Homologous Recombination Repair Mechanisms in Serous Endometrial Cancer

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient and Tumor Characteristics
2.2. Global Copy-Number Analyses
2.3. Analyses of HRD and DNA Double-Strand Break Repair Genes
2.4. Gene Amplifications and Deletions
2.5. Targeted Sequencing
2.6. Copy-Number Signatures
2.7. Immunohistochemistry (IHC)
2.8. Statistics
3. Results
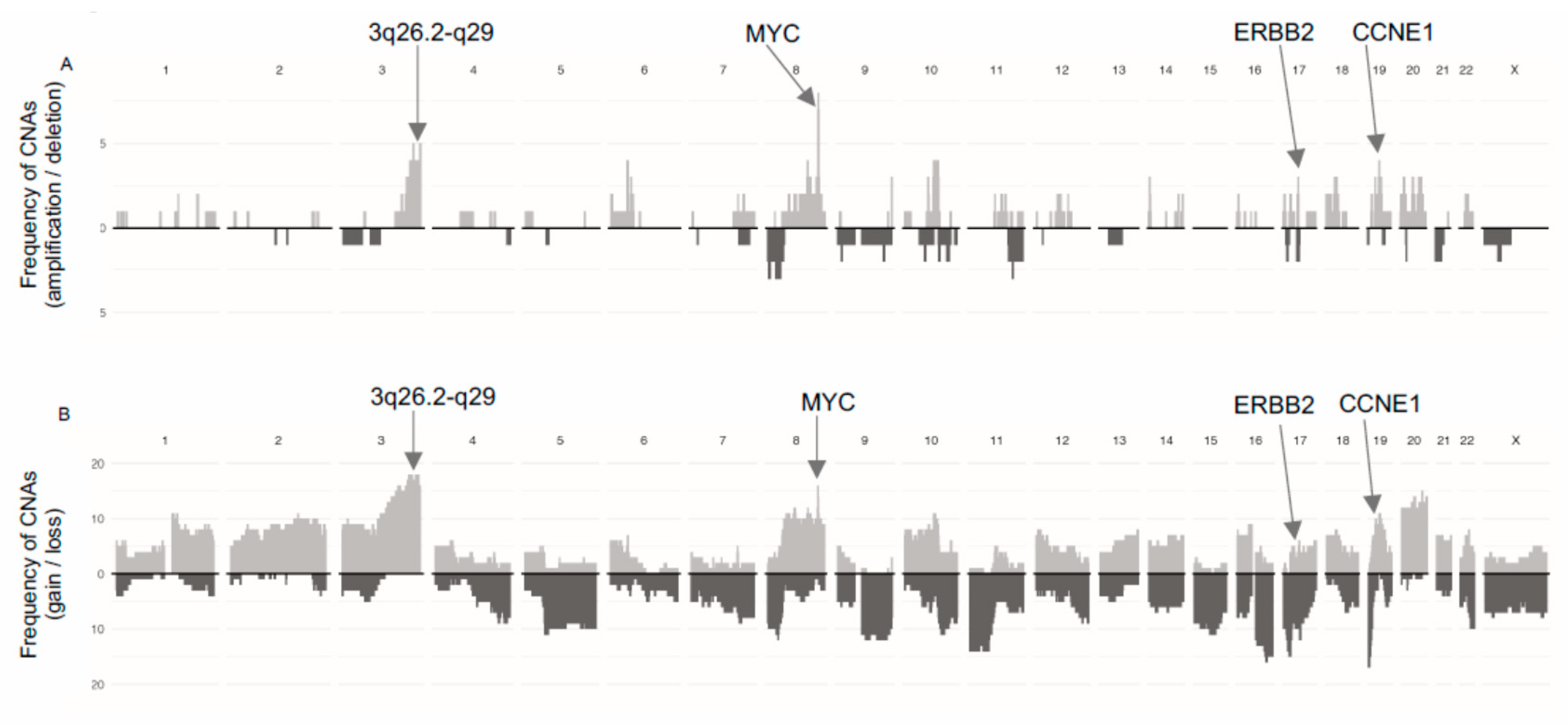
3.1. Global Copy-Number Analyses
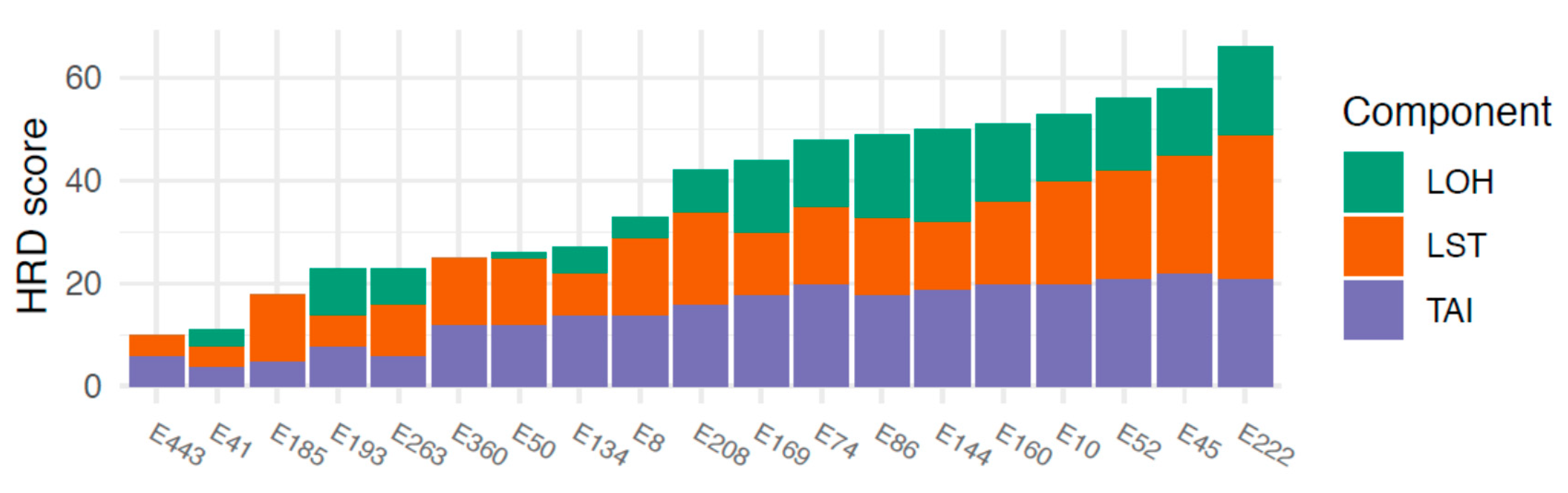
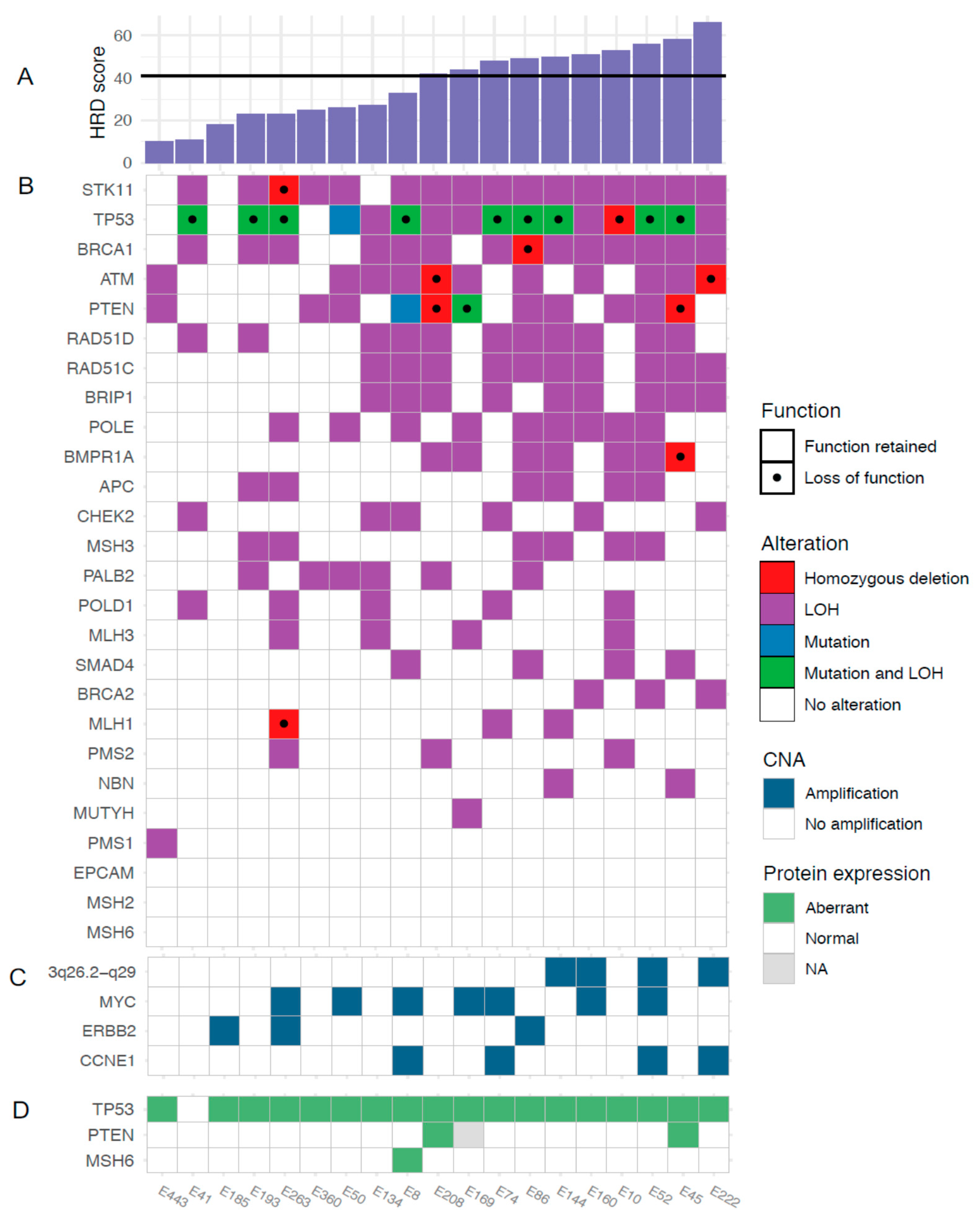
3.2. HRD and HRD-Associated Genes
- BRCA1 loss; mean HRD score 43 (loss) vs. 28 (no loss), (p = 0.048)
- BRCA1 LOH; mean 42 (LOH) vs. 29 (no LOH), (p = 0.12)
- BRCA2 loss; mean 50 vs. 34 (p = 0.18)
- BRCA2 LOH; mean 58 vs. 34 (p = 0.0070)
- RAD51C loss; mean 51 vs. 28 (p = 0.0010)
- RAD51C LOH; mean 48 vs. 26 (p = 0.0021).
3.3. HRD and NHEJ-Associated Genes
3.4. Gene Amplifications
3.5. Targeted Sequencing
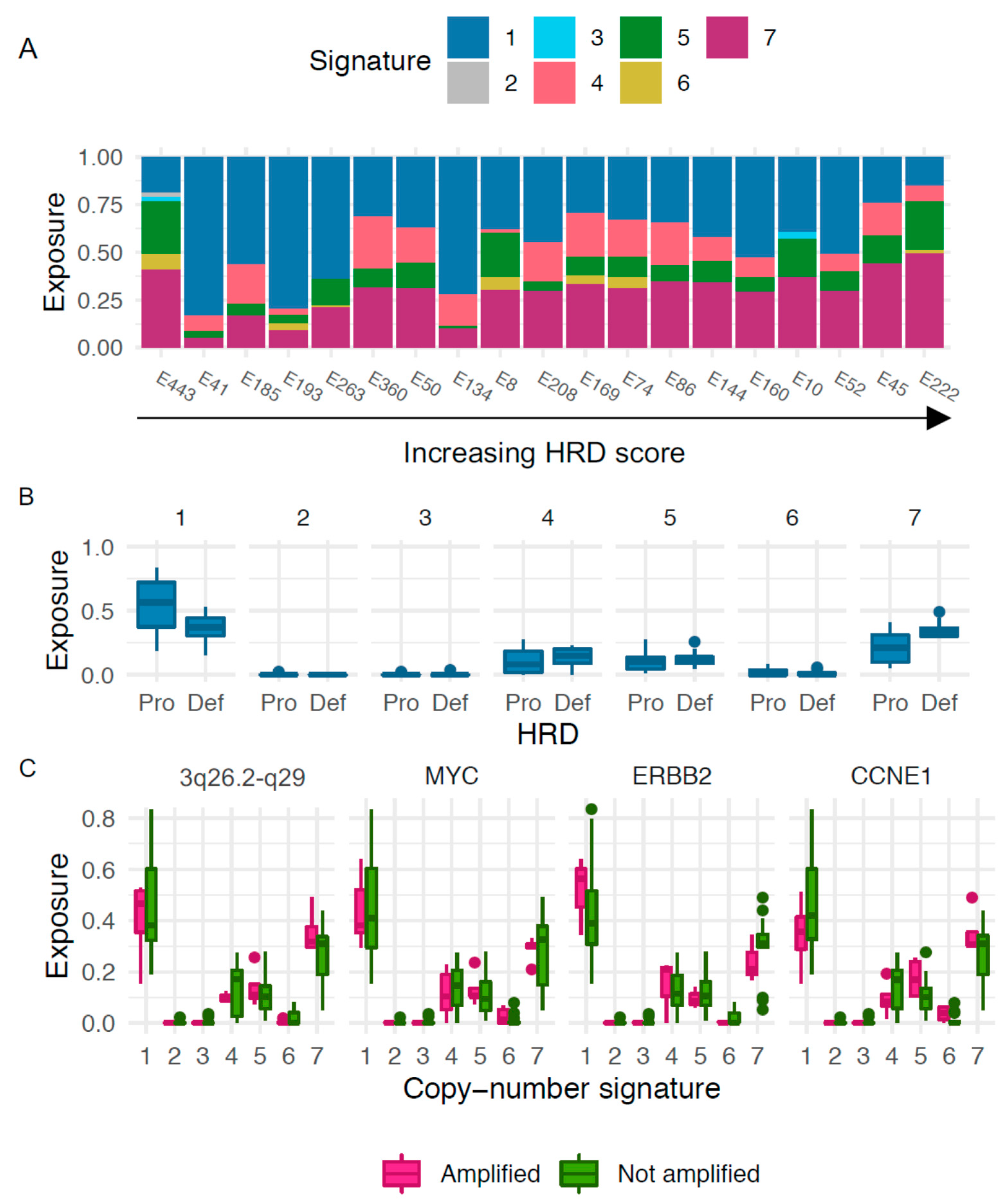
3.6. Copy-Number Signatures
3.7. Immunohistochemistry
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BER | Base Excision Repair |
| CI | Confidence Interval |
| CNA | Copy-Number Alteration |
| EC | Endometrial cancer |
| FGA | Fraction of Genome Altered |
| FIGO | International Federation of Gynecology and Obstetrics |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| GISTIC | Genomic Identification of Significant Targets in Cancer |
| H&E | Hematoxylin & Eosin |
| HGSOC | High-Grade Serous Ovarian Cancer |
| HR | Homologous Recombination Repair |
| HRD | Homologous Recombination Repair Deficiency |
| IHC | Immunohistochemistry |
| LOH | Loss of Heterozygosity |
| LST | Large-Scale State Transitions |
| MMR | Mismatch Repair |
| NER | Nucleotide Excision Repair |
| NHEJ | Non-Homologous End Joining |
| PARP | Poly (ADP-Ribose) Polymerase |
| SEC | Serous Endometrial Cancer |
| SI | Supplementary Information |
| SNP | Single Nucleotide Polymorphism |
| TAI | Telomeric Allelic Imbalance |
Appendix A
Apendix A. 1. Methods
Apendix A. 1. 1. Targeted Sequencing
Apendix A. 1. 2. Gene Amplifications and Deletions
Apendix A. 2. Extended Results
Targeted Sequencing and Immunohistochemistry
- For two cases displaying aberrant p53 overexpression without corresponding TP53 mutation, mutations of unknown significance, likely causing the aberrant IHC staining, were found
- For two cases displaying aberrant p53 overexpression without corresponding TP53 mutation, mutations were detected but were filtered out due to poor quality, likely because of high normal cell contamination
- For one case displaying aberrant p53 overexpression without corresponding TP53 mutation, amplification of c-Myc and loss of MDM2, regulators of p53 stability, were found
- For three cases displaying aberrant p53 overexpression without corresponding TP53 mutation, gain of c-Myc was found
- For one case, not regarded as p53 aberrant by IHC, a missense mutation was found which may confound IHC interpretation
- For one case with0020a missense PTEN mutation but retained protein expression in IHC, no LOH in the gene was detected which means that gene function may be retained
- For one case with a missense PTEN mutation data on protein expression was missing
- For two cases with loss of PTEN protein staining by IHC but no corresponding mutations, homozygous deletions in this region were found which may affect protein expression
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, A.C.; Cheung, M.K.; Osann, K.; Chen, L.; Teng, N.N.; Longacre, T.A.; Powell, M.A.; Hendrickson, M.R.; Kapp, D.S.; Chan, J.K. Uterine papillary serous and clear cell carcinomas predict for poorer survival compared to grade 3 endometrioid corpus cancers. Br. J. Cancer 2006, 94, 642–646. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robert-son, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Talhouk, A.; McConechy, M.K.; Leung, S.; Li-Chang, H.H.; Kwon, J.S.; Melnyk, N.; Yang, W.; Senz, J.; Boyd, N.F.; Karnezis, A.N.; et al. A clinically applicable molecular-based classification for endometrial cancers. Br. J. Cancer 2015, 113, 299–310. [Google Scholar] [CrossRef] [Green Version]
- Stelloo, E.; Bosse, T.; Nout, R.A.; Mackay, H.J.; Church, D.N.; Nijman, H.W.; Leary, A.; Edmondson, R.; Powell, M.E.; Crosbie, E.J.; et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod. Pathol. 2015, 28, 836–844. [Google Scholar] [CrossRef] [Green Version]
- Kommoss, S.; McConechy, M.; Leung, S.; Bunz, A.; Magrill, J.; Britton, H.; Grevenkamp, F.; Karnezis, A.; Yang, W.; Lum, A.; et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann. Oncol. 2018, 29, 1180–1188. [Google Scholar] [CrossRef]
- de Jonge, M.M.; Mooyaart, A.L.; Vreeswijk, M.P.; de Kroon, C.D.; van Wezel, T.; van Asperen, C.J.; Smit, V.T.; Dekkers, O.M.; Bosse, T. Linking uterine serous carcinoma to BRCA1/2-associated cancer syndrome: A meta-analysis and case report. Eur. J. Cancer 2017, 72, 215–225. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homolo-gous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.D.; Carey, M.; A Meyer, L.; Smithmccune, K.; Broaddus, R.R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [Green Version]
- Popova, T.; Manie, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 in-activation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.-Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA-Damaging Agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Kroeger, P.T., Jr.; Drapkin, R. Pathogenesis and heterogeneity of ovarian cancer. Curr. Opin. Obstet. Gynecol. 2017, 29, 26–34. [Google Scholar] [CrossRef]
- Takaya, H.; Nakai, H.; Takamatsu, S.; Mandai, M.; Matsumura, N. Homologous recombination deficiency status-based clas-sification of high-grade serous ovarian carcinoma. Sci. Rep. 2020, 10, 2757. [Google Scholar] [CrossRef] [Green Version]
- Fleury, H.; Carmona, E.; Morin, V.G.; Meunier, L.; Masson, J.-Y.; Tonin, P.N.; Provencher, D.; Mes-Masson, A.-M. Cumulative defects in DNA repair pathways drive the PARP inhibitor response in high-grade serous epithelial ovarian cancer cell lines. Oncotarget 2016, 8, 40152–40168. [Google Scholar] [CrossRef]
- de Jonge, M.M.; Ritterhouse, L.L.; de Kroon, C.D.; Vreeswijk, M.P.G.; Segal, J.P.; Puranik, R.; Hollema, H.; Rookus, M.A.; van Asperen, C.J.; van Leeuwen, F.E.; et al. Germline BRCA-Associated Endometrial Carcinoma is a Distinct Clinicopathologic Entity. Clin. Cancer Res. 2019, 25, 7517–7526. [Google Scholar] [CrossRef] [Green Version]
- Shu, C.A.; Pike, M.C.; Jotwani, A.R.; Friebel, T.M.; Soslow, R.A.; Levine, D.A.; Nathanson, K.L.; Konner, J.A.; Arnold, A.G.; Bogomolniy, F.; et al. Uterine Cancer After Risk-Reducing Salpingo-oophorectomy without Hysterectomy in Women with BRCA Mutations. JAMA Oncol. 2016, 2, 1434–1440. [Google Scholar] [CrossRef] [Green Version]
- Frimer, M.; Levano, K.S.; Rodriguez-Gabin, A.; Wang, Y.; Goldberg, G.L.; Horwitz, S.B.; Hou, J.Y. Germline mutations of the DNA repair pathways in uterine serous carcinoma. Gynecol. Oncol. 2016, 141, 101–107. [Google Scholar] [CrossRef]
- de Jonge, M.M.; Auguste, A.; van Wijk, L.M.; Schouten, P.C.; Meijers, M.; Ter Haar, N.T.; Smit, V.; Nout, R.A.; Glaire, M.A.; Church, D.N.; et al. Frequent Homologous Recombination Deficiency in High-grade Endometrial Carcinomas. Clin. Cancer Res. 2019, 25, 1087–1097. [Google Scholar] [CrossRef] [Green Version]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homolo-gous Recombination-Related Gene Mutations across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2, 1–13. [Google Scholar]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Ashley, C.W.; Paula, A.D.C.; Kumar, R.; Mandelker, D.; Pei, X.; Riaz, N.; Reis-Filho, J.S.; Weigelt, B. Analysis of mutational signatures in primary and metastatic endometrial cancer reveals distinct patterns of DNA repair defects and shifts during tumor progression. Gynecol. Oncol. 2019, 152, 11–19. [Google Scholar] [CrossRef] [PubMed]
- MacIntyre, G.; Goranova, T.; De Silva, D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.-A.; Hanif, A.; Wilson, C.; et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef]
- Kurman, R.J.; Carcangiu, M.L.; Herrington, C.S.; Young, R.H. WHO Classification of Tumours of the Female Reproductive Organs, 4th ed.; IARC Press: Lyon, France, 2014; Volume 6. [Google Scholar]
- Creasman, W.T. Revised FIGO staging for carcinoma of the endometrium. Int. J. Gynecol. Obstet. 2009, 105, 109. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.S.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat. Commun. 2017, 8, 857. [Google Scholar] [CrossRef] [PubMed]
- Sishc, B.J.; Davis, A.J. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers 2017, 9, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) in-hibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: https://www.R-project.org/ (accessed on 10 November 2020).
- Salvesen, H.B.; Carter, S.L.; Mannelqvist, M.; Dutt, A.; Getz, G.; Stefansson, I.M.; Raeder, M.B.; Sos, M.L.; Engelsen, I.B.; Trovik, J.; et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase ac-tivation. Proc. Natl. Acad. Sci. USA 2009, 106, 4834–4839. [Google Scholar] [CrossRef] [Green Version]
- Hussenet, T.; Dali, S.; Exinger, J.; Monga, B.; Jost, B.; Dembélé, D.; Martinet, N.; Thibault-Carpentier, C.; Huelsken, J.; Brambilla, E.; et al. SOX2 Is an Oncogene Activated by Recurrent 3q26.3 Amplifications in Human Lung Squamous Cell Carcinomas. PLoS ONE 2010, 5, e8960. [Google Scholar] [CrossRef] [Green Version]
- Fields, A.P.; Justilien, V.; Murray, N.R. The chromosome 3q26 OncCassette: A multigenic driver of human cancer. Adv. Biol. Regul. 2016, 60, 47–63. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Tweddle, D.A. p53, SKP2, and DKK3 as MYCN Target Genes and Their Potential Therapeutic Significance. Front. Oncol. 2012, 2, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Aimono, E.; Tanishima, S.; Imai, M.; Nagatsuma, A.K.; Hayashi, H.; Yoshimura, Y.; Nakayama, K.; Kyo, S.; Nishihara, H. Olaparib Monotherapy for BRIP1-Mutated High-Grade Serous Endometrial Cancer. JCO Precis. Oncol. 2020, 283–290. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar]
- Norquist, B.M.; Brady, M.F.; Harrell, M.I.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Burger, R.A.; Tewari, K.S.; et al. Mutations in Homologous Recombination Genes and Outcomes in Ovarian Carcinoma Patients in GOG 218: An NRG Oncology/Gynecologic Oncology Group Study. Clin. Cancer Res. 2018, 24, 777–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinopoulos, P.A.; Spentzos, D.; Karlan, B.Y.; Taniguchi, T.; Fountzilas, E.; Francoeur, N.; Levine, D.A.; Cannistra, S.A. Gene Expression Profile of BRCAness That Correlates with Responsiveness to Chemotherapy and with Outcome in Patients with Epithelial Ovarian Cancer. J. Clin. Oncol. 2010, 28, 3555–3561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Schultheis, A.M.; Martelotto, L.G.; De Filippo, M.R.; Piscuoglio, S.; Ng, C.K.Y.; Hussein, Y.R.; Reis-Filho, J.S.; Soslow, R.A.; Weigelt, B. TP53 Mutational Spectrum in Endometrioid and Serous Endometrial Cancers. Int. J. Gynecol. Pathol. 2016, 35, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Kanopiene, D.; Vidugiriene, J.; Valuckas, K.P.; Smailyte, G.; Uleckiene, S.; Bacher, J. Endometrial cancer and microsatellite in-stability status. Open Med. 2015, 10, 70–76. [Google Scholar]
- Auguste, A.; Genestie, C.; De Bruyn, M.; Adam, J.; Le Formal, A.; Drusch, F.; Pautier, P.; Crosbie, E.J.; Mackay, H.; Kitchener, H.C.; et al. Refinement of high-risk endometrial cancer classification using DNA damage response biomarkers: A TransPORTEC initiative. Mod. Pathol. 2018, 31, 1851–1861. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | HR Deficient * Subgroup | HR Proficient Subgroup |
|---|---|---|
| Number of patients (%) | 10 (53) | 9 (47) |
| Median HRD score (range) | 50.5 (42–66) | 23 (10–33) |
| Median age at diagnosis (range) | 76 (69–90) | 72 (56–90) |
| FIGO stage (%) | ||
| I | 6 (60) | 6 (67) |
| II | 0 | 0 |
| III | 3 (30) | 1 (11) |
| IV | 1 (10) | 2 (22) |
| Median follow-up, months (range) | 45.5 (2–60) | 48 (29–60) |
| Number deceased (%) | 2 (20) | 3 (33) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jönsson, J.-M.; Bååth, M.; Björnheden, I.; Sahin, I.D.; Måsbäck, A.; Hedenfalk, I. Homologous Recombination Repair Mechanisms in Serous Endometrial Cancer. Cancers 2021, 13, 254. https://doi.org/10.3390/cancers13020254
Jönsson J-M, Bååth M, Björnheden I, Sahin ID, Måsbäck A, Hedenfalk I. Homologous Recombination Repair Mechanisms in Serous Endometrial Cancer. Cancers. 2021; 13(2):254. https://doi.org/10.3390/cancers13020254
Chicago/Turabian StyleJönsson, Jenny-Maria, Maria Bååth, Ida Björnheden, Irem Durmaz Sahin, Anna Måsbäck, and Ingrid Hedenfalk. 2021. "Homologous Recombination Repair Mechanisms in Serous Endometrial Cancer" Cancers 13, no. 2: 254. https://doi.org/10.3390/cancers13020254
APA StyleJönsson, J. -M., Bååth, M., Björnheden, I., Sahin, I. D., Måsbäck, A., & Hedenfalk, I. (2021). Homologous Recombination Repair Mechanisms in Serous Endometrial Cancer. Cancers, 13(2), 254. https://doi.org/10.3390/cancers13020254