Overcoming Challenges for CD3-Bispecific Antibody Therapy in Solid Tumors

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Main Text
2.1. CD3-BsAbs in Hematological Malignancies
2.2. Historical Perspective and Current Status of CD3-BsAbs in Solid Cancers
2.3. Hurdles in Solid Tumors
2.4. Solutions and Opportunities
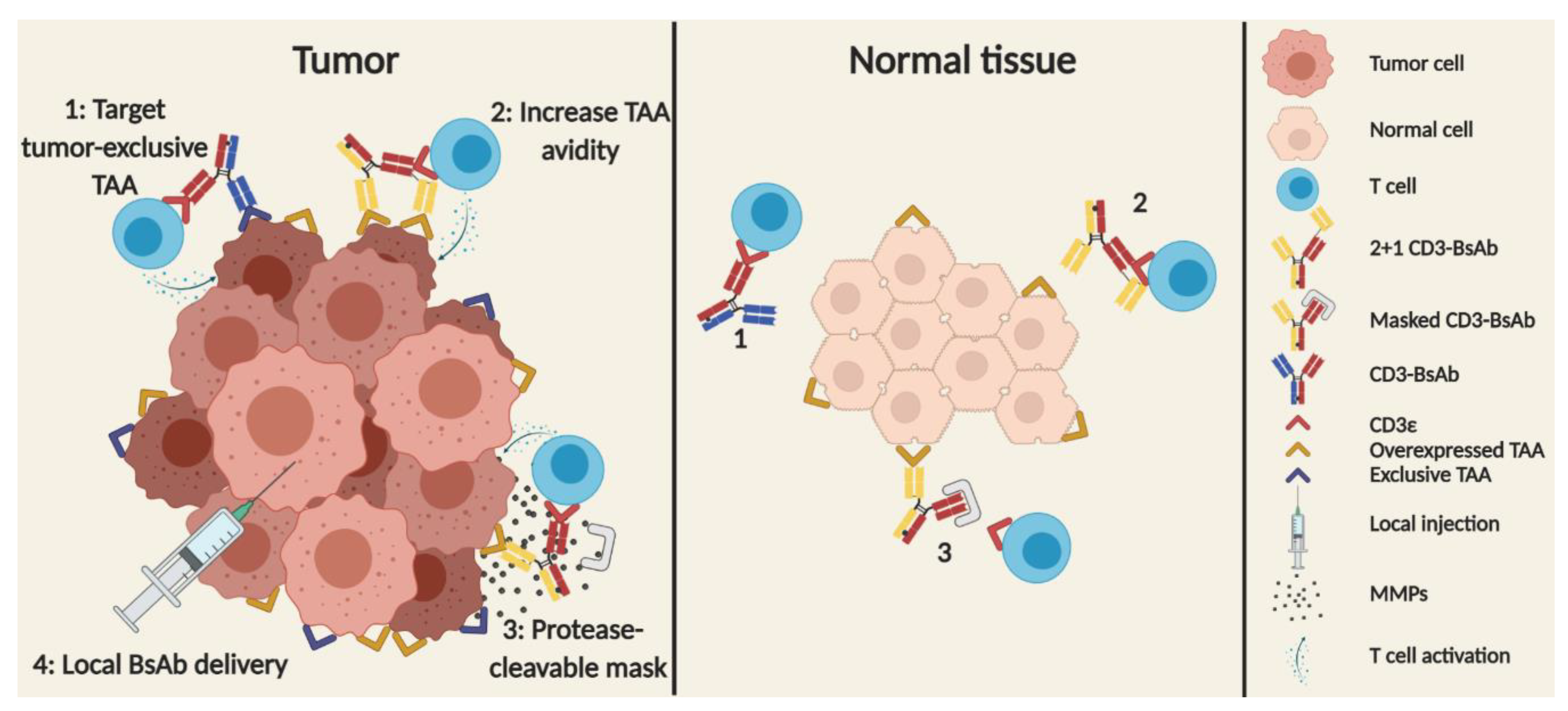
2.4.1. Mitigating of On-Target Off-Tumor Toxicities
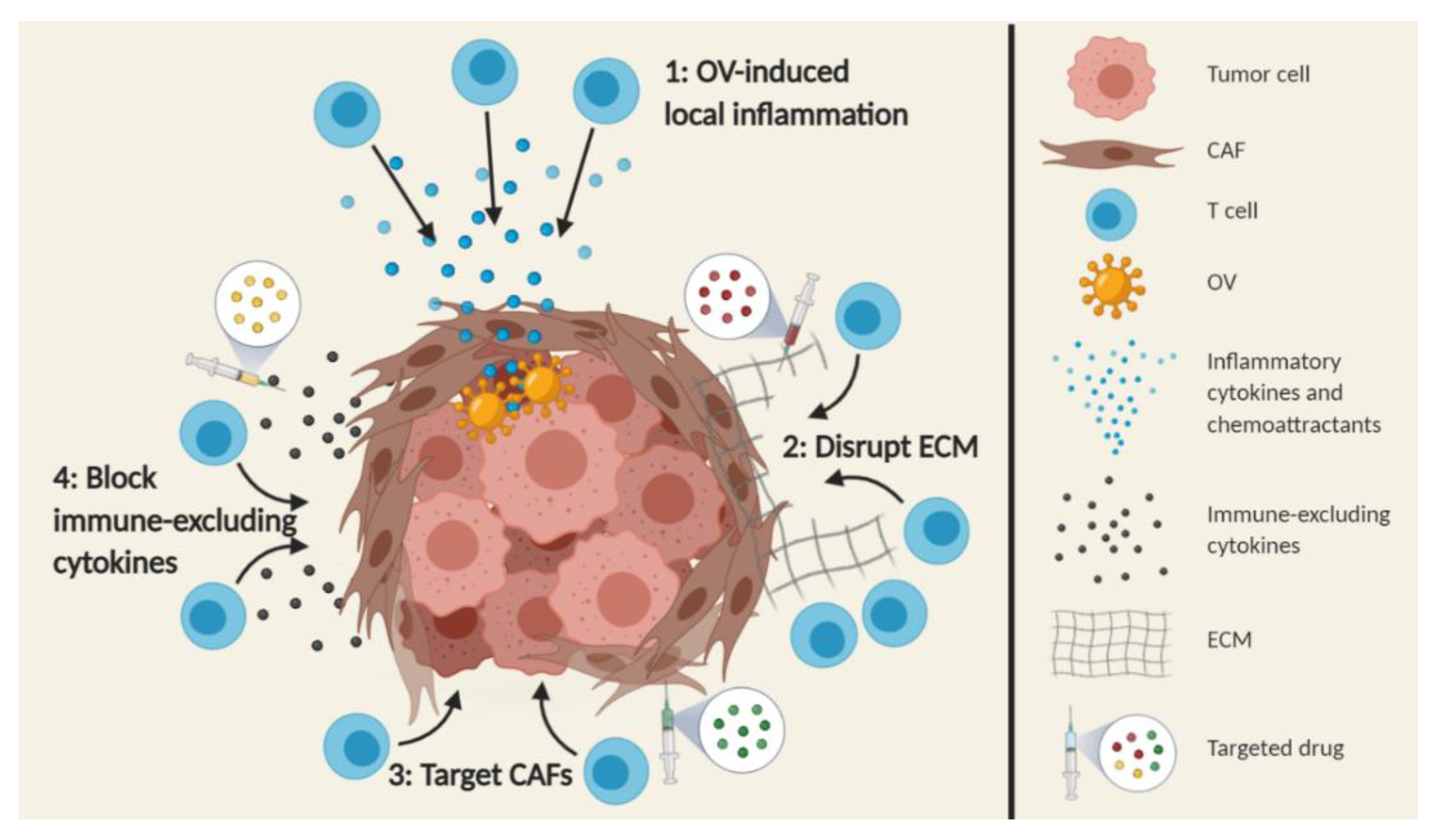
2.4.2. Increasing the Number of Intratumoral T cells
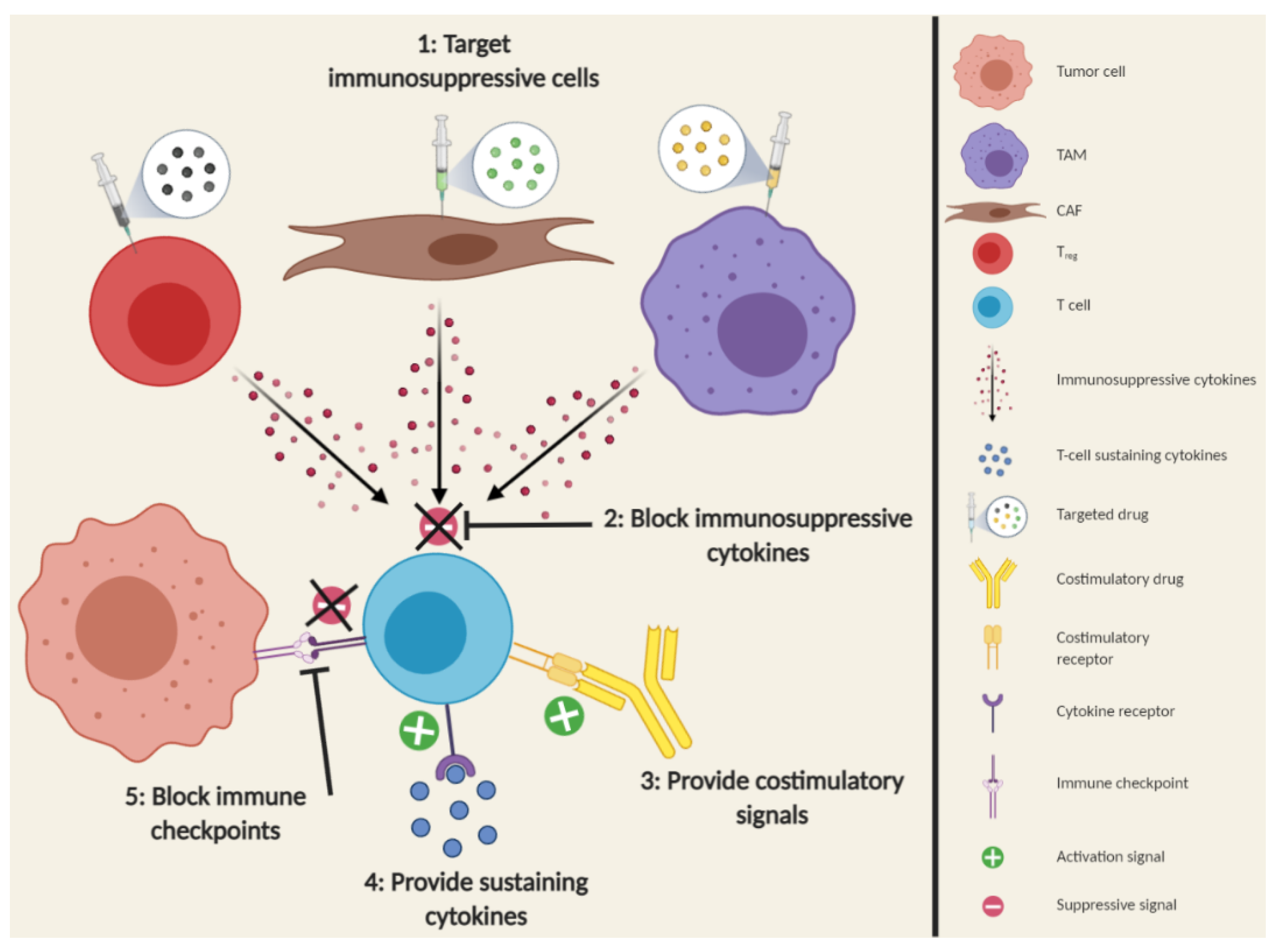
2.4.3. Improving the Quality of T-cell Responses
3. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van der Neut Kolfschoten, M.; Schuurman, J.; Losen, M.; Bleeker, W.K.; Martinez-Martinez, P.; Vermeulen, E.; den Bleker, T.H.; Wiegman, L.; Vink, T.; Aarden, L.A.; et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 2007, 317, 1554–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlen, E.; Veitonmaki, N.; Norlen, P. Bispecific antibodies in cancer immunotherapy. Ther. Adv. Vaccines Immunother. 2018, 6, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheng, M.; Guo, H.; Chen, Y.; Huse, M.; Cheung, N.K. Retargeting T cells to GD2 pentasaccharide on human tumors using Bispecific humanized antibody. Cancer Immunol. Res. 2015, 3, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Cheung, N.V. T cell engaging bispecific antibody (T-BsAb): From technology to therapeutics. Pharmacol. Ther. 2018, 182, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Guleria, I.; Gubbels Bupp, M.; Eagar, T.N.; Tang, Q.; Bour-Jordan, H.; Yagita, H.; Azuma, M.; Sayegh, M.H.; Bluestone, J.A. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J. Exp. Med. 2006, 203, 2737–2747. [Google Scholar] [CrossRef]
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef]
- Slaney, C.Y.; Wang, P.; Darcy, P.K.; Kershaw, M.H. CARs versus BiTEs: A Comparison between T Cell-Redirection Strategies for Cancer Treatment. Cancer Discov. 2018, 8, 924–934. [Google Scholar] [CrossRef] [Green Version]
- Sadelain, M. CD19 CAR T Cells. Cell 2017, 171, 1471. [Google Scholar] [CrossRef]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012, 4, 132ra153. [Google Scholar] [CrossRef] [Green Version]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Suurs, F.V.; Lub-de Hooge, M.N.; de Vries, E.G.E.; de Groot, D.J.A. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 2019, 201, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Borlak, J.; Langer, F.; Spanel, R.; Schondorfer, G.; Dittrich, C. Immune-mediated liver injury of the cancer therapeutic antibody catumaxomab targeting EpCAM, CD3 and Fcgamma receptors. Oncotarget 2016, 7, 28059–28074. [Google Scholar] [CrossRef] [PubMed]
- Clynes, R.A.; Desjarlais, J.R. Redirected T Cell Cytotoxicity in Cancer Therapy. Annu. Rev. Med. 2019, 70, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Przepiorka, D.; Ko, C.W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, G.; Dombret, H.; Chevallier, P.; Ottmann, O.G.; Goekbuget, N.; Topp, M.S.; Fielding, A.K.; Sterling, L.R.; Benjamin, J.; Stein, A.S. Complete Molecular and Hematologic Response in Adult Patients with Relapsed/Refractory (R/R) Philadelphia Chromosome-Positive B-Precursor Acute Lymphoblastic Leukemia (ALL) Following Treatment with Blinatumomab: Results from a Phase 2 Single-Arm, Multicenter Study (ALCANTARA). Blood 2015, 126, 679. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foa, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Franquiz, M.J.; Short, N.J. Blinatumomab for the Treatment of Adult B-Cell Acute Lymphoblastic Leukemia: Toward a New Era of Targeted Immunotherapy. Biologics 2020, 14, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, K.M.; Saunders, I.M.; Goodman, A.M. Characterization of relapse patterns in patients with acute lymphoblastic leukemia treated with blinatumomab. J. Oncol. Pharm. Pract. 2020. [Google Scholar] [CrossRef]
- Zhao, Y.; Aldoss, I.; Qu, C.; Crawford, J.C.; Gu, Z.; Allen, E.K.; Zamora, A.E.; Alexander, T.B.; Wang, J.; Goto, H.; et al. Tumor intrinsic and extrinsic determinants of response to blinatumomab in adults with B-ALL. Blood 2020. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.; Emadi, A.; et al. Flotetuzumab as Salvage Immunotherapy for Refractory Acute Myeloid Leukemia. Blood 2020. [Google Scholar] [CrossRef]
- Hutchings, M.; Lugtenburg, P.; Mous, R.; Clausen, M.R.; Chamuleau, M.; Linton, K.; Rule, S.; Lopez, J.S.; Oliveri, R.S.; DeMarco, D.; et al. Epcoritamab (GEN3013; DuoBody-CD3×CD20) to induce complete response in patients with relapsed/refractory B-cell non-Hodgkin lymphoma (B-NHL): Complete dose escalation data and efficacy results from a phase I/II trial. J. Clin. Oncol. 2020, 38, 8009. [Google Scholar] [CrossRef]
- Bannerji, R.; Allan, J.N.; Arnason, J.E.; Brown, J.R.; Advani, R.H.; Barnes, J.A.; Ansell, S.M.; O’Brien, S.M.; Chavez, J.; Duell, J.; et al. Clinical Activity of REGN1979, a Bispecific Human, Anti-CD20 x Anti-CD3 Antibody, in Patients with Relapsed/Refractory (R/R) B-Cell Non-Hodgkin Lymphoma (B-NHL). Blood 2019, 134, 762. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bartlett, N.L.; Assouline, S.; Yoon, S.-S.; Bosch, F.; Sehn, L.H.; Cheah, C.Y.; Shadman, M.; Gregory, G.P.; Ku, M.; et al. Mosunetuzumab Induces Complete Remissions in Poor Prognosis Non-Hodgkin Lymphoma Patients, Including Those Who Are Resistant to or Relapsing After Chimeric Antigen Receptor T-Cell (CAR-T) Therapies, and Is Active in Treatment through Multiple Lines. Blood 2019, 134, 6. [Google Scholar] [CrossRef]
- Varghese, B.; Menon, J.; Rodriguez, L.; Haber, L.; Olson, K.; Duramad, P.; Oyejide, A.; Smith, E.; Thurston, G.; Kirshner, J. A Novel CD20xCD3 Bispecific Fully Human Antibody Induces Potent Anti-Tumor Effects Against B Cell Lymphoma in Mice. Blood 2014, 124, 4501. [Google Scholar] [CrossRef]
- Teachey, D.T.; Rheingold, S.R.; Maude, S.L.; Zugmaier, G.; Barrett, D.M.; Seif, A.E.; Nichols, K.E.; Suppa, E.K.; Kalos, M.; Berg, R.A.; et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood 2013, 121, 5154–5157. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Godel, P.; Subklewe, M.; Stemmler, H.J.; Schlosser, H.A.; Schlaak, M.; Kochanek, M.; Boll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [Green Version]
- Strohl, W.R.; Naso, M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Piskol, R.; Ybarra, R.; Chen, Y.J.; Li, J.; Slaga, D.; Hristopoulos, M.; Clark, R.; Modrusan, Z.; Totpal, K.; et al. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Sasaki, M.; Harada, A.; Taketo, J.; Hara, T.; Akai, S.; Ishiguro, T.; Narita, A.; Kaneko, A.; Mishima, M. Daily ascending dosing in cynomolgus monkeys to mitigate cytokine release syndrome induced by ERY22, surrogate for T-cell redirecting bispecific antibody ERY974 for cancer immunotherapy. Toxicol. Appl. Pharmacol. 2019, 379, 114657. [Google Scholar] [CrossRef] [PubMed]
- Deppisch, N.; Ruf, P.; Eissler, N.; Neff, F.; Buhmann, R.; Lindhofer, H.; Mocikat, R. Efficacy and Tolerability of a GD2-Directed Trifunctional Bispecific Antibody in a Preclinical Model: Subcutaneous Administration Is Superior to Intravenous Delivery. Mol. Cancer Ther. 2015, 14, 1877–1883. [Google Scholar] [CrossRef] [Green Version]
- Trinklein, N.D.; Pham, D.; Schellenberger, U.; Buelow, B.; Boudreau, A.; Choudhry, P.; Clarke, S.C.; Dang, K.; Harris, K.E.; Iyer, S.; et al. Efficient tumor killing and minimal cytokine release with novel T-cell agonist bispecific antibodies. MAbs 2019, 11, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.R.; Sukumaran, S.; Hristopoulos, M.; Totpal, K.; Stainton, S.; Lu, E.; Wong, A.; Tam, L.; Newman, R.; Vuillemenot, B.R.; et al. An anti-CD3/anti-CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood 2017, 129, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Staflin, K.; Zuch de Zafra, C.L.; Schutt, L.K.; Clark, V.; Zhong, F.; Hristopoulos, M.; Clark, R.; Li, J.; Mathieu, M.; Chen, X.; et al. Target arm affinities determine preclinical efficacy and safety of anti-HER2/CD3 bispecific antibody. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Vafa, O.; Trinklein, N.D. Perspective: Designing T-Cell Engagers With Better Therapeutic Windows. Front. Oncol. 2020, 10, 446. [Google Scholar] [CrossRef] [Green Version]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef]
- Riesenberg, R.; Buchner, A.; Pohla, H.; Lindhofer, H. Lysis of prostate carcinoma cells by trifunctional bispecific antibodies (alpha EpCAM x alpha CD3). J. Histochem. Cytochem. 2001, 49, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Hoseini, S.S.; Xu, H.; Ponomarev, V.; Cheung, N.K. Silencing Fc Domains in T cell-Engaging Bispecific Antibodies Improves T-cell Trafficking and Antitumor Potency. Cancer Immunol. Res. 2019, 7, 2013–2024. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Meesters, J.I.; Bunce, M.; Armstrong, A.A.; Somani, S.; Nesspor, T.C.; Chiu, M.L.; Altintas, I.; Verploegen, S.; Schuurman, J.; et al. Efficient Generation of Bispecific Murine Antibodies for Pre-Clinical Investigations in Syngeneic Rodent Models. Sci. Rep. 2017, 7, 2476. [Google Scholar] [CrossRef] [Green Version]
- Oates, J.; Hassan, N.J.; Jakobsen, B.K. ImmTACs for targeted cancer therapy: Why, what, how, and which. Mol. Immunol. 2015, 67, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, Y.; Zhong, K.; Jiang, C.; Wang, L.; Yuan, Z.; Nie, C.; Xu, J.; Guo, G.; Zhou, L.; et al. Frequent B7-H3 overexpression in craniopharyngioma. Biochem. Biophys. Res. Commun. 2019, 514, 379–385. [Google Scholar] [CrossRef]
- Crawford, A.; Haber, L.; Kelly, M.P.; Vazzana, K.; Canova, L.; Ram, P.; Pawashe, A.; Finney, J.; Jalal, S.; Chiu, D.; et al. A Mucin 16 bispecific T cell-engaging antibody for the treatment of ovarian cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Fisher, T.S.; Hooper, A.T.; Lucas, J.; Clark, T.H.; Rohner, A.K.; Peano, B.; Elliott, M.W.; Tsaparikos, K.; Wang, H.; Golas, J.; et al. A CD3-bispecific molecule targeting P-cadherin demonstrates T cell-mediated regression of established solid tumors in mice. Cancer Immunol. Immunother. 2018, 67, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; He, Q.; Guo, Z.; Zhou, X.; Li, H.; Zhao, L.; Tang, H.; Zhou, X.; Zhu, H.; Shen, G.; et al. Therapeutic Bispecific T-Cell Engager Antibody Targeting the Transferrin Receptor. Front. Immunol. 2019, 10, 1396. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.P.; Guo, Z.L.; Tang, H.L.; Zhu, H.F.; Shen, G.X.; He, Y.; Lei, P. Selection for Anti-transferrin Receptor Bispecific T-cell Engager in Different Molecular Formats. Curr. Med. Sci. 2020, 40, 28–34. [Google Scholar] [CrossRef]
- Hettich, M.; Lahoti, J.; Prasad, S.; Niedermann, G. Checkpoint Antibodies but not T Cell-Recruiting Diabodies Effectively Synergize with TIL-Inducing gamma-Irradiation. Cancer Res. 2016, 76, 4673–4683. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Xie, K.; Li, H.; Wang, R.; Xu, X.; Chen, K.; Gu, H.; Fang, J. Suppression of c-Met-Overexpressing Tumors by a Novel c-Met/CD3 Bispecific Antibody. Drug Des. Dev. Ther. 2020, 14, 3201–3214. [Google Scholar] [CrossRef]
- Iizuka, A.; Nonomura, C.; Ashizawa, T.; Kondou, R.; Ohshima, K.; Sugino, T.; Mitsuya, K.; Hayashi, N.; Nakasu, Y.; Maruyama, K.; et al. A T-cell-engaging B7-H4/CD3-bispecific Fab-scFv Antibody Targets Human Breast Cancer. Clin. Cancer Res. 2019, 25, 2925–2934. [Google Scholar] [CrossRef] [Green Version]
- Kamada, H.; Taki, S.; Nagano, K.; Inoue, M.; Ando, D.; Mukai, Y.; Higashisaka, K.; Yoshioka, Y.; Tsutsumi, Y.; Tsunoda, S. Generation and characterization of a bispecific diabody targeting both EPH receptor A10 and CD3. Biochem. Biophys. Res. Commun. 2015, 456, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, N.; Wakata, Y.; Ida, K.; Midorikawa, A.; Isobe, M. High throughput development of TCR-mimic antibody that targets survivin-2B80-88/HLA-A*A24 and its application in a bispecific T-cell engager. Sci. Rep. 2019, 9, 9827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Huang, C.; Zhang, Z.; Feng, Y.; Wang, Z.; Tang, X.; Zhong, K.; Hu, Y.; Guo, G.; Zhou, L.; et al. MEK Inhibitor Augments Antitumor Activity of B7-H3-Redirected Bispecific Antibody. Front. Oncol. 2020, 10, 1527. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Shang, T.; Ma, P.; Sun, X.; Zhao, J.; Sun, X.; Zhang, M. Bispecific anti-CD3 x anti-B7-H3 antibody mediates T cell cytotoxic ability to human melanoma in vitro and in vivo. Investig. New Drugs 2019, 37, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Ma, J.; Lei, T.; Zhao, M.; Zhang, M. Targeting immunotherapy for bladder cancer by using anti-CD3x CD155 bispecific antibody. J. Cancer 2019, 10, 5153–5161. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Ma, J.; Ma, P.; Lei, T.; Zhao, M.; Zhang, M. Targeting immunotherapy for bladder cancer using anti-CD3x B7-H3 bispecific antibody. Cancer Med. 2018, 7, 5167–5177. [Google Scholar] [CrossRef]
- Martini, S.; Figini, M.; Croce, A.; Frigerio, B.; Pennati, M.; Gianni, A.M.; De Marco, C.; Daidone, M.G.; Argueta, C.; Landesman, Y.; et al. Selinexor Sensitizes TRAIL-R2-Positive TNBC Cells to the Activity of TRAIL-R2xCD3 Bispecific Antibody. Cells 2020, 9, 2231. [Google Scholar] [CrossRef]
- Mathur, D.; Root, A.R.; Bugaj-Gaweda, B.; Bisulco, S.; Tan, X.; Fang, W.; Kearney, J.C.; Lucas, J.; Guffroy, M.; Golas, J.; et al. A Novel GUCY2C-CD3 T-Cell Engaging Bispecific Construct (PF-07062119) for the Treatment of Gastrointestinal Cancers. Clin. Cancer Res. 2020, 26, 2188–2202. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Hymel, D.; Nelson, C.G.; Burke, T.R., Jr.; Rader, C. Conventional and Chemically Programmed Asymmetric Bispecific Antibodies Targeting Folate Receptor 1. Front. Immunol. 2019, 10, 1994. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Li, X.; Peng, H.; Cook, E.M.; Dadashian, E.L.; Wiestner, A.; Park, H.; Rader, C. Potent and selective antitumor activity of a T cell-engaging bispecific antibody targeting a membrane-proximal epitope of ROR1. Proc. Natl. Acad. Sci. USA 2018, 115, E5467–E5476. [Google Scholar] [CrossRef] [Green Version]
- Root, A.R.; Cao, W.; Li, B.; LaPan, P.; Meade, C.; Sanford, J.; Jin, M.; O’Sullivan, C.; Cummins, E.; Lambert, M.; et al. Development of PF-06671008, a Highly Potent Anti-P-cadherin/Anti-CD3 Bispecific DART Molecule with Extended Half-Life for the Treatment of Cancer. Antibodies 2016, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Ruan, S.; Lin, M.; Zhu, Y.; Lum, L.; Thakur, A.; Jin, R.; Shao, W.; Zhang, Y.; Hu, Y.; Huang, S.; et al. Integrin beta4-Targeted Cancer Immunotherapies Inhibit Tumor Growth and Decrease Metastasis. Cancer Res. 2020, 80, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Satta, A.; Grazia, G.; Caroli, F.; Frigerio, B.; Di Nicola, M.; Raspagliesi, F.; Mezzanzanica, D.; Zaffaroni, N.; Gianni, A.M.; Anichini, A.; et al. A Bispecific Antibody to Link a TRAIL-Based Antitumor Approach to Immunotherapy. Front. Immunol. 2019, 10, 2514. [Google Scholar] [CrossRef] [Green Version]
- Satta, A.; Mezzanzanica, D.; Caroli, F.; Frigerio, B.; Di Nicola, M.; Kontermann, R.E.; Iacovelli, F.; Desideri, A.; Anichini, A.; Canevari, S.; et al. Design, selection and optimization of an anti-TRAIL-R2/anti-CD3 bispecific antibody able to educate T cells to recognize and destroy cancer cells. MAbs 2018, 10, 1084–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, C.R.; Bahr-Mahmud, H.; Plum, L.M.; Schmoldt, K.; Kolsch, A.C.; Tureci, O.; Sahin, U. Characterization of the first-in-class T-cell-engaging bispecific single-chain antibody for targeted immunotherapy of solid tumors expressing the oncofetal protein claudin 6. Oncoimmunology 2016, 5, e1091555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taki, S.; Kamada, H.; Inoue, M.; Nagano, K.; Mukai, Y.; Higashisaka, K.; Yoshioka, Y.; Tsutsumi, Y.; Tsunoda, S. A Novel Bispecific Antibody against Human CD3 and Ephrin Receptor A10 for Breast Cancer Therapy. PLoS ONE 2015, 10, e0144712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Ma, J.; Lei, T.; Ma, W.; Zhang, M. The bispecific anti-CD3 x anti-CD155 antibody mediates T cell immunotherapy for human prostate cancer. Investig. New Drugs 2019, 37, 810–817. [Google Scholar] [CrossRef]
- Zhao, L.; Yang, Y.; Zhou, P.; Ma, H.; Zhao, X.; He, X.; Wang, T.; Zhang, J.; Liu, Y.; Zhang, T. Targeting CD133high Colorectal Cancer Cells In Vitro and In Vivo with an Asymmetric Bispecific Antibody. J. Immunother. 2015, 38, 217–228. [Google Scholar] [CrossRef]
- Zhou, Y.; Zong, H.; Han, L.; Xie, Y.; Jiang, H.; Gilly, J.; Zhang, B.; Lu, H.; Chen, J.; Sun, R.; et al. A novel bispecific antibody targeting CD3 and prolactin receptor (PRLR) against PRLR-expression breast cancer. J. Exp. Clin. Cancer Res. 2020, 39, 87. [Google Scholar] [CrossRef]
- Kebenko, M.; Goebeler, M.E.; Wolf, M.; Hasenburg, A.; Seggewiss-Bernhardt, R.; Ritter, B.; Rautenberg, B.; Atanackovic, D.; Kratzer, A.; Rottman, J.B.; et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE(R)) antibody construct, in patients with refractory solid tumors. Oncoimmunology 2018, 7, e1450710. [Google Scholar] [CrossRef]
- Pishvaian, M.; Morse, M.A.; McDevitt, J.; Norton, J.D.; Ren, S.; Robbie, G.J.; Ryan, P.C.; Soukharev, S.; Bao, H.; Denlinger, C.S. Phase 1 Dose Escalation Study of MEDI-565, a Bispecific T-Cell Engager that Targets Human Carcinoembryonic Antigen, in Patients With Advanced Gastrointestinal Adenocarcinomas. Clin. Colorectal. Cancer 2016, 15, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Melero, I.; Ros, W.; Argiles, G.; Marabelle, A.; Rodriguez-Ruiz, M.E.; Albanell, J.; Calvo, E.; Moreno, V.; Cleary, J.M.; et al. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: Preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2017, 35, 3002. [Google Scholar] [CrossRef]
- Middleton, M.R.; McAlpine, C.; Woodcock, V.K.; Corrie, P.; Infante, J.R.; Steven, N.M.; Evans, T.R.J.; Anthoney, A.; Shoushtari, A.N.; Hamid, O.; et al. Tebentafusp, A TCR/Anti-CD3 Bispecific Fusion Protein Targeting gp100, Potently Activated Antitumor Immune Responses in Patients with Metastatic Melanoma. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hummel, H.-D.; Kufer, P.; Grüllich, C.; Deschler-Baier, B.; Chatterjee, M.; Goebeler, M.-E.; Miller, K.; Santis, M.D.; Loidl, W.C.; Buck, A.; et al. Phase 1 study of pasotuxizumab (BAY 2010112), a PSMA-targeting Bispecific T cell Engager (BiTE) immunotherapy for metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2019, 37, 5034. [Google Scholar] [CrossRef]
- Lutterbuese, R.; Raum, T.; Kischel, R.; Hoffmann, P.; Mangold, S.; Rattel, B.; Friedrich, M.; Thomas, O.; Lorenczewski, G.; Rau, D.; et al. T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 12605–12610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellerman, D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 2019, 154, 102–117. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Lanitis, E.; Dangaj, D.; Irving, M.; Coukos, G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. 2017, 28, xii18–xii32. [Google Scholar] [CrossRef]
- Groeneveldt, C.; van Hall, T.; van der Burg, S.H.; Ten Dijke, P.; van Montfoort, N. Immunotherapeutic Potential of TGF-beta Inhibition and Oncolytic Viruses. Trends Immunol. 2020, 41, 406–420. [Google Scholar] [CrossRef]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.L.; Carretta, M.; Kalvisa, A.; Siersbaek, M.S.; Simoes, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen density regulates the activity of tumor-infiltrating T cells. J. Immunother. Cancer 2019, 7, 68. [Google Scholar] [CrossRef] [Green Version]
- Strohlein, M.A.; Lefering, R.; Bulian, D.R.; Heiss, M.M. Relative lymphocyte count is a prognostic parameter in cancer patients with catumaxomab immunotherapy. Med. Hypotheses 2014, 82, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halin, C.; Scimone, M.L.; Bonasio, R.; Gauguet, J.M.; Mempel, T.R.; Quackenbush, E.; Proia, R.L.; Mandala, S.; von Andrian, U.H. The S1P-analog FTY720 differentially modulates T-cell homing via HEV: T-cell-expressed S1P1 amplifies integrin activation in peripheral lymph nodes but not in Peyer patches. Blood 2005, 106, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Chen, R.; Wang, X.; Hu, K.; Huang, L.; Lu, M.; Hu, Q. CCL19 and CCR7 Expression, Signaling Pathways, and Adjuvant Functions in Viral Infection and Prevention. Front. Cell Dev. Biol. 2019, 7, 212. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R.; Luster, A.D. CXCR3 in T cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Iijima, N.; Iwasaki, A. Tissue instruction for migration and retention of TRM cells. Trends Immunol. 2015, 36, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Amsen, D.; van Gisbergen, K.; Hombrink, P.; van Lier, R.A.W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol. 2018, 19, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Tormoen, G.W.; Crittenden, M.R.; Gough, M.J. Role of the immunosuppressive microenvironment in immunotherapy. Adv. Radiat. Oncol. 2018, 3, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Grywalska, E.; Pasiarski, M.; Gozdz, S.; Rolinski, J. Immune-checkpoint inhibitors for combating T-cell dysfunction in cancer. Onco Targets Ther. 2018, 11, 6505–6524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Vigneron, N. Human Tumor Antigens and Cancer Immunotherapy. Biomed. Res. Int. 2015, 2015, 948501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, A.; Singh, M.P.; Rai, B. Human papillomavirus-associated cancers: A growing global problem. Int. J. Appl. Basic Med. Res. 2016, 6, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Zhao, W.; Zhou, B.; Su, Z.; Gu, X.; Zhou, Z.; Chen, S. TSNAdb: A Database for Tumor-specific Neoantigens from Immunogenomics Data Analysis. Genomics Proteomics Bioinform. 2018, 16, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Koster, J.; Plasterk, R.H.A. A library of Neo Open Reading Frame peptides (NOPs) as a sustainable resource of common neoantigens in up to 50% of cancer patients. Sci. Rep. 2019, 9, 6577. [Google Scholar] [CrossRef]
- Kraus, M.H.; Popescu, N.C.; Amsbaugh, S.C.; King, C.R. Overexpression of the EGF receptor-related proto-oncogene erbB-2 in human mammary tumor cell lines by different molecular mechanisms. EMBO J. 1987, 6, 605–610. [Google Scholar] [CrossRef]
- Imrich, S.; Hachmeister, M.; Gires, O. EpCAM and its potential role in tumor-initiating cells. Cell Adh. Migr. 2012, 6, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Trenevska, I.; Li, D.; Banham, A.H. Therapeutic Antibodies against Intracellular Tumor Antigens. Front. Immunol. 2017, 8, 1001. [Google Scholar] [CrossRef] [Green Version]
- Bunk, S.; Hofmann, M.; Unverdorben, F.; Hutt, M.; Pszolla, G.; Schwöbel, F.; Wagner, C.; Yousef, S.; Schuster, H.; Missel, S.; et al. Effective Targeting of PRAME-Positive Tumors with Bispecific T Cell-Engaging Receptor (TCER®) Molecules. Blood 2019, 134, 3368. [Google Scholar] [CrossRef]
- Maruta, M.; Ochi, T.; Tanimoto, K.; Asai, H.; Saitou, T.; Fujiwara, H.; Imamura, T.; Takenaka, K.; Yasukawa, M. Direct comparison of target-reactivity and cross-reactivity induced by CAR- and BiTE-redirected T cells for the development of antibody-based T-cell therapy. Sci. Rep. 2019, 9, 13293. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.; Pankov, D.; Scott, A.; Korontsvit, T.; Zakhaleva, V.; Xu, Y.; Xiang, J.; Yan, S.; de Morais Guerreiro, M.D.; Veomett, N.; et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat. Biotechnol. 2015, 33, 1079–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marijt, K.A.; Doorduijn, E.M.; van Hall, T. TEIPP antigens for T-cell based immunotherapy of immune-edited HLA class I(low) cancers. Mol. Immunol. 2019, 113, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ressing, M.E.; de Jong, J.H.; Brandt, R.M.; Drijfhout, J.W.; Benckhuijsen, W.E.; Schreuder, G.M.; Offringa, R.; Kast, W.M.; Melief, C.J. Differential binding of viral peptides to HLA-A2 alleles. Implications for human papillomavirus type 16 E7 peptide-based vaccination against cervical carcinoma. Eur. J. Immunol. 1999, 29, 1292–1303. [Google Scholar] [CrossRef]
- Bacac, M.; Klein, C.; Umana, P. CEA TCB: A novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology 2016, 5, e1203498. [Google Scholar] [CrossRef] [Green Version]
- Slaga, D.; Ellerman, D.; Lombana, T.N.; Vij, R.; Li, J.; Hristopoulos, M.; Clark, R.; Johnston, J.; Shelton, A.; Mai, E.; et al. Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [Green Version]
- Panowski, S.H.; Kuo, T.C.; Zhang, Y.; Chen, A.; Geng, T.; Aschenbrenner, L.; Kamperschroer, C.; Pascua, E.; Chen, W.; Delaria, K.; et al. Preclinical Efficacy and Safety Comparison of CD3 Bispecific and ADC Modalities Targeting BCMA for the Treatment of Multiple Myeloma. Mol. Cancer Ther. 2019, 18, 2008–2020. [Google Scholar] [CrossRef] [Green Version]
- Mazor, Y.; Sachsenmeier, K.F.; Yang, C.; Hansen, A.; Filderman, J.; Mulgrew, K.; Wu, H.; Dall’Acqua, W.F. Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci. Rep. 2017, 7, 40098. [Google Scholar] [CrossRef] [PubMed]
- DeClerck, Y.A.; Mercurio, A.M.; Stack, M.S.; Chapman, H.A.; Zutter, M.M.; Muschel, R.J.; Raz, A.; Matrisian, L.M.; Sloane, B.F.; Noel, A.; et al. Proteases, extracellular matrix, and cancer: A workshop of the path B study section. Am. J. Pathol. 2004, 164, 1131–1139. [Google Scholar] [CrossRef]
- Stubbs, M.; McSheehy, P.M.; Griffiths, J.R.; Bashford, C.L. Causes and consequences of tumour acidity and implications for treatment. Mol. Med. Today 2000, 6, 15–19. [Google Scholar] [CrossRef]
- Boustany, L.M.; Wong, L.; White, C.W.; Diep, L.; Huang, Y.; Liu, S.; Richardson, J.H.; Kavanaugh, W.M.; Irving, B.A. Abstract A164: EGFR-CD3 bispecific Probody™ therapeutic induces tumor regressions and increases maximum tolerated dose >60-fold in preclinical studies. Mol. Cancer Ther. 2018, 17, A164. [Google Scholar] [CrossRef]
- Panchal, A.; Seto, P.; Wall, R.; Hillier, B.J.; Zhu, Y.; Krakow, J.; Datt, A.; Pongo, E.; Bagheri, A.; Chen, T.T.; et al. COBRA: A highly potent conditionally active T cell engager engineered for the treatment of solid tumors. MAbs 2020, 12, 1792130. [Google Scholar] [CrossRef]
- Geiger, M.; Stubenrauch, K.-G.; Sam, J.; Richter, W.F.; Jordan, G.; Eckmann, J.; Hage, C.; Nicolini, V.; Freimoser-Grundschober, A.; Ritter, M.; et al. Protease-activation using anti-idiotypic masks enables tumor specificity of a folate receptor 1-T cell bispecific antibody. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Banaszek, A.; Bumm, T.G.P.; Nowotny, B.; Geis, M.; Jacob, K.; Wolfl, M.; Trebing, J.; Kucka, K.; Kouhestani, D.; Gogishvili, T.; et al. On-target restoration of a split T cell-engaging antibody for precision immunotherapy. Nat. Commun. 2019, 10, 5387. [Google Scholar] [CrossRef]
- Minogue, E.; Millar, D.; Chuan, Y.; Zhang, S.; Grauwet, K.; Guo, M.; Langenbucher, A.; Benes, C.H.; Heather, J.; Minshull, J.; et al. Redirecting T-Cells Against AML in a Multidimensional Targeting Space Using T-Cell Engaging Antibody Circuits (TEAC). Blood 2019, 134, 2653. [Google Scholar] [CrossRef]
- Mandikian, D.; Takahashi, N.; Lo, A.A.; Li, J.; Eastham-Anderson, J.; Slaga, D.; Ho, J.; Hristopoulos, M.; Clark, R.; Totpal, K.; et al. Relative Target Affinities of T-Cell-Dependent Bispecific Antibodies Determine Biodistribution in a Solid Tumor Mouse Model. Mol. Cancer Ther. 2018, 17, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Kroesen, B.J.; ter Haar, A.; Spakman, H.; Willemse, P.; Sleijfer, D.T.; de Vries, E.G.; Mulder, N.H.; Berendsen, H.H.; Limburg, P.C.; The, T.H.; et al. Local antitumour treatment in carcinoma patients with bispecific-monoclonal-antibody-redirected T cells. Cancer Immunol. Immunother. 1993, 37, 400–407. [Google Scholar] [CrossRef]
- Blanco, B.; Ramirez-Fernandez, A.; Alvarez-Vallina, L. Engineering Immune Cells for in vivo Secretion of Tumor-Specific T Cell-Redirecting Bispecific Antibodies. Front. Immunol. 2020, 11, 1792. [Google Scholar] [CrossRef] [PubMed]
- Iwahori, K.; Kakarla, S.; Velasquez, M.P.; Yu, F.; Yi, Z.; Gerken, C.; Song, X.T.; Gottschalk, S. Engager T cells: A new class of antigen-specific T cells that redirect bystander T cells. Mol. Ther. 2015, 23, 171–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, B.; Holliger, P.; Vile, R.G.; Alvarez-Vallina, L. Induction of human T lymphocyte cytotoxicity and inhibition of tumor growth by tumor-specific diabody-based molecules secreted from gene-modified bystander cells. J. Immunol. 2003, 171, 1070–1077. [Google Scholar] [CrossRef]
- Compte, M.; Blanco, B.; Serrano, F.; Cuesta, A.M.; Sanz, L.; Bernad, A.; Holliger, P.; Alvarez-Vallina, L. Inhibition of tumor growth in vivo by in situ secretion of bispecific anti-CEA x anti-CD3 diabodies from lentivirally transduced human lymphocytes. Cancer Gene Ther. 2007, 14, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Everts, B.; van der Poel, H.G. Replication-selective oncolytic viruses in the treatment of cancer. Cancer Gene Ther. 2005, 12, 141–161. [Google Scholar] [CrossRef]
- Fajardo, C.A.; Guedan, S.; Rojas, L.A.; Moreno, R.; Arias-Badia, M.; de Sostoa, J.; June, C.H.; Alemany, R. Oncolytic Adenoviral Delivery of an EGFR-Targeting T-cell Engager Improves Antitumor Efficacy. Cancer Res. 2017, 77, 2052–2063. [Google Scholar] [CrossRef] [Green Version]
- Speck, T.; Heidbuechel, J.P.W.; Veinalde, R.; Jaeger, D.; von Kalle, C.; Ball, C.R.; Ungerechts, G.; Engeland, C.E. Targeted BiTE Expression by an Oncolytic Vector Augments Therapeutic Efficacy Against Solid Tumors. Clin. Cancer Res. 2018, 24, 2128–2137. [Google Scholar] [CrossRef] [Green Version]
- Freedman, J.D.; Hagel, J.; Scott, E.M.; Psallidas, I.; Gupta, A.; Spiers, L.; Miller, P.; Kanellakis, N.; Ashfield, R.; Fisher, K.D.; et al. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol. Med. 2017, 9, 1067–1087. [Google Scholar] [CrossRef]
- Porter, C.E.; Rosewell Shaw, A.; Jung, Y.; Yip, T.; Castro, P.D.; Sandulache, V.C.; Sikora, A.; Gottschalk, S.; Ittman, M.M.; Brenner, M.K.; et al. Oncolytic Adenovirus Armed with BiTE, Cytokine, and Checkpoint Inhibitor Enables CAR T Cells to Control the Growth of Heterogeneous Tumors. Mol. Ther. 2020, 28, 1251–1262. [Google Scholar] [CrossRef]
- Yu, F.; Wang, X.; Guo, Z.S.; Bartlett, D.L.; Gottschalk, S.M.; Song, X.T. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol. Ther. 2014, 22, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Gujar, S.; Pol, J.G.; Kroemer, G. Heating it up: Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. Oncoimmunology 2018, 7, e1442169. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Daeffler, L.; Pozdeev, V.I.; Angelova, A.; Rommelaere, J. Immune Conversion of Tumor Microenvironment by Oncolytic Viruses: The Protoparvovirus H-1PV Case Study. Front. Immunol. 2019, 10, 1848. [Google Scholar] [CrossRef]
- Russell, L.; Peng, K.W.; Russell, S.J.; Diaz, R.M. Oncolytic Viruses: Priming Time for Cancer Immunotherapy. BioDrugs 2019, 33, 485–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groeneveldt, C.; Kinderman, P.; van den Wollenberg, D.J.M.; van den Oever, R.L.; Middelburg, J.; Mustafa, D.A.M.; Hoeben, R.C.; van der Burg, S.H.; van Hall, T.; van Montfoort, N. Preconditioning of the tumor microenvironment with oncolytic reovirus converts CD3-bispecific antibody treatment into effective immunotherapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergun, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2019, 6, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulouarn, C.; Clement, B. Stellate cells and the development of liver cancer: Therapeutic potential of targeting the stroma. J. Hepatol. 2014, 60, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Luo, J.; He, L.; Cheng, X.; Yan, G.; Wang, J.; Tang, R. Hybrid pH-sensitive nanogels surface-functionalized with collagenase for enhanced tumor penetration. J. Colloid Interface Sci. 2018, 525, 269–281. [Google Scholar] [CrossRef]
- Yoshida, E.; Kudo, D.; Nagase, H.; Suto, A.; Shimoda, H.; Suto, S.; Kakizaki, I.; Endo, M.; Hakamada, K. 4-Methylumbelliferone Decreases the Hyaluronan-rich Extracellular Matrix and Increases the Effectiveness of 5-Fluorouracil. Anticancer Res. 2018, 38, 5799–5804. [Google Scholar] [CrossRef]
- Eikenes, L.; Tari, M.; Tufto, I.; Bruland, O.S.; de Lange Davies, C. Hyaluronidase induces a transcapillary pressure gradient and improves the distribution and uptake of liposomal doxorubicin (Caelyx) in human osteosarcoma xenografts. Br. J. Cancer 2005, 93, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Chen, J.; Hu, Y.; Lin, L.; Sun, P.; Tian, H.; Chen, X. Highly enhanced cancer immunotherapy by combining nanovaccine with hyaluronidase. Biomaterials 2018, 171, 198–206. [Google Scholar] [CrossRef]
- Wen, Y.; Wang, C.T.; Ma, T.T.; Li, Z.Y.; Zhou, L.N.; Mu, B.; Leng, F.; Shi, H.S.; Li, Y.O.; Wei, Y.Q. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. 2010, 101, 2325–2332. [Google Scholar] [CrossRef] [PubMed]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshio, Y.; Teramoto, K.; Hanaoka, J.; Tezuka, N.; Itoh, Y.; Asai, T.; Daigo, Y.; Ogasawara, K. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci. 2015, 106, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.D.; Duffy, M.R.; Lei-Rossmann, J.; Muntzer, A.; Scott, E.M.; Hagel, J.; Campo, L.; Bryant, R.J.; Verrill, C.; Lambert, A.; et al. An Oncolytic Virus Expressing a T-cell Engager Simultaneously Targets Cancer and Immunosuppressive Stromal Cells. Cancer Res. 2018, 78, 6852–6865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Sostoa, J.; Fajardo, C.A.; Moreno, R.; Ramos, M.D.; Farrera-Sal, M.; Alemany, R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J. Immunother. Cancer 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Hong, B.; Song, X.-T. A T-cell engager-armed oncolytic vaccinia virus to target the tumor stroma. Cancer Transl. Med. 2017, 3, 122–132. [Google Scholar] [CrossRef]
- Harryvan, T.J.; Verdegaal, E.M.E.; Hardwick, J.C.H.; Hawinkels, L.; van der Burg, S.H. Targeting of the Cancer-Associated Fibroblast-T-Cell Axis in Solid Malignancies. J. Clin. Med. 2019, 8, 1989. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Fromming, A.; Vater, A. Increasing Tumor-Infiltrating T Cells through Inhibition of CXCL12 with NOX-A12 Synergizes with PD-1 Blockade. Cancer Immunol. Res. 2017, 5, 950–956. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A.; Jin, J.Y.; Lemaire, V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann. Oncol. 2018, 29, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, L.; van der Burg, S.H.; van Hall, T. The NKG2A-HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 5549–5556. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Crabill, G.A.; Pritchard, T.S.; McMiller, T.L.; Wei, P.; Pardoll, D.M.; Pan, F.; Topalian, S.L. Mechanisms regulating PD-L1 expression on tumor and immune cells. J. Immunother. Cancer 2019, 7, 305. [Google Scholar] [CrossRef]
- Chikuma, S.; Terawaki, S.; Hayashi, T.; Nabeshima, R.; Yoshida, T.; Shibayama, S.; Okazaki, T.; Honjo, T. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J. Immunol. 2009, 182, 6682–6689. [Google Scholar] [CrossRef] [Green Version]
- Kobold, S.; Pantelyushin, S.; Rataj, F.; Vom Berg, J. Rationale for Combining Bispecific T Cell Activating Antibodies With Checkpoint Blockade for Cancer Therapy. Front. Oncol. 2018, 8, 285. [Google Scholar] [CrossRef]
- Schreiner, J.; Thommen, D.S.; Herzig, P.; Bacac, M.; Klein, C.; Roller, A.; Belousov, A.; Levitsky, V.; Savic, S.; Moersig, W.; et al. Expression of inhibitory receptors on intratumoral T cells modulates the activity of a T cell-bispecific antibody targeting folate receptor. Oncoimmunology 2016, 5, e1062969. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Apasov, S.; Koshiba, M.; Sitkovsky, M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood 1997, 90, 1600–1610. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.; Cekic, C. Regulation of lymphocyte function by adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2097–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osada, T.; Patel, S.P.; Hammond, S.A.; Osada, K.; Morse, M.A.; Lyerly, H.K. CEA/CD3-bispecific T cell-engaging (BiTE) antibody-mediated T lymphocyte cytotoxicity maximized by inhibition of both PD1 and PD-L1. Cancer Immunol. Immunother. 2015, 64, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Singer, K.; Gottfried, E.; Kreutz, M.; Mackensen, A. Suppression of T-cell responses by tumor metabolites. Cancer Immunol. Immunother. 2011, 60, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Mittal, D.; Stagg, J.; Smyth, M.J. Targeting cancer-derived adenosine: New therapeutic approaches. Cancer Discov. 2014, 4, 879–888. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Bai, L.; Li, W.; Zeng, T.; Tian, H.; Cui, J. Targeting T cell metabolism in the tumor microenvironment: An anti-cancer therapeutic strategy. J. Exp. Clin. Cancer Res. 2019, 38, 403. [Google Scholar] [CrossRef]
- Zhao, E.; Maj, T.; Kryczek, I.; Li, W.; Wu, K.; Zhao, L.; Wei, S.; Crespo, J.; Wan, S.; Vatan, L.; et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat. Immunol. 2016, 17, 95–103. [Google Scholar] [CrossRef]
- Lee, G.K.; Park, H.J.; Macleod, M.; Chandler, P.; Munn, D.H.; Mellor, A.L. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology 2002, 107, 452–460. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Gorelik, L.; Constant, S.; Flavell, R.A. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J. Exp. Med. 2002, 195, 1499–1505. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Jung, S.M.; Park, S.H.; Kato, M.; Yamashita, T.; Lee, I.K.; Sudo, K.; Nakae, S.; Han, J.S.; Kim, O.H.; et al. Activin receptor-like kinase5 inhibition suppresses mouse melanoma by ubiquitin degradation of Smad4, thereby derepressing eomesodermin in cytotoxic T lymphocytes. EMBO Mol. Med. 2013, 5, 1720–1739. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.K.; Cho, K.J.; Ishido, S.; Roche, P.A. Interleukin 10 (IL-10)-mediated Immunosuppression: March-i induction regulates antigen presentation by macrophages but not Dendritic cells. J. Biol. Chem. 2015, 290, 27158–27167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbrink, K.; Graulich, E.; Kubsch, S.; Knop, J.; Enk, A.H. CD4(+) and CD8(+) anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood 2002, 99, 2468–2476. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Zea, A.H.; DeSalvo, J.; Culotta, K.S.; Zabaleta, J.; Quiceno, D.G.; Ochoa, J.B.; Ochoa, A.C. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J. Immunol. 2003, 171, 1232–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taheri, F.; Ochoa, J.B.; Faghiri, Z.; Culotta, K.; Park, H.J.; Lan, M.S.; Zea, A.H.; Ochoa, A.C. L-Arginine regulates the expression of the T-cell receptor zeta chain (CD3zeta) in Jurkat cells. Clin. Cancer Res. 2001, 7, 958s–965s. [Google Scholar] [PubMed]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Ni, G.; Wang, T.; Walton, S.; Zhu, B.; Chen, S.; Wu, X.; Wang, Y.; Wei, M.Q.; Liu, X. Manipulating IL-10 signalling blockade for better immunotherapy. Cell Immunol. 2015, 293, 126–129. [Google Scholar] [CrossRef]
- Hausler, S.F.; Del Barrio, I.M.; Diessner, J.; Stein, R.G.; Strohschein, J.; Honig, A.; Dietl, J.; Wischhusen, J. Anti-CD39 and anti-CD73 antibodies A1 and 7G2 improve targeted therapy in ovarian cancer by blocking adenosine-dependent immune evasion. Am. J. Transl. Res. 2014, 6, 129–139. [Google Scholar]
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552. [Google Scholar] [CrossRef] [Green Version]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef]
- Spranger, S.; Koblish, H.K.; Horton, B.; Scherle, P.A.; Newton, R.; Gajewski, T.F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J. Immunother. Cancer 2014, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, R.; Zhou, Y.; Tian, X.; Wang, L.; Wu, X. Selective inhibition of IDO1, D-1-methyl-tryptophan (D-1MT), effectively increased EpCAM/CD3-bispecific BiTE antibody MT110 efficacy against IDO1(hi)breast cancer via enhancing immune cells activity. Int. Immunopharmacol. 2018, 54, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Kapoor, V.; Jassar, A.S.; Kaiser, L.R.; Albelda, S.M. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevko, A.; Michels, T.; Vrohlings, M.; Umansky, L.; Beckhove, P.; Kato, M.; Shurin, G.V.; Shurin, M.R.; Umansky, V. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J. Immunol. 2013, 190, 2464–2471. [Google Scholar] [CrossRef]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rebe, C.; Ghiringhelli, F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, E.; Wenthe, J.; Irenaeus, S.; Loskog, A.; Ullenhag, G. Gemcitabine reduces MDSCs, tregs and TGFbeta-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl. Med. 2016, 14, 282. [Google Scholar] [CrossRef]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra267. [Google Scholar] [CrossRef]
- Sun, L.; Clavijo, P.E.; Robbins, Y.; Patel, P.; Friedman, J.; Greene, S.; Das, R.; Silvin, C.; Van Waes, C.; Horn, L.A.; et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. J. CI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Lerman, B.; Sakamaki, I.; Wei, G.; Cha, S.C.; Rao, S.S.; Qian, J.; Hailemichael, Y.; Nurieva, R.; Dwyer, K.C.; et al. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat. Med. 2014, 20, 676–681. [Google Scholar] [CrossRef]
- Scott, E.M.; Jacobus, E.J.; Lyons, B.; Frost, S.; Freedman, J.D.; Dyer, A.; Khalique, H.; Taverner, W.K.; Carr, A.; Champion, B.R.; et al. Bi- and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J. Immunother. Cancer 2019, 7, 320. [Google Scholar] [CrossRef]
- Cheng, P.; Eksioglu, E.; Chen, X.; Wei, M.; Guenot, J.; Fox, J.; List, A.F.; Wei, S. Immunodepletion of MDSC By AMV564, a Novel Tetravalent Bispecific CD33/CD3 T Cell Engager Restores Immune Homeostasis in MDS in Vitro. Blood 2017, 130, 51. [Google Scholar] [CrossRef]
- Jitschin, R.; Saul, D.; Braun, M.; Tohumeken, S.; Volkl, S.; Kischel, R.; Lutteropp, M.; Dos Santos, C.; Mackensen, A.; Mougiakakos, D. CD33/CD3-bispecific T-cell engaging (BiTE(R)) antibody construct targets monocytic AML myeloid-derived suppressor cells. J. Immunother. Cancer 2018, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Koristka, S.; Cartellieri, M.; Arndt, C.; Feldmann, A.; Seliger, B.; Ehninger, G.; Bachmann, M.P. Tregs activated by bispecific antibodies: Killers or suppressors? Oncoimmunology 2015, 4, e994441. [Google Scholar] [CrossRef] [Green Version]
- Koristka, S.; Cartellieri, M.; Theil, A.; Feldmann, A.; Arndt, C.; Stamova, S.; Michalk, I.; Topfer, K.; Temme, A.; Kretschmer, K.; et al. Retargeting of human regulatory T cells by single-chain bispecific antibodies. J. Immunol. 2012, 188, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Kvarnhammar, A.M.; Veitonmaki, N.; Hagerbrand, K.; Dahlman, A.; Smith, K.E.; Fritzell, S.; von Schantz, L.; Thagesson, M.; Werchau, D.; Smedenfors, K.; et al. The CTLA-4 x OX40 bispecific antibody ATOR-1015 induces anti-tumor effects through tumor-directed immune activation. J. Immunother. Cancer 2019, 7, 103. [Google Scholar] [CrossRef] [Green Version]
- Dao, T.; Mun, S.S.; Scott, A.C.; Jarvis, C.A.; Korontsvit, T.; Yang, Z.; Liu, L.; Klatt, M.G.; Guerreiro, M.; Selvakumar, A.; et al. Depleting T regulatory cells by targeting intracellular Foxp3 with a TCR mimic antibody. Oncoimmunology 2019, 8, 1570778. [Google Scholar] [CrossRef]
- Morse, M.A.; Hobeika, A.C.; Osada, T.; Serra, D.; Niedzwiecki, D.; Lyerly, H.K.; Clay, T.M. Depletion of human regulatory T cells specifically enhances antigen-specific immune responses to cancer vaccines. Blood 2008, 112, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Onda, M.; Kobayashi, K.; Pastan, I. Depletion of regulatory T cells in tumors with an anti-CD25 immunotoxin induces CD8 T cell-mediated systemic antitumor immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 4575–4582. [Google Scholar] [CrossRef] [Green Version]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: Influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef] [Green Version]
- Yano, H.; Thakur, A.; Tomaszewski, E.N.; Choi, M.; Deol, A.; Lum, L.G. Ipilimumab augments antitumor activity of bispecific antibody-armed T cells. J. Transl. Med. 2014, 12, 191. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Li, Y.; Shao, Y.; Zhang, Y. Gene modification strategies for next-generation CAR T cells against solid cancers. J. Hematol. Oncol. 2020, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Correnti, C.E.; Laszlo, G.S.; de van der Schueren, W.J.; Godwin, C.D.; Bandaranayake, A.; Busch, M.A.; Gudgeon, C.J.; Bates, O.M.; Olson, J.M.; Mehlin, C.; et al. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia 2018, 32, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Jiang, W.; Yang, M.; Guo, H.; Zhang, Y.; Wang, J.; Zhu, H.; Shi, R.; Fan, D.; Yang, C.; et al. Efficient inhibition of human B-cell lymphoma in SCID mice by synergistic antitumor effect of human 4-1BB ligand/anti-CD20 fusion proteins and anti-CD3/anti-CD20 diabodies. J. Immunother. 2010, 33, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Chiu, D.; Tavare, R.; Haber, L.; Aina, O.H.; Vazzana, K.; Ram, P.; Danton, M.; Finney, J.; Jalal, S.; Krueger, P.; et al. A PSMA-Targeting CD3 Bispecific Antibody Induces Antitumor Responses that Are Enhanced by 4-1BB Costimulation. Cancer Immunol. Res. 2020, 8, 596–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubrot, J.; Milheiro, F.; Alfaro, C.; Palazon, A.; Martinez-Forero, I.; Perez-Gracia, J.L.; Morales-Kastresana, A.; Romero-Trevejo, J.L.; Ochoa, M.C.; Hervas-Stubbs, S.; et al. Treatment with anti-CD137 mAbs causes intense accumulations of liver T cells without selective antitumor immunotherapeutic effects in this organ. Cancer Immunol. Immunother. 2010, 59, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Strahotin, S.; Hewes, B.; Zhang, B.; Zhang, Y.; Archer, D.; Spencer, T.; Dillehay, D.; Kwon, B.; Chen, L.; et al. Cytokine-mediated disruption of lymphocyte trafficking, hemopoiesis, and induction of lymphopenia, anemia, and thrombocytopenia in anti-CD137-treated mice. J. Immunol. 2007, 178, 4194–4213. [Google Scholar] [CrossRef]
- Skokos, D.; Waite, J.C.; Haber, L.; Crawford, A.; Hermann, A.; Ullman, E.; Slim, R.; Godin, S.; Ajithdoss, D.; Ye, X.; et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Daniel, P.T.; Kroidl, A.; Kopp, J.; Sturm, I.; Moldenhauer, G.; Dorken, B.; Pezzutto, A. Immunotherapy of B-cell lymphoma with CD3x19 bispecific antibodies: Costimulation via CD28 prevents “veto” apoptosis of antibody-targeted cytotoxic T cells. Blood 1998, 92, 4750–4757. [Google Scholar] [CrossRef]
- Rossi, E.A.; Rossi, D.L.; Cardillo, T.M.; Chang, C.H.; Goldenberg, D.M. Redirected T-cell killing of solid cancers targeted with an anti-CD3/Trop-2-bispecific antibody is enhanced in combination with interferon-alpha. Mol. Cancer Ther. 2014, 13, 2341–2351. [Google Scholar] [CrossRef] [Green Version]
- Schmohl, J.U.; Felices, M.; Taras, E.; Miller, J.S.; Vallera, D.A. Enhanced ADCC and NK Cell Activation of an Anticarcinoma Bispecific Antibody by Genetic Insertion of a Modified IL-15 Cross-linker. Mol. Ther. 2016, 24, 1312–1322. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TAA | Disease | Phase |
|---|---|---|
| Completed Clinical Trials | ||
| CEA | CEA-positive tumors | Phase I (NCT02324257, completed) |
| CEA | Gastrointestinal adenocarcinomas | Phase I NCT01284231, completed) |
| CEA | Advanced CEA-positive solid tumors | Phase I (NCT02291614, completed) |
| CEA | Advanced CEA-positive solid tumors | Phase I (NCT02650713, completed) |
| EGFR | Brain and central nervous system tumors | Phase I (NCT00005813, completed) |
| EpCAM | Solid tumors | Phase I (NCT00635596, completed) |
| EpCAM | Ascites, ovarian cancer, fallopian tube cancer, peritoneal cancer | Phase II (NCT00326885, completed) |
| EpCAM | Ovarian cancer, fallopian tube cancer, peritoneal cancer | Phase II (NCT00377429, completed) |
| EpCAM | Recurrent ovarian cancer, fallopian tube cancer, peritoneal carcinomatosis | Phase II (NCT01815528, completed) |
| EpCAM | Ovarian cancer, fallopian tube cancer, peritoneal cancer | Phase II (NCT01246440, completed) |
| EpCAM | Ovarian cancer | Phase II (NCT00563836, completed) |
| EpCAM | Ascites, carcinoma, epithelial cancer | Phase II (NCT01065246, completed) |
| EpCAM | Ovarian cancer, gastric cancer, pancreatic cancer, malignant ascites | Phase II (2005-001700-39, completed) |
| EpCAM | Gastric cancer and gastric adenocarcinoma | Phase II (NCT00352833, completed) |
| EpCAM | Peritoneal carcinomatosis and gastric adenocarcinoma | Phase II (NCT01504256, completed) |
| EpCAM | Ovarian cancer, fallopian tube cancer, peritoneal cancer | Phase II (NCT00189345, completed) |
| EpCAM | Gastric cancer and gastric adenocarcinoma | Phase II (NCT00464893, completed) |
| EpCAM | Malignant ascites and EpCAM-positive tumors | Phase II/III, (NCT00836654, completed) |
| EpCAM | EpCAM-positive solid cancers | Phase III (NCT00822809, completed) |
| GD2 | Neuroblastoma | Phase I (NCT00877110, completed) |
| gpA33 | Colorectal carcinoma | Phase I (NCT02248805, completed) |
| GPC3 | Solid tumors | Phase I (NCT02748837, completed) |
| HER2 | Breast cancer, metastatic breast cancer | Phase I (NCT00027807, completed) |
| HLA-A*02:01:gp100 | Melanoma, advanced melanoma | Phase I (NCT01209676, completed) |
| HLA-A*02:01:gp100 | Malignant melanoma | Phase I (NCT01211262, completed) |
| PSMA | Prostate cancer | Phase I (NCT02262910, completed) |
| PSMA | Prostatic neoplasms | Phase I (NCT01723475, completed) |
| Active clinical trials * | ||
| 5T4 | Malignant solid tumors | Phase I/II (NCT04424641, recruiting) |
| B7-H3 | Advanced solid tumors, metastatic solid tumors | Phase I (NCT03406949, active not recruiting) |
| CEA | Colorectal cancers | Phase I (NCT03866239, recruiting) |
| CEA | NSCLC | Phase I/II (NCT03337698, recruiting) |
| CEA, EGFR, GPC3, HER2, MUC1 | Malignant solid tumors | Phase I (NCT04076137, recruiting) |
| CEA, EpCAM, GPC3, MUC1 | Advanced liver cancer | Phase II (NCT03146637, recruiting) |
| CLDN18.2 | Gastric and gastroesophageal junction adenocarcinoma | Phase I (NCT04260191, recruiting) |
| DLL3 | Small cell lung carcinoma | Phase I (NCT03319940, recruiting) |
| DLL3 | Small cell lung cancer, advanced cancers | Phase I/II (NCT04471727, not yet recruiting) |
| EGFR | Multiple solid gastrointestinal tumors | Phase I (NCT01420874, active not recruiting) |
| EGFR | Glioblastoma multiforme, gliosarcoma | Phase I (NCT03344250, recruiting) |
| EGFR | Pancreatic cancer | Phase I (NCT04137536, recruiting) |
| EGFR | Advanced pancreatic cancer | Phase Ib/II (NCT02620865, active not recruiting) |
| EGFR | Advanced and metastatic pancreatic adenocarcinoma | Phase Ib/II (NCT03269526, recruiting) |
| EGFRv3 | Glioblastoma multiforme, malignant glioma | Phase I (NCT03296696, active not recruiting) |
| EpCAM | Large bowel (colon) cancer, colorectal cancer | Phase U (ChiCTR-ROC-16008620, not yet recruiting) |
| EpCAM | Malignant ascites, advanced solid tumors | Phase I (CTR20181212, recruiting) |
| EpCAM | Ascites, advanced solid tumors | Phase I (ChiCTR1900024144, recruiting) |
| EpCAM | Malignant ascites | Phase I (NCT04501744, recruiting) |
| EpCAM | Gastric adenocarcinoma, peritoneal carcinomatosis, colorectal adenocarcinoma | Phase II (2010-022810-26, recruiting) |
| EpCAM | Advanced gastric cancer, stomach cancer, gastric cancer | Phase III (NCT04222114, recruiting) |
| GD2 | Neuroblastoma | Phase I (NCT02650648, active not recruiting) |
| GD2 | Neuroblastoma, osteosarcoma, other solid tumors | Phase I/II (NCT03860207, recruiting) |
| GD2 | Neuroblastoma, osteosarcoma | Phase I/II (NCT02173093, recruiting) |
| gpA33 | Metastatic colorectal cancer | Phase I/II (NCT03531632, active not recruiting) |
| GPC3 | Advanced solid tumors, recurrent solid tumors | Phase I (JapicCTI-194805, recruiting) |
| GUCY2C | Gastrointestinal malignancies, esophageal cancer | Phase I (NCT04171141, recruiting) |
| HER2 | Breast cancer | Phase U (ChiCTR-ROC-16008650, not yet recruiting) |
| HER2 | HER2-positive solid tumors | Phase I (NCT04501770, recruiting) |
| HER2 | Breast cancer and leptomeningeal metastases | Phase I (NCT03661424, recruiting) |
| HER2 | Esophageal, gastric, pancreatic, liver, gallbladder and bowel cancer | Phase I (NCT02662348, unknown status) |
| HER2 | Advanced solid tumors | Phase I (NCT03448042, recruiting) |
| HER2 | Advanced solid tumors | Phase I (CTR20171194, recruiting) |
| HER2 | Solid tumors, advanced solid tumors | Phase I (ChiCTR1900024128, recruiting) |
| HER2 | Breast cancer | Phase I/II (NCT03983395, recruiting) |
| HER2 | Metastatic breast cancer | Phase I/II NCT03272334, recruiting) |
| HER2 | Metastatic castration resistant prostate cancer | Phase II (NCT03406858, status unknown) |
| HER2 | Breast cancer | Phase II (NCT01147016, status unknown) |
| HER2 | Breast cancer | Phase II (NCT01022138, status unknown) |
| HLA-A*02:01:gp100 | Uveal melanoma | Phase I/II (NCT02570308, active not recruiting) |
| HLA-A*02:01:gp100 | Melanoma | Phase I/II (NCT02535078, active not recruiting) |
| HLA-A*02:01:gp100 | Uveal melanoma, metastatic uveal melanoma, advanced uveal melanoma | Phase II (NCT03070392, active not recruiting) |
| HLA-A*02:MAGE-A4 | Advanced solid tumors, metastatic solid tumors | Phase I/II (NCT03973333, recruiting) |
| MSLN | Mesotheliomas, ovarian cancers, pancreatic cancers | Phase I/II (NCT03872206, recruiting) |
| MUC16 | Ovarian cancer fallopian tube cancer, peritoneal cancer | Phase I/II (NCT04590326, not yet recruiting) |
| MUC16 | Ovarian cancer fallopian tube cancer, peritoneal cancer | Phase I/II, (NCT03564340, recruiting) |
| MUC17 | Gastric and gastroesophageal junction cancer | Phase I (NCT04117958, recruiting) |
| NY-ESO1 | NY-ESO1-positive tumors | Phase I/II (NCT03515551, recruiting) |
| PRAME | Advanced solid tumors, cancer indications | Phase I/II (NCT04262466, recruiting) |
| PSCA | NSCLC, breast cancer, pancreatic cancer, urogenital cancer | Phase I NCT(03927573, recruiting) |
| PSMA | Prostate cancer | Phase I (NCT04077021, recruiting) |
| PSMA | Prostate cancers, advanced solid tumors, neoplasms, renal cancers, small cell lung cancer | Phase I (NCT03926013, recruiting) |
| PSMA | Castration-resistant prostate carcinoma | Phase I (NCT04104607, recruiting) |
| PSMA | Metastatic castration-resistant prostate cancer | Phase I (NCT03792841, recruiting) |
| PSMA | Squamous cell lung carcinoma | Phase I/II NCT04496674, not yet recruiting) |
| PSMA | Prostate cancer | Phase I/II (NCT03577028, recruiting) |
| SSTR2 | Neuroendocrine tumors and gastrointestinal neoplasms | Phase I (NCT03411915, recruiting) |
| SSTR2 | Merkel cell carcinoma and small cell lung cancer | Phase I/II (NCT04590781, not yet recruiting) |
| STEAP1 | Metastatic castration-resistant prostate cancer | Phase I (NCT04221542, recruiting) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Middelburg, J.; Kemper, K.; Engelberts, P.; Labrijn, A.F.; Schuurman, J.; van Hall, T. Overcoming Challenges for CD3-Bispecific Antibody Therapy in Solid Tumors. Cancers 2021, 13, 287. https://doi.org/10.3390/cancers13020287
Middelburg J, Kemper K, Engelberts P, Labrijn AF, Schuurman J, van Hall T. Overcoming Challenges for CD3-Bispecific Antibody Therapy in Solid Tumors. Cancers. 2021; 13(2):287. https://doi.org/10.3390/cancers13020287
Chicago/Turabian StyleMiddelburg, Jim, Kristel Kemper, Patrick Engelberts, Aran F. Labrijn, Janine Schuurman, and Thorbald van Hall. 2021. "Overcoming Challenges for CD3-Bispecific Antibody Therapy in Solid Tumors" Cancers 13, no. 2: 287. https://doi.org/10.3390/cancers13020287
APA StyleMiddelburg, J., Kemper, K., Engelberts, P., Labrijn, A. F., Schuurman, J., & van Hall, T. (2021). Overcoming Challenges for CD3-Bispecific Antibody Therapy in Solid Tumors. Cancers, 13(2), 287. https://doi.org/10.3390/cancers13020287