ T-cell inactivation rate by quiescent tumor cells | estimated from [51] |
| Regions of Phase Map | Quiescent Subpopulation | Proliferative Subpopulation | Heterogeneous Tumor State |
|---|---|---|---|
| Region 1 | Eliminated | Eliminated | Eliminated |
| Region 2 | Dormant | Eliminated | Dormant |
| Region 3 | Eliminated | Escaped | Escaped |
| Region 4 | Eliminated | Eliminated | Eliminated |
| Region 5 | Eliminated | Escaped | Escaped |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehdizadeh, R.; Shariatpanahi, S.P.; Goliaei, B.; Peyvandi, S.; Rüegg, C. Dormant Tumor Cell Vaccination: A Mathematical Model of Immunological Dormancy in Triple-Negative Breast Cancer. Cancers 2021, 13, 245. https://doi.org/10.3390/cancers13020245
Mehdizadeh R, Shariatpanahi SP, Goliaei B, Peyvandi S, Rüegg C. Dormant Tumor Cell Vaccination: A Mathematical Model of Immunological Dormancy in Triple-Negative Breast Cancer. Cancers. 2021; 13(2):245. https://doi.org/10.3390/cancers13020245
Chicago/Turabian StyleMehdizadeh, Reza, Seyed Peyman Shariatpanahi, Bahram Goliaei, Sanam Peyvandi, and Curzio Rüegg. 2021. "Dormant Tumor Cell Vaccination: A Mathematical Model of Immunological Dormancy in Triple-Negative Breast Cancer" Cancers 13, no. 2: 245. https://doi.org/10.3390/cancers13020245
APA StyleMehdizadeh, R., Shariatpanahi, S. P., Goliaei, B., Peyvandi, S., & Rüegg, C. (2021). Dormant Tumor Cell Vaccination: A Mathematical Model of Immunological Dormancy in Triple-Negative Breast Cancer. Cancers, 13(2), 245. https://doi.org/10.3390/cancers13020245