1. Introduction
Angiogenesis encompasses the formation of new blood vessels from an already mature vasculature. It comprises endothelial cell sprouting from the vessel wall in response to angiogenic stimuli, followed by degradation of the surrounding extracellular matrix (ECM) and migration through poorly-perfused tissues to extend the existing endothelial network. In physiological conditions, this is a well-orchestrated and controlled process balanced by pro- versus anti-angiogenic signals. On the other hand, in pathological angiogenesis, the tight control and balance is lost. This leads to immature, leaky vessels as observed in cancer and is a prerequisite not only for persistent growth of the tumor mass, but also for distant metastasis. Some of the angiogenic signals modulating neo-angiogenesis include vascular endothelial growth factor-A (VEGF-A), basic fibroblast growth factor (FGF-2) and epidermal growth factor (EGF) that bind with high specificity to their respective receptor tyrosine kinases (RTKs) and initiate downstream signaling cascades [
1,
2,
3,
4].
Over the years, it has become evident that these angiogenic signaling events not only require RTKs, but also involve active participation of proteoglycans [
5]. Proteoglycans (PGs) consist of a protein core to which long, linear, negatively charged heteropolysaccharides, known as glycosaminoglycans (GAGs), are covalently linked. Based on the polysaccharide composition and sulfation pattern, GAGs are subdivided into four major families: heparin/heparan sulfate (Hep/HS), chondroitin/dermatan sulfate (CS/DS), keratan sulfate (KS) and hyaluronan (HA) [
6]. This molecular superfamily is characterized by a high structural and spatial diversity, which makes its members highly promiscuous in their interactions and allows them to modulate a plethora of molecular processes [
7,
8]. Over the years, the role and importance of heparan sulfate proteoglycans (HSPGs) in angiogenesis and tumor progression has become clearer and efforts have been made to better delineate the specific processes in which they are involved [
9,
10]. Cell surface HSPGs aid in growth factor signaling by, e.g., interacting with both the growth factor (GF) and its receptor (GFR), which is reported for VEGF and FGF. As such, they facilitate the formation of a stable HS:GF:GFR complex allowing more potent and prolonged downstream signaling initiating differentiation, proliferation, migration, cell-cell and cell-matrix adhesion [
11,
12,
13,
14]. In addition, HSPGs have also been reported to facilitate signaling cascades via integrins, which adds to the complexity of their interactome [
15]. EGF itself is not classified as a heparin-binding growth factor, but its receptor, EGFR, was shown to associate with the cell surface HSPG, syndecan-4, and integrins α
6β
4 and α
3β
1 on epithelial cells. This signaling complex mediated EGF-induced cell motility [
16]. Genetic studies have underlined the importance of HS in signaling networks of several heparin-binding pro-tumoral growth factors such as VEGF, FGF, platelet-derived growth factor B (PDGF-B), transforming growth factor-beta (TGF-β), but also bone morphogenetic protein (BMP) and sonic hedgehog (Shh) [
17]. For example, endothelial-specific targeting of HS in mice resulted in altered tumor angiogenesis with smaller tumors and vascular networks with reduced density and branching [
18]. As a consequence of reduced HS production, FGF-2 and VEGF binding to mutant endothelial cells propagated in vitro was also dramatically reduced and exhibited reduced sprouting and extracellular signal-regulated kinase (ERK) phosphorylation [
18].
Since angiogenesis takes a leading role in the progression of cancer, targeting its molecular players has been an area of intense investigation. Current anti-angiogenic strategies in cancer mainly target RTKs and sequestration of the growth factor itself, however, with limited success in terms of survival in cancer patients due to the acquired resistance and insufficient efficacy of therapy [
19,
20]. This emphasizes the need for other agents that target the vasculature [
21]. Considering the obvious role of HS in tumor pathophysiology, HS-targeting drugs have become an appealing new strategy [
10].
Our laboratory recently discovered that the COOH-terminal tail of the chemokine CXCL9, which is unusually long compared to most chemokines, is highly hydrophilic and has a high affinity for GAGs [
22,
23,
24]. A COOH-terminal fragment comprising 30 amino acids, CXCL9(74-103), was considered the most potent GAG binder. This chemokine-derived peptide was characterized by a loss of CXCR3 receptor binding and signaling capacities. It could however effectively displace chemokines from GAGs, leading to a reduced chemokine-mediated neutrophil recruitment in various models of inflammation in vivo [
22,
23,
24,
25]. To further explore the applicability of CXCL9(74-103), and in light of the potential to target HS to reduce angiogenesis, we decided to investigate the anti-angiogenic properties of this high affinity GAG-binding peptide. We hypothesized that CXCL9(74-103) would interrupt the binding of angiogenic growth factors and/or their receptors to HSPGs and reduce or prevent the activation of their downstream signaling cascades.
Therefore, we first examined the effect of CXCL9(74-103) in a number of angiogenic growth factor- (FGF-2, VEGF165, EGF) mediated tumorigenic processes such as in vitro endothelial cell proliferation, migration and spheroid sprouting. As a negative control, we included a shorter CXCL9 COOH-terminal fragment, CXCL9(86-103), lacking 12 amino acids important for GAG binding. As a result, this CXCL9-derived peptide has a rather low affinity for GAGs. We studied the molecular mechanism of action through ELISA-like GAG and growth factor-binding assays and grating-coupled interferometry experiments. We then evaluated the anti-angiogenic potential of CXCL9(74-103) in various in vivo models, including the Matrigel plug assay, the corneal cauterization assay and an MDA-MB-231 breast cancer xenograft model.
2. Materials and Methods
2.1. Cell Culture and Reagents
Human microvascular endothelial cells (HMVECs) (Cell Systems, Kirkland, WA, USA) were cultured in endothelial cell basal medium-2 (EBM™-2) supplemented with the EGM™-2 MV SingleQuots kit (both Lonza, Basel, Switzerland) in an atmosphere of 5% CO
2 at 37 °C. Mouse endothelial cells (MECs) were cultured in Dulbecco’s modified eagle medium (DMEM) (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 20 mM HEPES, 1 mM sodium pyruvate and 10% (
v/v) fetal calf serum (FCS) in 5% CO
2 at 37 °C. Growth factors used were human recombinant VEGF165 (Biolegend, San Diego, CA, USA), human recombinant FGF-2 and EGF (both R&D Systems, Minneapolis, MN, USA) and murine recombinant VEGF165 (PeproTech, Cranbury, NJ, USA). When VEGF165 is mentioned as a stimulus, human VEGF165 is implied, unless specified otherwise. CXCL9 COOH-terminal peptides CXCL9(86-103) and CXCL9(74-103) were chemically synthesized using 9-fluorenyl methoxycarbonyl (Fmoc) solid-phase synthesis on an Activo-P11 automated peptide synthesizer (Activotec, Cambridge, UK), as previously described [
26]. CXCL9(74-103) was site-specifically biotinylated or fluorescently labeled at the NH
2-terminus using biotin-p-nitrophenyl ester (Novabiochem, Darmstadt, Germany), or 5(6)-carboxytetramethylrhodamine (TAMRA; Merck Millipore, Darmstadt, Germany), respectively, as described earlier [
23,
26]. The CXCL9 chemokine-based peptide sequences were KKVLKVRKSQRSRQKKTT and KKKQKNGKKHQKKKVLKVRKSQRSRQKKTT for CXCL9(86-103) and CXCL9(74-103), respectively.
2.2. Proliferation Assay
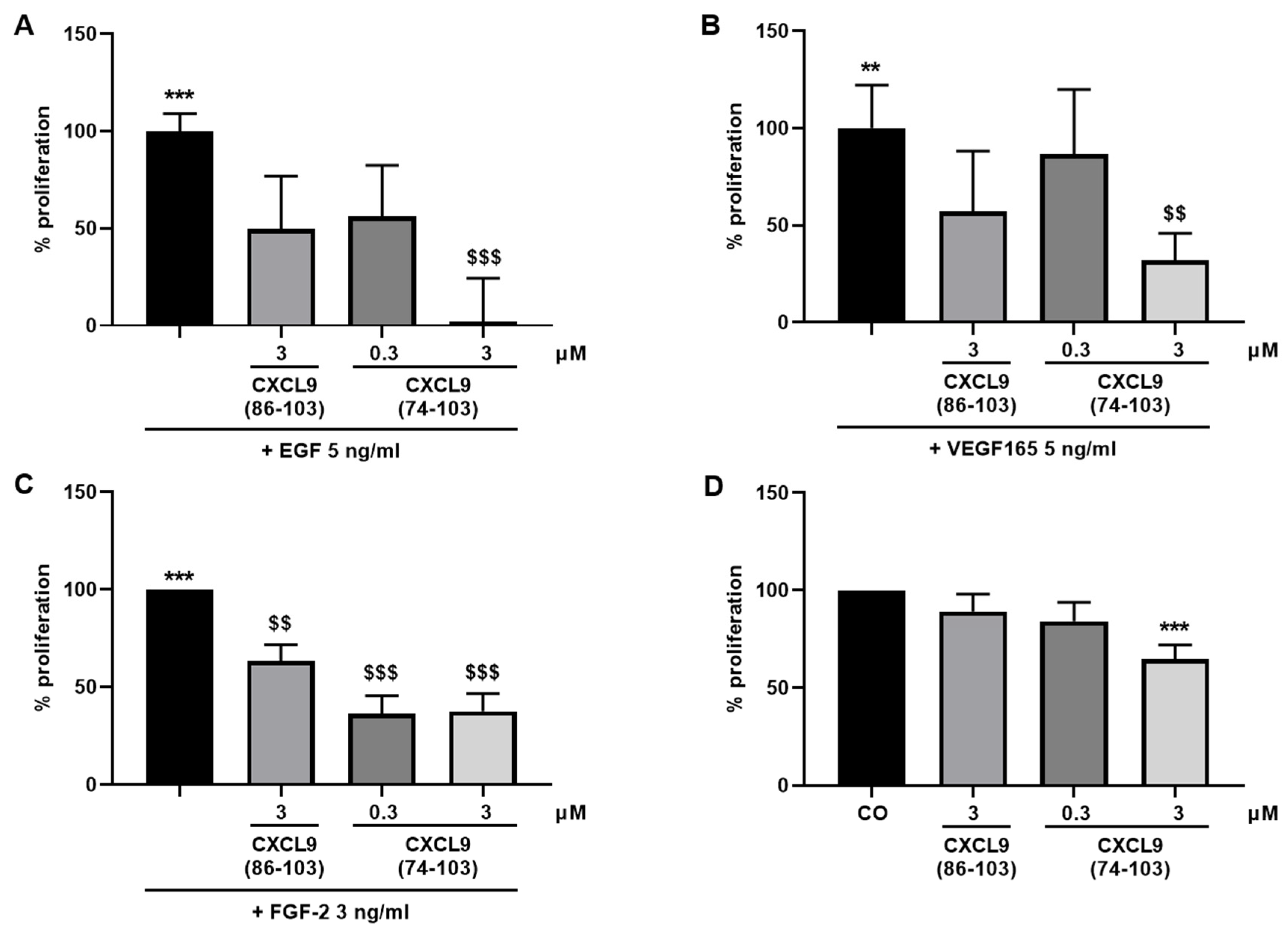
The ability of the CXCL9-derived peptides to inhibit growth factor-induced proliferation of endothelial cells was assessed. HMVECs were seeded at 5 × 103 cells/well in EBM™-2 medium containing 1% (v/v) FCS (proliferation medium). After settling of the cells, they were stimulated with EGF, VEGF165 (both 5 ng/mL) or FGF-2 (3 ng/mL) as a single stimulus or in combination with CXCL9(86-103) or CXCL9(74-103) at 0.3 or 3 µM in proliferation medium or left untreated. HMVECs were also treated with 0.3 or 3 µM of CXCL9(74-103) or 3 µM of CXCL9(86-103) alone to assess the effect of CXCL9-derived peptide treatment on spontaneous proliferation. After 3 to 4 days, a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) or ATP lite assay was performed. The ATP lite assay was performed according to the manufacturer’s instructions (Perkin Elmer, Waltham, MA). When MTT was used as substrate, the medium was replaced with 200 µL of MTT solution (RPMI without phenol red containing 0.4 g/L MTT) and the cells were incubated at 37 °C and 5% CO2 in the dark for 4 hours. Then, the MTT solution was discarded and the formed formazan crystals were dissolved in 200 µL acidic propanol [0.04 M HCl, 0.1% (v/v) NP-40 in isopropanol] under continuous shaking in the dark for 10 min. The optical densities were measured at 570 nm and 630 nm (reference wavelength).
2.3. xCELLigence Chemotaxis Assay
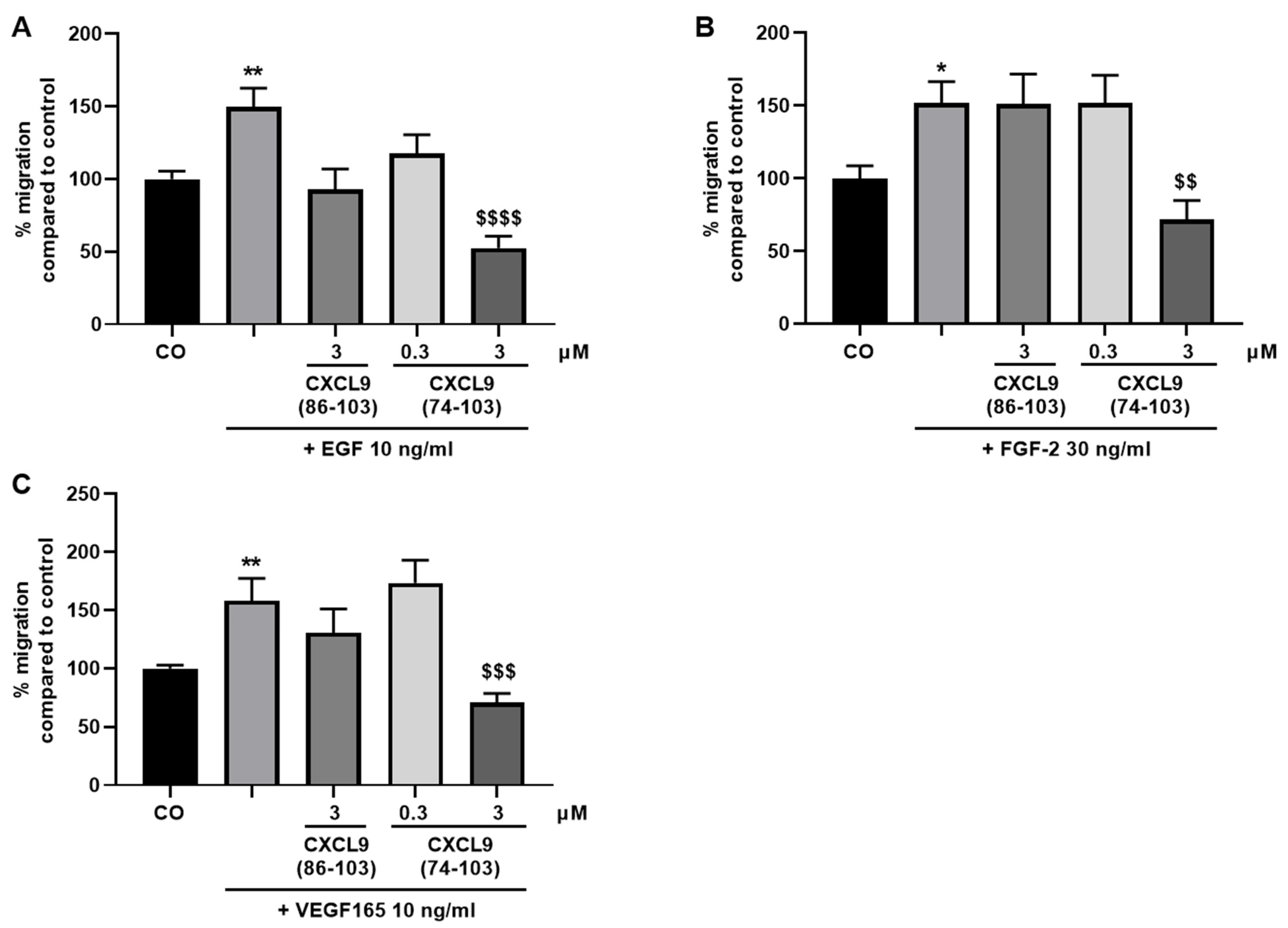
To evaluate the effect of the CXCL9-derived peptides on growth factor-induced endothelial cell migration, the xCELLigence® real-time cell analyzer (RTCA DP) system (ACEA Biosciences, Inc.; San Diego, CA, USA) was used. First, 160 µL MCDB131 medium (Gibco) supplemented with 0.4% (v/v) FCS (control medium) or 160 µL control medium with 30 ng/mL FGF-2, 10 ng/mL VEGF165 or 10 ng/mL EGF were added to the lower chamber of a cell invasion/migration (CIM)-Plate (ACEA Biosciences, Inc.). After assembly of the lower and upper chamber, 50 µL of serum-free MCDB131 medium was added in the upper wells. Following equilibration of the plate at 37 °C for 1 h, HMVECs were added in the upper chamber at 4 × 104 cells in 100 µL/well. CXCL9(74-103) or CXCL9(86-103) was added together with the cells in the upper compartment at a concentration of 0.3 or 3 µM. The assembled CIM-Plate was left at room temperature for 30 min to allow the cells to settle onto the membrane. Finally, the plate was placed in the instrument at 37 °C to monitor cell migration for 15 h. Cell migration from one compartment to the other was recorded (one recording every minute) as changes in electrical impedance. These changes were converted into cell indices, as a measure of cell migration.
2.4. xCELLigence Adhesion Assay
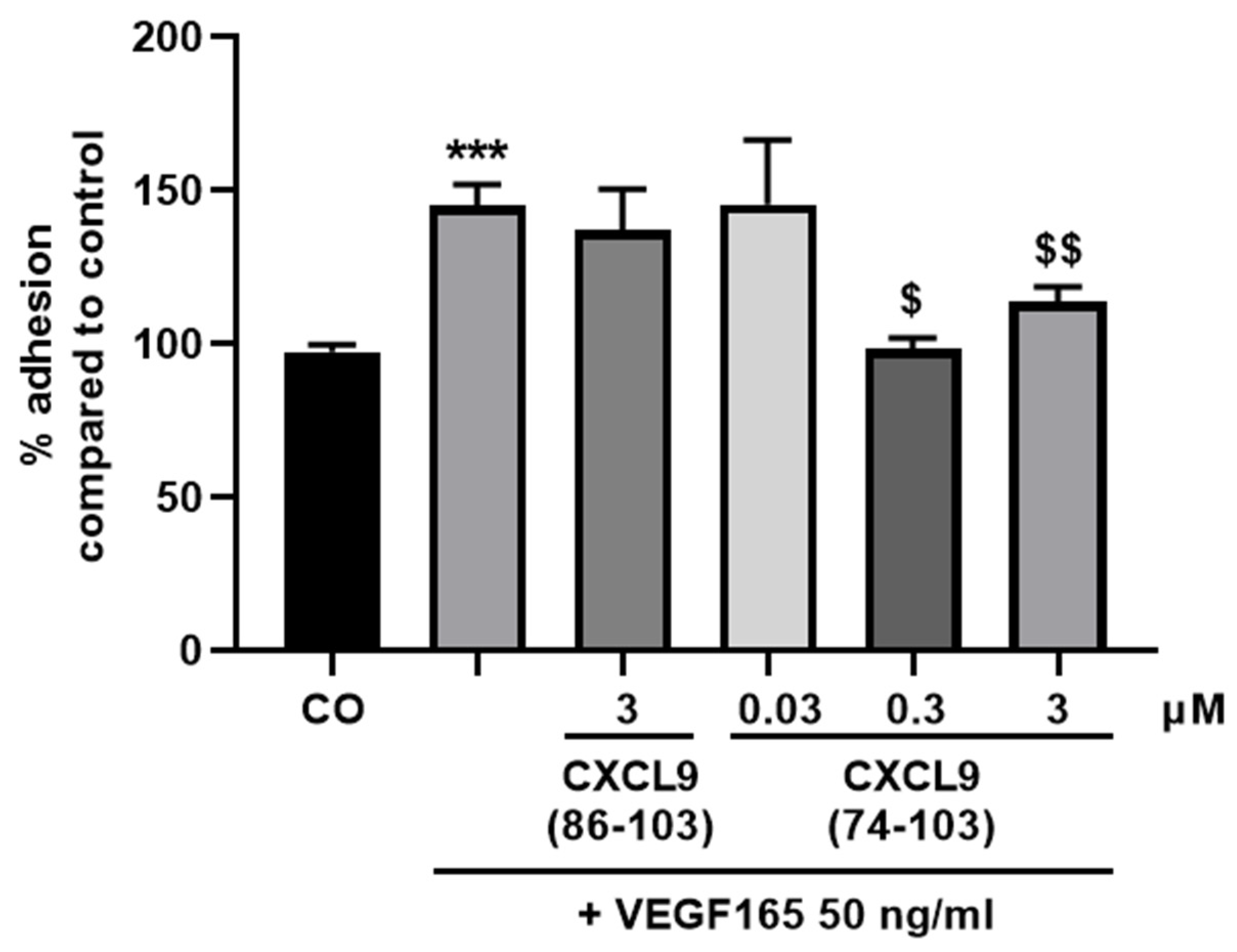
The wells of an E-plate (ACEA Biosciences, Inc.) were washed and coated with 0.1% (v/v) gelatin in phosphate-buffered saline (PBS) at 37 °C for 1 h. Following a washing step, wells were incubated with 0.1% (v/v) bovine serum albumin (BSA) in PBS at 37 °C for 1 h. After washing, 50 µL basal MCDB131 medium was added and the plate was allowed to equilibrate at 37 °C. HMVECs were harvested and seeded at 4 × 104 cells/well together with 50 ng/mL VEGF165 with or without 0.03, 0.3 or 3 µM of CXCL9(74-103) or 3 µM of CXCL9(86-103) in 100 µL control medium (vide supra). Real-time changes in electrical impedance depicted as cell indices, as measure for cell adhesion and spreading, were analyzed using the xCELLigence® RTCA DP System. The adhesion of HMVECs was analyzed 1 h after seeding.
2.5. Spheroid Sprouting Assay
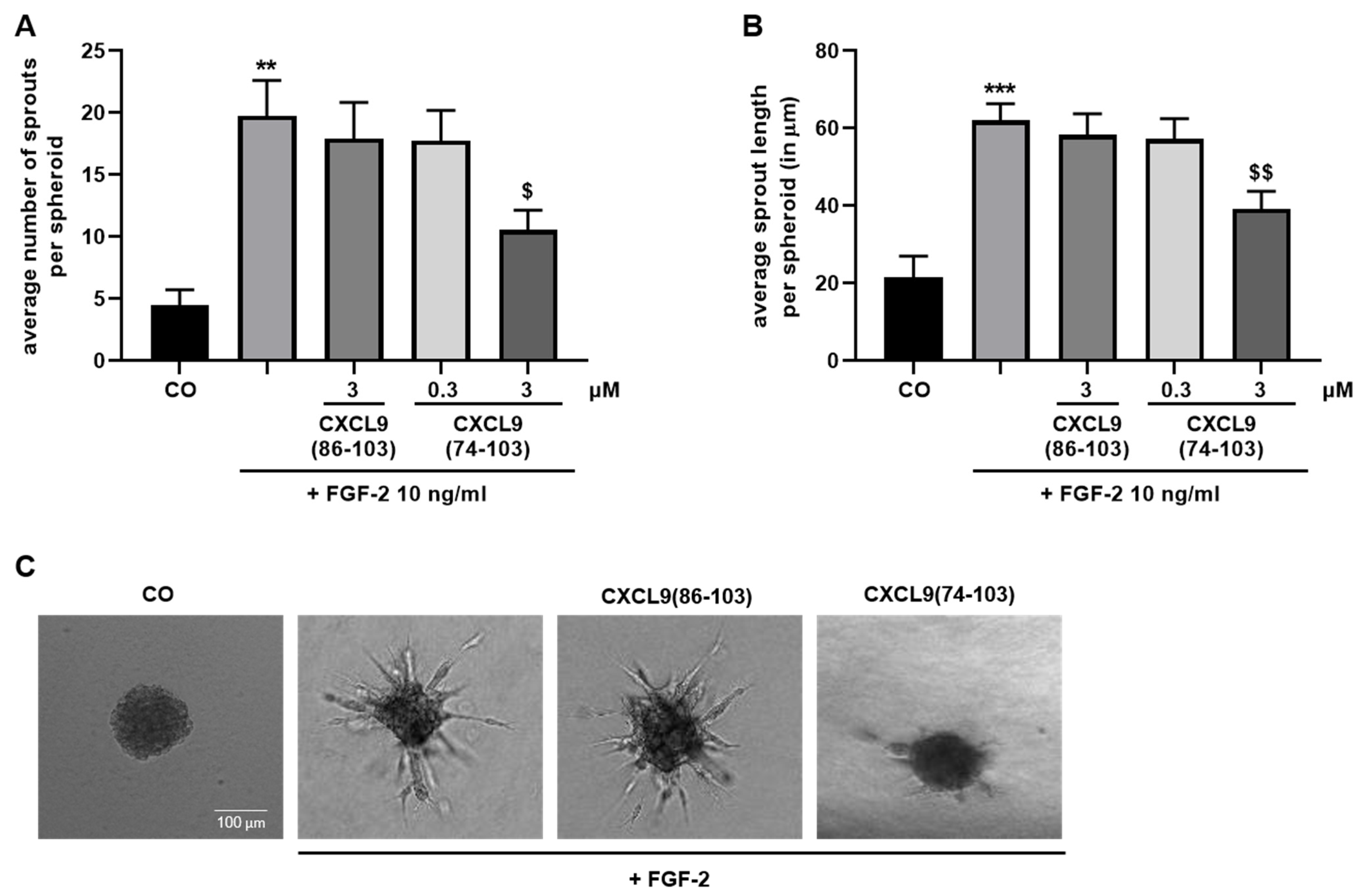
A 1:5 mixture of endothelial cells (4 × 104 cells/mL) and methylcellulose (Sigma-Aldrich, Saint Louis, MO, USA; 12 mg/mL in EBM™-2 basal medium) in EBM™-2 cell culture medium was prepared. Drops of 1 × 103 HMVECs were plated on a petri dish and allowed to form single spheroids in hanging droplets at 37 °C, 5% CO2 for 24 h. Spheroids were collected, sedimented by centrifugation and resuspended in a mixture of 24 mg/mL methylcellulose in basal EBM™-2, 1 mg/mL collagen type I (BD Biosciences, San Jose, CA, USA), 2.5 mg/mL NaHCO3 and 10 mM NaOH. The suspension was plated in a 96-well plate and the collagen was allowed to polymerize at 37 °C, 5% CO2 for 30 min. Spheroids were left untreated or stimulated with 0.3 or 3 µM of CXCL9(74-103) or 3 µM of CXCL9(86-103) for 15 min prior to addition of 10 ng/mL FGF-2 or 10 ng/mL EGF in EBM™-2 + 3% (v/v) FCS. In some experiments, HMVECs were pretreated with heparinase II (vide infra). After 16 hours, spheroid sprouting was assessed with an inverted Axiovert 200M microscope (Zeiss, Germany) through a 10× objective. The average number of sprouts was calculated and the average sprout length per spheroid was measured using Fiji software.
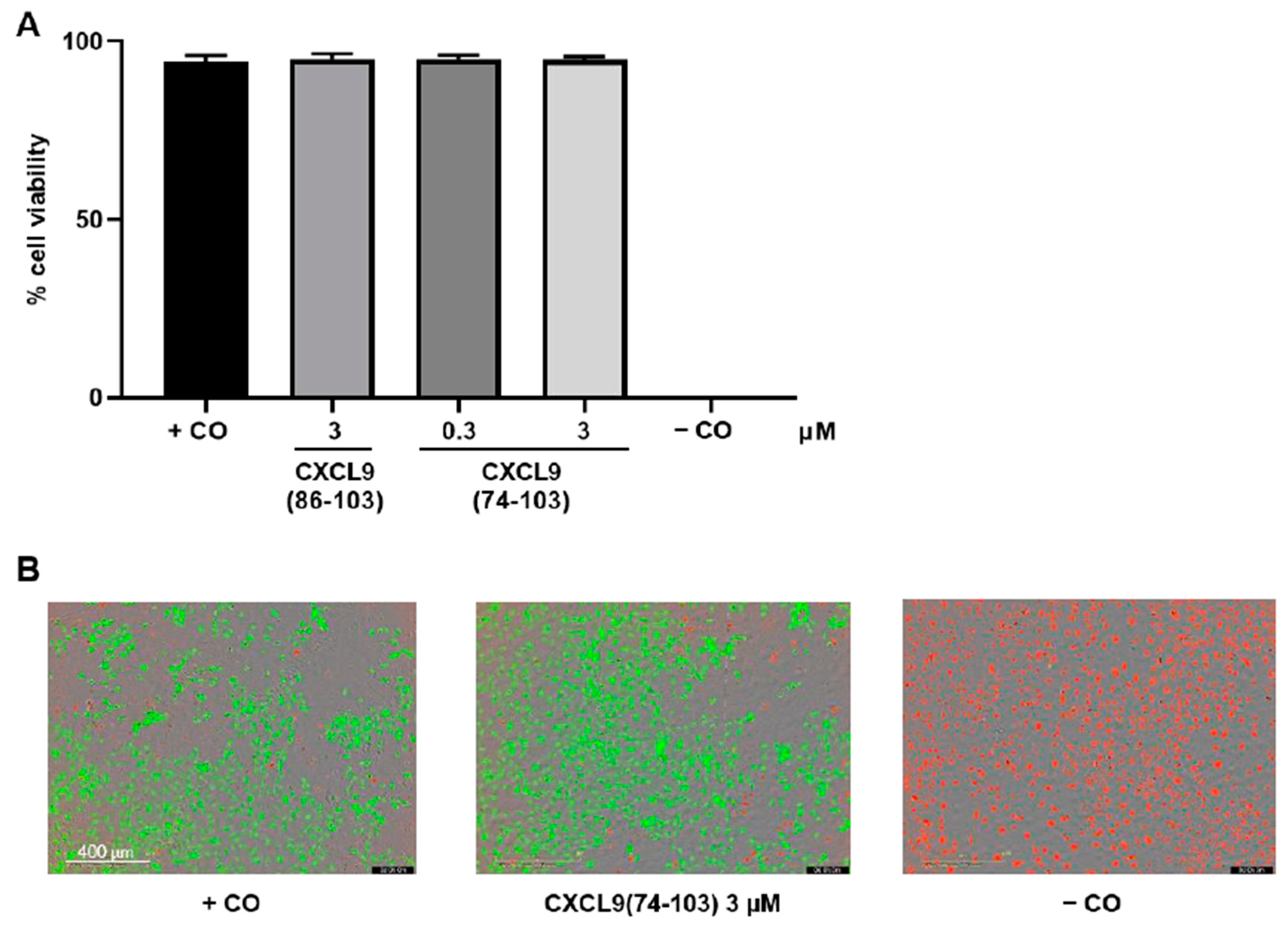
2.6. In Vitro Toxicity Assay
HMVECs were seeded in MCDB131 medium supplemented with 3% (v/v) FCS at 8 × 103 cells/well in a black, clear bottom 96-well plate coated with 0.1% (v/v) gelatin in PBS. The next day, cells were washed and incubated with control medium (vide supra) alone or in the presence of 0.3 or 3 µM of CXCL9(74-103) or CXCL9(86-103) for 24 h. The negative control cells were treated with 2% (v/v) Triton™ X-100 to induce cell death 21 h after the start of the experiment, i.e., 1 h prior to the addition of the LIVE/DEAD stain. The toxicity of the peptides was assessed using the LIVE/DEAD viability/cytotoxicity kit (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. At 24 h, calcein-acetoxymethyl ester (AM) and ethidium homodimer-1 (EthD-1) in 0.4% (v/v) FCS in DMEM FluoroBrite (Gibco) were added to all wells and the fluorescence was measured in the IncuCyte S3 live cell imaging system (Essen BioScience, Ltd.; Newark, UK).
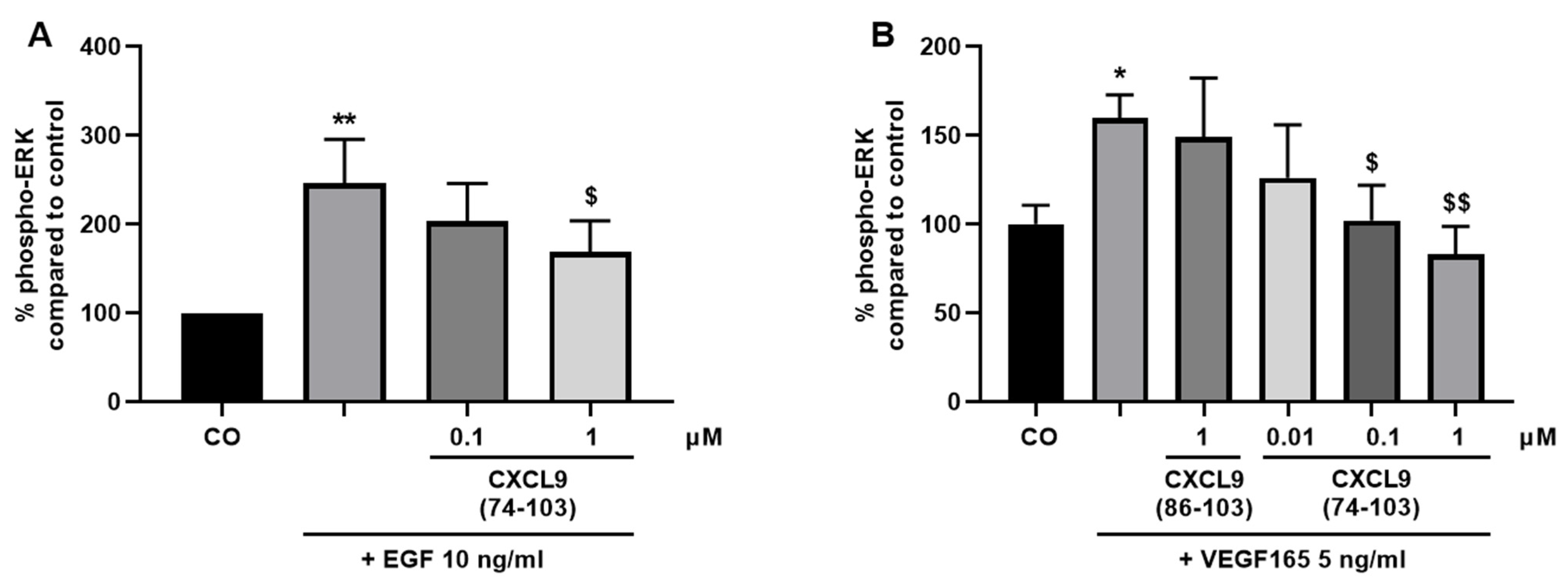
2.7. Signal Transduction Assays
HMVECs and MECs were seeded in 6-well plates at 1.25 × 105 cells/well in their respective culture medium. Once the cell density reached 70–80% confluency, the cells were starved in serum-free medium overnight. Fifteen minutes prior to stimulation, cells were incubated with various concentrations of CXCL9(74-103) or CXCL9(86-103) in 0.5% (v/v) BSA in MCDB131 or DMEM medium (assay medium for HMVECs and MECs, respectively) at 37 °C. HMVECs and MECs were stimulated with assay medium alone or supplemented with 10 ng/mL EGF or 5 ng/mL murine VEGF165 at 37 °C for 15 or 5 min, respectively. Then, the cells were placed on ice, washed with ice-cold PBS and 90 µL of lysis buffer [1% (v/v) protease inhibitor cocktail, phosphatase inhibitor cocktail 2 and 3 (all Sigma-Aldrich) in 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5% (v/v) Triton™ X-100, 5 mM NaF, 6 M urea in PBS] was added per well. After a 15-minute incubation on ice, cells were collected using a cell scraper, transferred to pre-cooled tubes and incubated on ice for another 5 min. The cell lysates were centrifuged at 558 g and 4 °C for 5 min and the samples were stored at −20 °C until analysis. The amount of phospho-ERK1/2 in the cell lysates was determined using an ERK1 (Thr202/Tyr204)/ERK2 (Thr185/Tyr187) ELISA duoset (R&D Systems) according to the manufacturer’s instructions. The amount of phosphorylated ERK was normalized to the total protein content in the samples measured by the Pierce™ bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific).
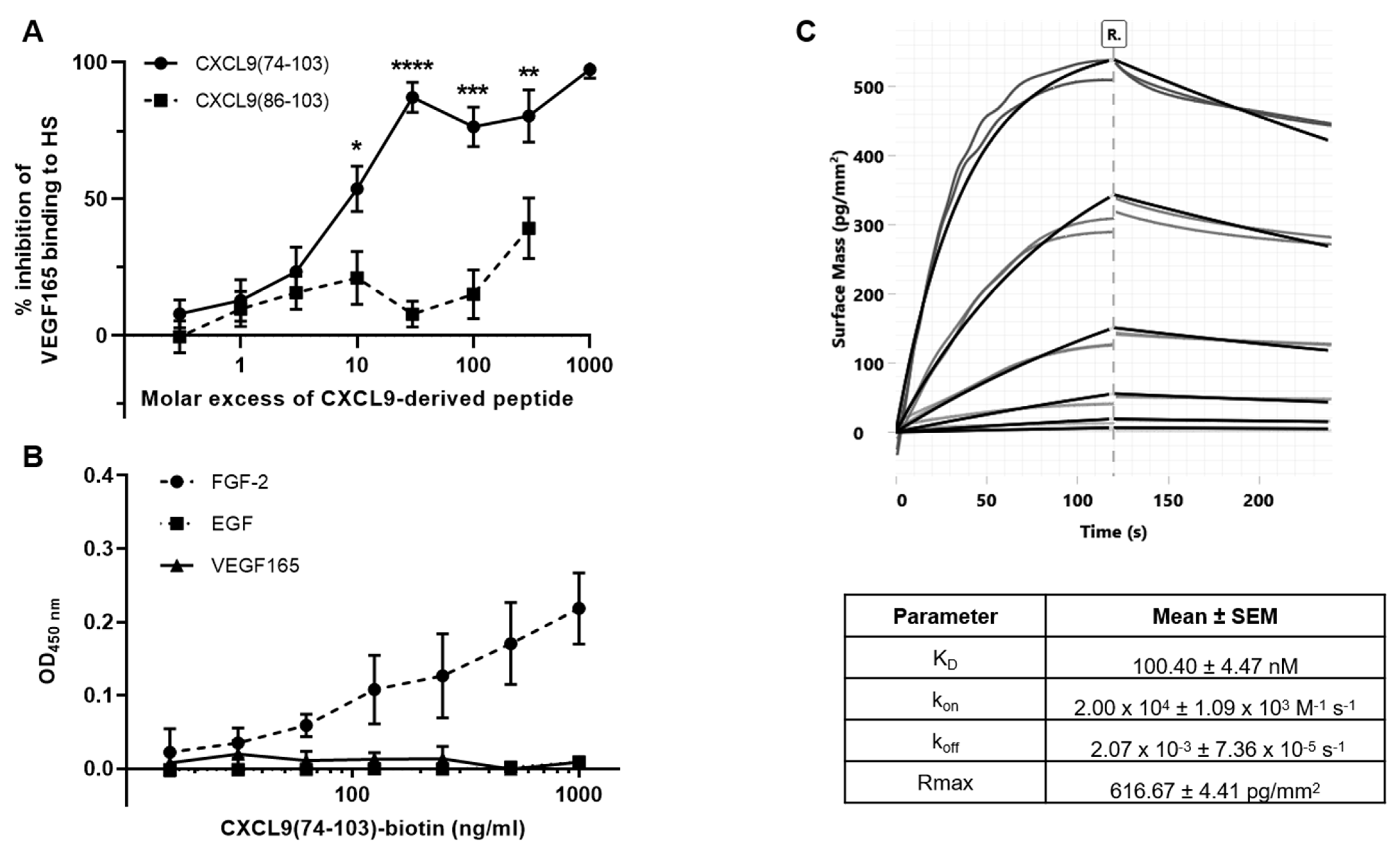
2.8. ELISA-Like GAG Binding Assay
The ability of the CXCL9-derived peptides to compete with VEGF165 for HS binding was assessed in an ELISA-like GAG binding assay. First, the GAG binding plates (Galen Laboratory Supplies, North Haven, CT, USA) were coated with 25 µg/mL HS (Iduron, Alderley Edge, UK) in standard assay buffer (SAB) [100 mM NaCl, 50 mM sodium acetate, 0.2% (v/v) Tween® 20 in ultrapure water, pH 7.2] at room temperature overnight. The plates were washed with SAB and blocked with blocking buffer [SAB supplemented with 0.2% (w/v) gelatin] at 37 °C for 1 h to prevent nonspecific binding. Then, dilutions of VEGF165 and the peptides, CXCL9(74-103) or CXCL9(86-103), were added and allowed to interact at 37 °C for 2 h. Unbound material was removed through washing and biotinylated polyclonal goat anti-human VEGF165 antibody (PeproTech) in blocking buffer was added to interact with HS-bound VEGF165 at 37 °C for 1 h. Following additional washing steps, detection was performed with streptavidin-horseradish peroxidase (HRP) (R&D Systems) in blocking buffer at 37 °C for 30 min and conversion of 3,3’,5,5’-tetramethylbenzidine (TMB) to a colorimetric signal in the presence of 0.015% (v/v) H2O2. The reaction was stopped by the addition of H2SO4 and the absorbance was measured at 450 nm. All incubation steps were performed in the dark.
2.9. ELISA-Like Direct Binding Assay
Direct interaction of CXCL9(74-103) and growth factors was assessed in an ELISA-like direct binding assay. The 96-well plates were coated with 50 ng/mL VEGF165, FGF-2 or EGF in PBS at 4 °C overnight. After washing with 0.05% (v/v) Tween® 20 in PBS (washing buffer), the plate was incubated with washing buffer supplemented with 0.1% (w/v) casein (blocking buffer) at 37 °C for 1 h to prevent nonspecific binding. Dilutions of biotinylated CXCL9(74-103) in blocking buffer (1–1000 ng/mL) were added and allowed to interact at 37 °C for 1 h. Following additional washing steps, detection of the bound peptide was accomplished with streptavidin-HRP, as described above.
2.10. Grating-Coupled Interferometry (GCI)
GCI experiments were performed on a Creoptix WAVEdelta system (Creoptix AG, Wädenswil, Switzerland), which is a label-free surface biosensor system for the characterization of molecular interactions. 4PCP-STA chips (Creoptix AG) were used, which contain a quasi-planar polycarboxyl-matrix with pre-immobilized streptavidin. The running buffer for the experiments was PBS supplemented with 30 mM NaCl, 15 mM L-Arginine, 0.015% (v/v) Tween® 20 and 0.05 mg/mL BSA at pH 7.4 for reducing nonspecific binding. Chips were conditioned using borate buffer (100 mM sodium borate, 1 M NaCl, pH 9.0). Capture was performed by injecting biotinylated CXCL9(74-103) at a concentration of 2 µg/mL and a flow rate of 10 µL/minute over the chip surface, until a density of +/− 160 was reached. After capture of biotinylated CXCL9(74-103), binding with FGF-2 was studied by injecting an FGF-2 concentration series over the chip surface. Since FGF-2 showed nonspecific binding on the surface, biotinylated antibodies (biotinylated polyclonal goat anti-rabbit immunoglobulins; Dako, Agilent, Santa Clara, CA, USA) were injected at a concentration of 10 µg/mL for 60 seconds for blocking the surface. A reference channel was blocked in the same way. Injections of the FGF-2 analyte were performed at 25 °C, with an association and dissociation time of 120 seconds and a flow rate of 40 µL/min. Absence of mass-transport limitation was verified by repeating the experiment at 50 µL/minute. A 1:3 dilution series with six dilutions (4.53 nM, 13.6 nM, 40.7 nM, 122 nM, 367 nM, 1.10 µM; all in duplicate) was analyzed. After each FGF-2 injection, a regeneration step was included, consisting of an injection of 2.5 M NaCl (40 µL/min flow rate, 30 sec injection time and 120 sec rinsing), followed by three injections of running buffer (same parameters as NaCl injection). Blank injections for referencing were included every 3rd sample cycle. Data adjustment (X and Y offset, dimethyl sulfoxide calibration and double referencing) and analysis (1:1 binding model with bulk correction) were performed using the Creoptix WAVEcontrol software.
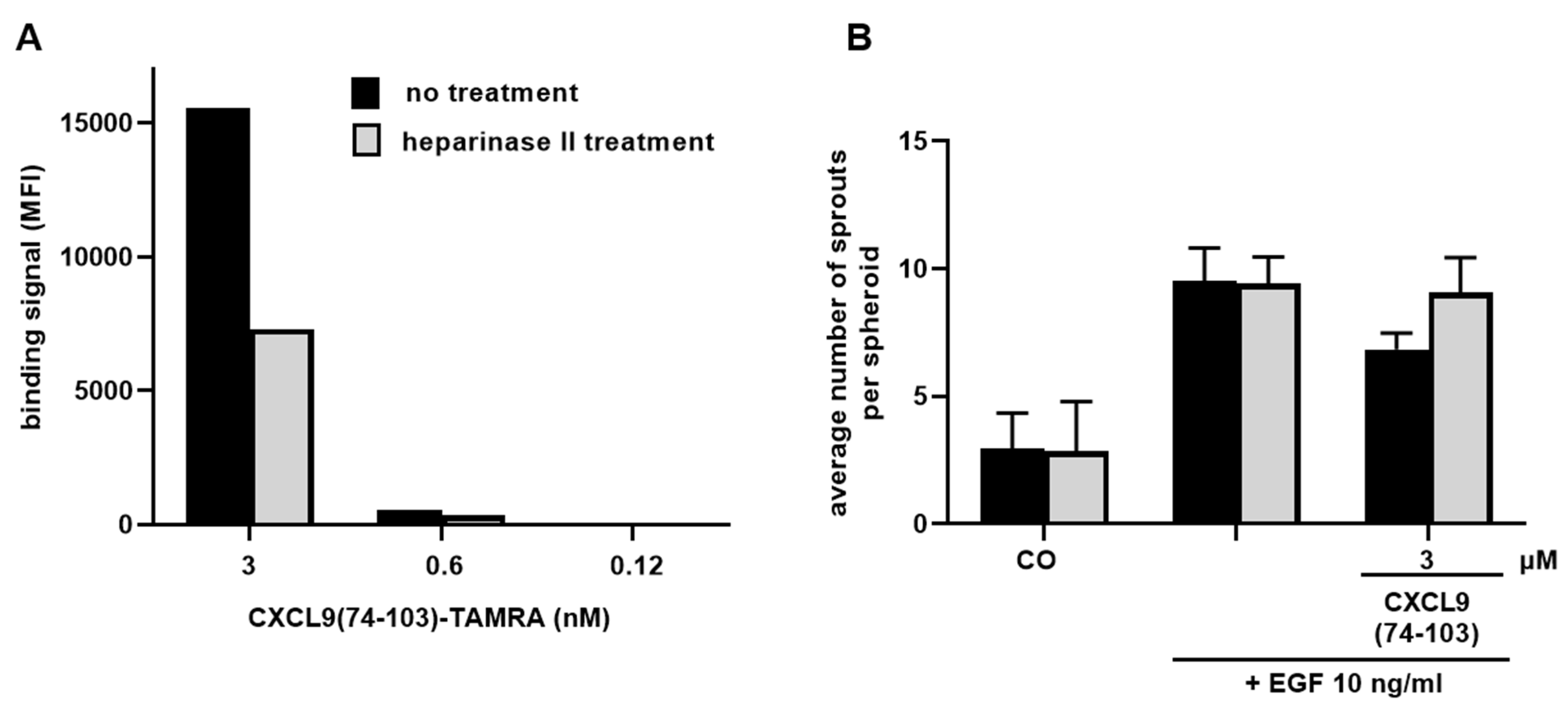
2.11. Analysis of Cellular HS Binding of CXCL9(74-103)
CXCL9(74-103) binding to cellular HS was assessed via flow cytometry. HMVECs were either left untreated or incubated with 0.75 U/mL heparinase II (Sigma-Aldrich) at 37 °C for 2 h. Cells were harvested and left at room temperature for 1 h to recuperate, they were then placed on ice and centrifuged. After centrifugation (300 g, 4 °C, 7 min) and resuspension in PBS, 1 × 105 cells per condition were stained with 3, 0.6 or 0.12 nM TAMRA-labeled CXCL9(74-103). To verify downregulation of HS expression, cells were stained with mouse anti-HS Ab (clone F58-10E4, Cat No 370255-S; Amsbio, Abingdon, UK) and R-PE goat anti-mouse IgM Ab (Cat No 115-116-075; Jackson ImmunoResearch, Westgrove, PA, USA). Flow cytometric analysis was performed using the BD LSRFortessa™ X-20 flow cytometer and FlowJo software (both BD Biosciences).
Sprouting experiments examining the dependency on HS binding for the inhibitory action of CXCL9(74-103) were performed as described above (vide supra). Heparinase II treatment (0.75 U/mL) of spheroids was carried out 4 hours before starting the experiment and dilutions of EGF (10 ng/mL) and CXCL9(74-103) (3 µM) were additionally supplemented with 0.75 U/mL heparinase II.
2.12. Analysis of Retinal Vascular Permeability in the Rat Streptozotocin-Induced Diabetes Model
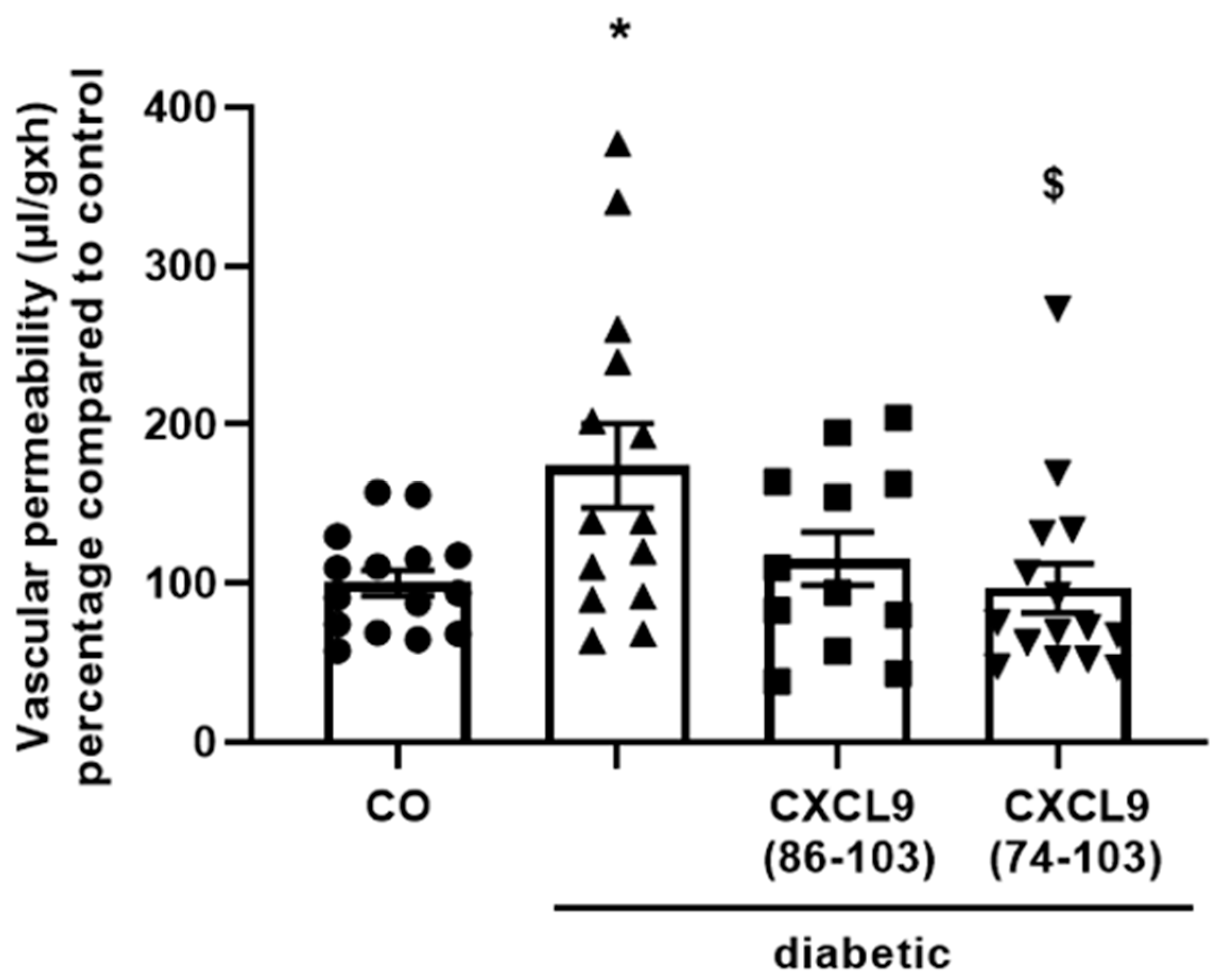
Male Sprague-Dawley rats (210–240 g) were fasted overnight and injected intraperitoneally (i.p.) with streptozotocin (55 mg/kg body weight, dissolved in citrate buffer, pH 4.5) to induce diabetes. After 3 days, the animals were anesthetized, diabetes was confirmed and the vitreous of the right eye was injected with equimolar amounts of CXCL9(74-103) (30 µg), CXCL9(86-103) (18 µg) or PBS (5 µL/injection). Diabetes-induced breakdown of the blood-retinal barrier as a consequence of increased vascular permeability was evaluated in retinas two weeks after streptozotocin injection, as previously described [
27]. Briefly, 30 min prior to sacrifice, fluorescein isothiocyanate (FITC)-conjugated dextran of 3-5 kDa (Sigma-Aldrich) was intravenously (i.v.) injected. After collection of a blood sample, rats were perfused with PBS. The retinas were excised, weighed and homogenized. The fluorescence in the retina lysates was measured using a SpectraMax
® Gemini™ XPS system. For normalization of the data, the following equation was used to express the obtained results:
Rat experiments were approved by the Institutional Animal Care and Committee of the College of Pharmacy, King Saud University.
2.13. In Vivo Angiogenesis Matrigel Plug Assay
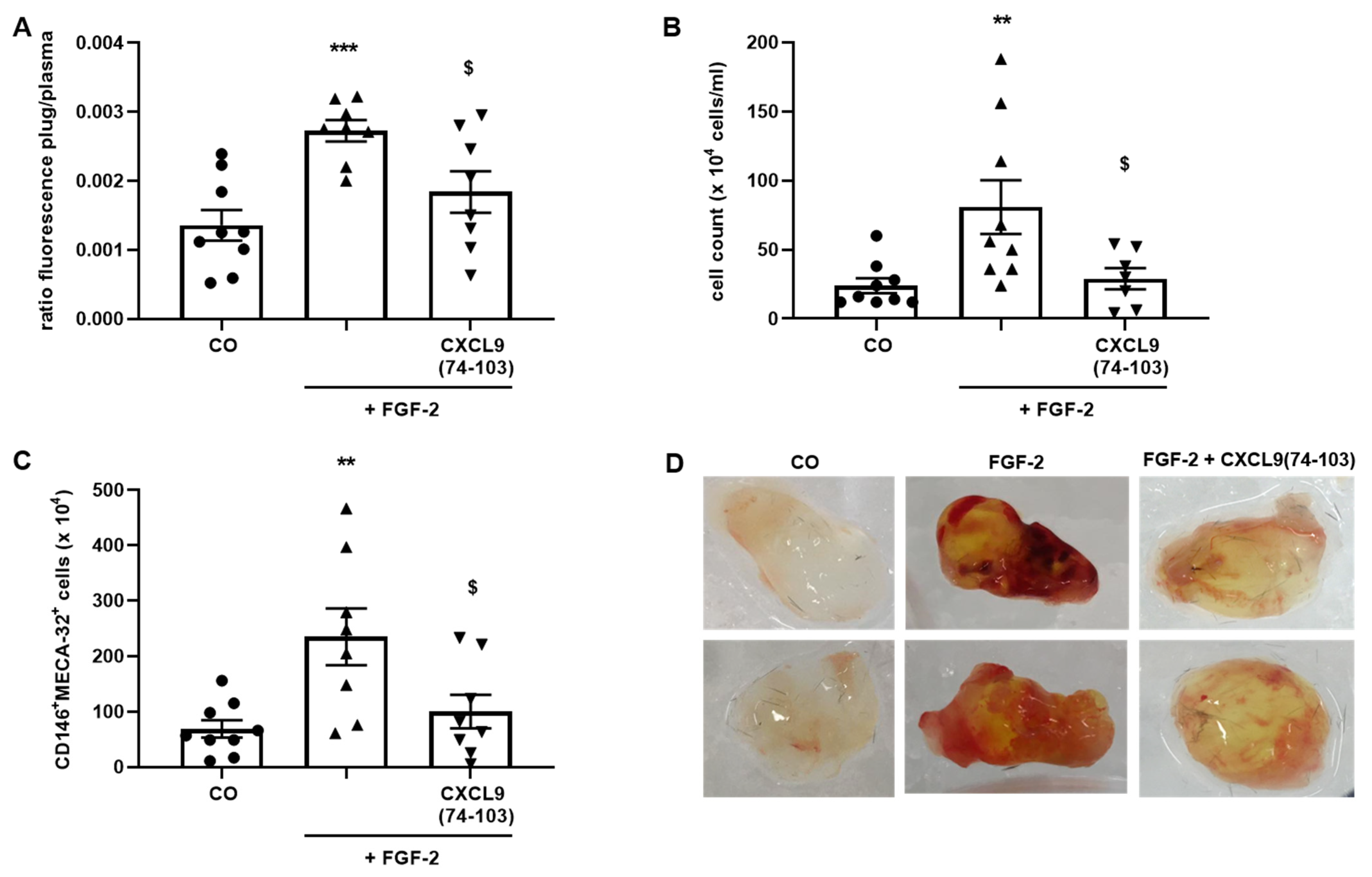
Six-to-eight-week-old female C57BL/6 mice were anesthetized with a mixture of 2.5 mg/mL xylazine (V.M.D, Inovet, Arendonk, Belgium) and 37.5 mg/mL ketamine (Dechra, Northwich, UK) in PBS by i.p. injection. A vertical dorsal incision was made and an ALZET osmotic pump (model 1007D, DURECT Corporation, Cupertine, CA, USA) filled with 400 µg/100 µL CXCL9(74-103) or 100 µL PBS was subcutaneously (s.c.) implanted. The incision was closed with a surgical suture. Growth factor-reduced Matrigel (Corning Matrigel Growth Factor (GFR) Basement Membrane Matrix, Phenol Red-Free, LDEV-Free; Corning Life Sciences, Amsterdam, The Netherlands) was s.c. injected in the dorsal flank forming a firm plug (600 µL/plug). The Matrigel was either injected as such (control plugs) or supplemented with 300 µg CXCL9(74-103) and/or 180 ng FGF-2. Seven days later, the mice were sacrificed. Thirty minutes before sacrifice, the mice were i.v. injected with 200 µL (25 mg/kg) FITC-labeled 2000 kDa dextran (Sigma-Aldrich) to quantify vessel formation in the Matrigel plugs. Blood was collected through cardiac puncture into heparinized tubes containing 20 µL heparin (5.000 IE/mL, Leo Pharma, Ballerup, Denmark). The blood was centrifuged at 200 g and 4 °C for 10 min, whereafter the supernatant was centrifuged at 3000 g and 4 °C for 10 min. The collected plasma was stored away from light at 4 °C until analysis. The Matrigel plugs were removed and enzymatically and mechanically digested in 1.2 mL dispase (Corning Life Sciences) using an incubation at 37 °C for 1.5 h and the gentleMACS™ Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany). The digested Matrigel plugs were filtered through a 70 µm cell strainer, washed with 1.5 mL FACS buffer [2% (v/v) FCS + 2 mM EDTA in PBS] and centrifuged at 300 g and 4 °C for 7 min. The supernatant (SN) was centrifuged again at 300 g and 4 °C for 7 min and the SN was retained away from light at 4 °C until analysis. The pellet was resuspended in 1 mL FACS buffer, the cells were counted and used for flow cytometry. The antibodies used to identify endothelial cells were anti-mouse CD45-APC (clone 30-F11, eBioscience, Thermo Fisher Scientific), anti-mouse CD146-PE (clone ME-9F1) and anti-mouse Pan-endothelial Cell Antigen-BV711 (clone MECA-32) antibodies (both BD Biosciences). Mice experiments were approved by the Ethical Committee of KU Leuven (project number P199/2018).
2.14. Corneal Cauterization Angiogenesis Assay
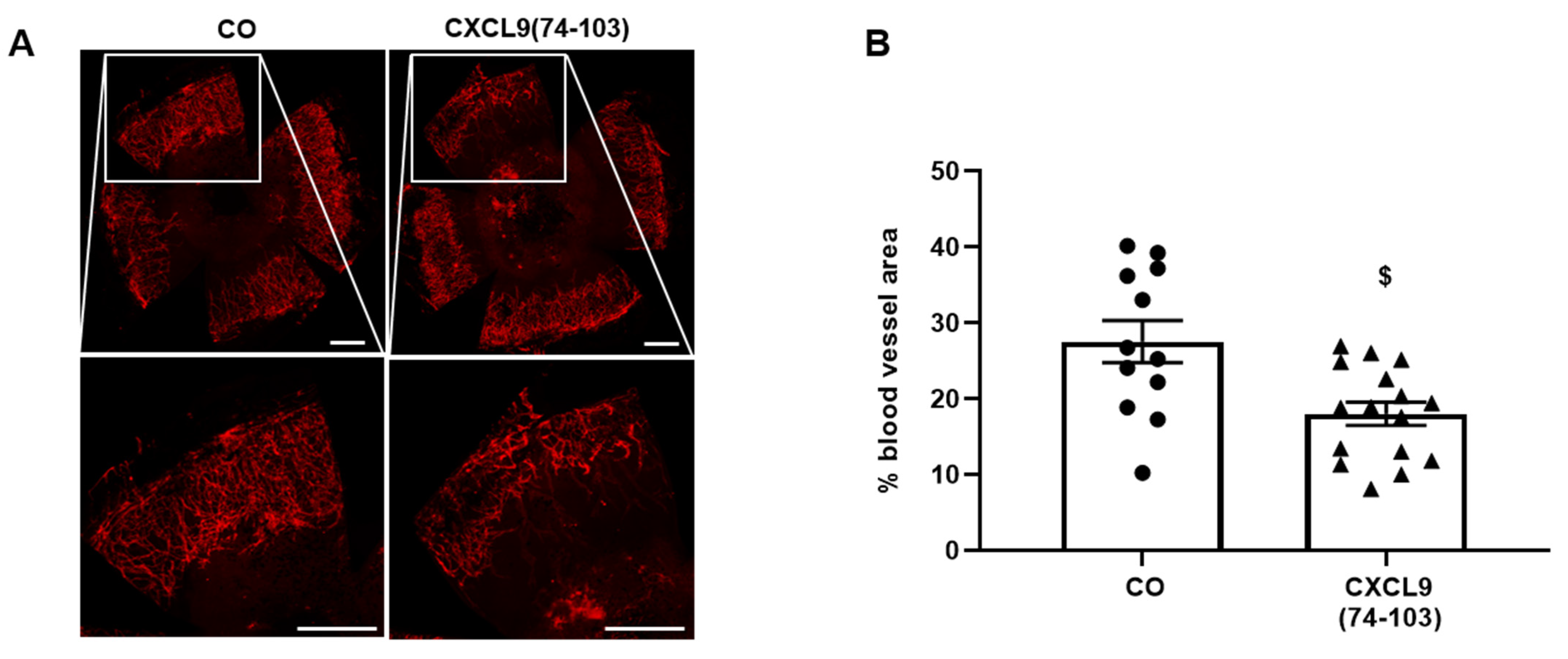
The corneal cauterization assay was performed, as previously described [
28]. Eight-week-old female C57BL/6 mice were anesthetized with a mixture of xylazine (10 mg/kg) and ketamine (100 mg/kg). After application of a local anesthetic (Unicaïne, 0.4%) to the eye, the cornea was thermally cauterized using an ophthalmic cautery. One day after cauterization, drops of 10 µL of CXCL9(74-103) (100 µg/mL) or PBS were applied daily for 4 days. On day 5, the mice were sacrificed, and the corneas were removed. Fixation and blocking of whole-mounted corneas were performed in 70% (
v/v) ethanol for 1 h and 3% (
w/v) BSA in PBS for 1 h, respectively. To stain for blood vessels, the corneas were incubated with rat anti-mouse CD31 antibody (clone MEC13.3, BD Biosciences) overnight and AlexaFluor568 goat anti-rat secondary antibody (Cat No A-11077, Invitrogen) for 2 h. Corneas were flat mounted on microscope glasses overlaid with Prolong Gold antifade mounting medium and imaged using a Leica DMI6000 microscope (Leica Microsystems, Wetzlar, Germany). The cornea blood vessel area was quantified using Leica MM AF morphometric analysis software (Leica Microsystems) and expressed as the percentage of the total corneal area. Ethical approval for animal experiments was obtained from the Ethical Committee of KU Leuven (number LA1210604).
2.15. In Vivo MDA-MB-231 Breast Cancer Mouse Model
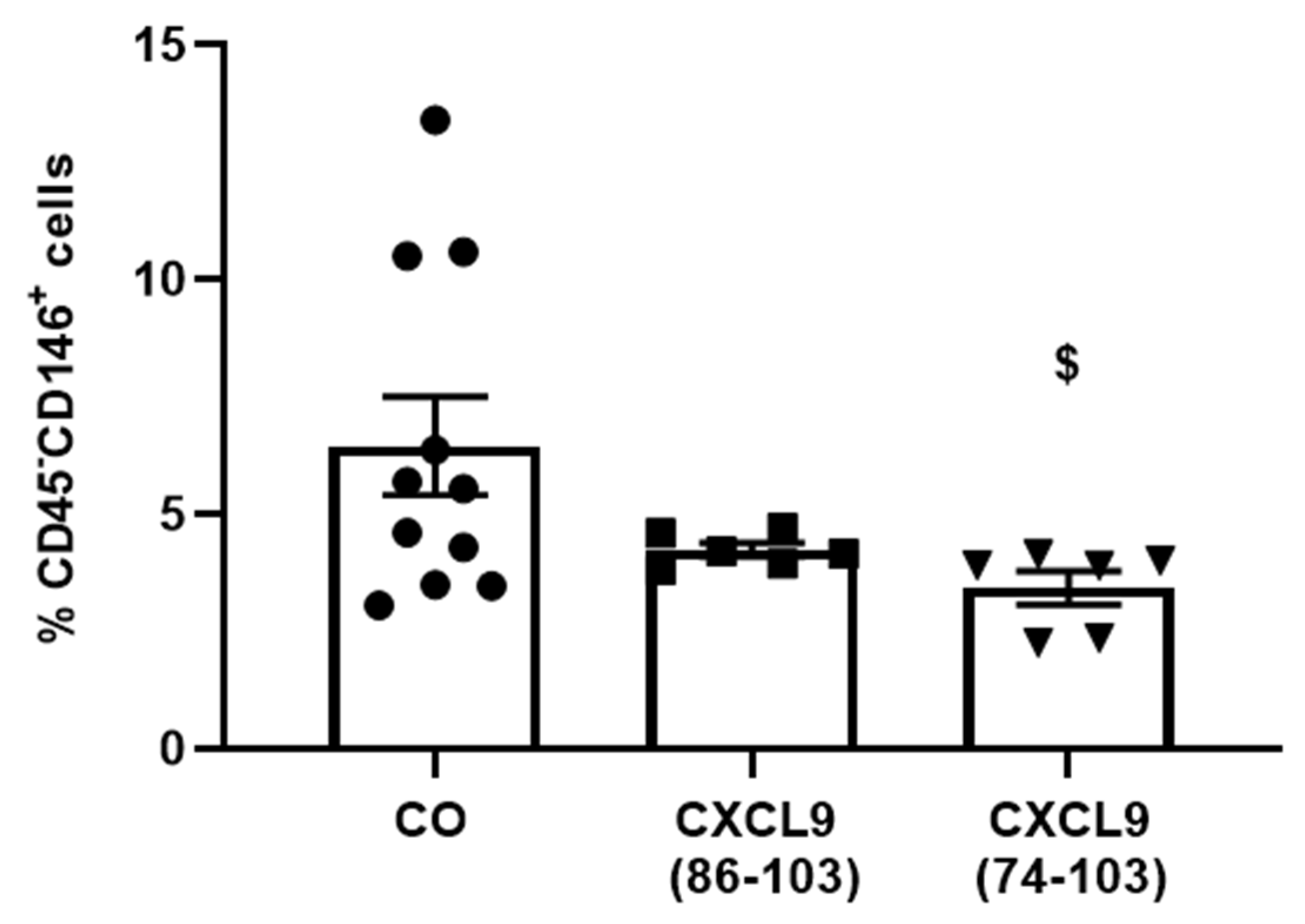
Six-to-eight-week-old female SCID mice were s.c. injected with 6 × 106 MDA-MB-231 breast tumor cells (American Type Culture Collection, Manassas, VA) in 200 µL PBS. Three days later, mice were anesthetized as described (vide supra), a dorsal incision was made and an ALZET osmotic pump (model 1002, DURECT Corporation) containing 800 µg/100 µL CXCL9(74-103) or CXCL9(86-103) or 100 µL PBS was s.c. implanted, which delivered a continuous dose over the course of two weeks. Three weeks after tumor cell injection, mice were sacrificed. Tumors were isolated, minced and added to 2 mL of tumor cell medium [TCM; 5% (v/v) FCS in RPMI 1640 + Glutamax (Gibco)]. After centrifugation (500 g, room temperature, 5 min), minced tumors were incubated at 37 °C for 30 min in 3 mL of digestion medium [2 mg/mL collagenase D, 0.1 mg/mL DNAse and 5 U/mL dispase (Corning Life Sciences) in TCM]. Subsequently, tumor fragments were further disrupted using a 1 mL syringe and 18G needle, and 2 mL of digestion medium was added for an additional incubation at 37 °C for 15 min. Finally, mechanical disruption was repeated, followed by centrifugation of the suspension (500 g, room temperature, 5 min). To stop the digestion, the pellet was placed on ice and resuspended in 1 mL 10 mM EDTA in PBS. Then, 2 mL of 2% (v/v) FCS in PBS was added and the samples were centrifuged at 500 g and 4 °C for 5 min. The pellet was resuspended in 1 mL ACK lysis buffer (Gibco) to remove red blood cells and the reaction was stopped after 3 min by adding 4 mL of 2% (v/v) FCS in PBS. To remove debris, the sample was run through a 70 µm cell strainer, which was washed with 5 mL of 2% (v/v) FCS in PBS. After centrifugation (500 g, 4 °C, 5 min), the pellet was resuspended in 500 µL of 2% (v/v) FCS in PBS and the cells were counted. To assess the anti-angiogenic potential of the CXCL9-derived peptides, the population of endothelial cells within the tumor samples was determined via flow cytometry (vide supra).
2.16. Statistical Analysis
The data are represented as mean ± SEM. Statistical analysis was carried out by first performing a Kruskal-Wallis test. Comparisons between groups were performed using a Mann–Whitney U test for nonpaired data or a Wilcoxon test for paired data.
4. Discussion
Glycosaminoglycans and their associated protein-bound form, proteoglycans, are implicated in numerous physiological and pathological processes. The importance of cell surface HSPGs, as integrators of growth factor signaling in angiogenesis has already been established in a number of studies [
17]. In addition, deficiency in the major lymphatic HSPG, syndecan-4, was associated with a dysfunctional VEGF-C:VEGFR3 complex formation required for adequate VEGFR3 signaling and VEGF-C-mediated pathological lymphangiogenesis in vivo [
37]. Given the importance of proteoglycans in pathological angiogenesis and the pressing need for additional angiogenesis inhibitors, targeting HS has become a field of intense investigation. In practice, however, the number of HS-targeting drugs is limited.
In our present study, we wanted to explore the potential of CXCL9(74-103), a high affinity GAG-binding peptide, to interfere with angiogenesis by targeting growth factor-induced signaling cascades and associated processes. We found that CXCL9(74-103) efficiently inhibited multiple angiogenic factor-mediated processes in vitro such as proliferation (VEGF165, FGF-2, EGF), chemotaxis (VEGF165, FGF-2, EGF), adhesion (VEGF) and spheroid sprouting (FGF-2, EGF) of endothelial cells, without inducing any cell toxicity. These findings were corroborated by the observation that CXCL9(74-103) reduced neovascularization in several in vivo models, wherein one or more of the growth factors studied in vitro is the principal actor inducing angiogenesis. We opted for diabetic retinopathy for VEGF, the Matrigel plug assay for FGF-2 and a xenograft breast cancer model for EGF. In the corneal cauterization model, beside VEGF, TGF-β and CCL2 are also involved [
33,
34,
35]. In our study, we made use of two CXCL9-derived COOH-terminal peptides. CXCL9(74-103) is characterized by a high affinity for GAGs, whereas CXCL9(86-103) has low GAG affinity. Our laboratory reported dissociation constants (K
D) of 61 nM [low molecular weight (LMW) heparin], 4.8 nM (HS) and 4.4 nM (DS) for CXCL9(74-103) [
22,
38]. Interestingly, CXCL9(74-103) showed a 12-fold higher affinity for HS compared with LMW heparin. These data also demonstrate that CXCL9(74-103) binds with a high but different affinity to these GAGs, which confers it a certain degree of binding specificity. In contrast, CXCL9(86-103) demonstrated a reduced affinity for GAGs with a K
D of 820 nM for heparin binding. This was also reflected in our results, where CXCL9(86-103) showed no overall effect on angiogenesis, despite seeming to affect FGF-2-induced proliferation. CXCL9(86-103) could not significantly improve diabetes-induced vascular leakage and had also no anti-angiogenic activity in several in vivo models, including tumor-associated angiogenesis and FGF-2-induced angiogenesis in the Matrigel plug assay (data not shown).
Earlier studies that report on targeting HS-mediated interactions most commonly rely on HS mimetics that either mask HS for GF binding or directly interact with the GF, preventing it from binding to HS [
39]. For example, LHT7, a chemically modified heparin was shown to inhibit angiogenesis by reducing tyrosine receptor activation by FGF-2 and VEGF165 via direct interaction with the two said growth factors [
40]. Pagano et al. reported that the small molecule inhibitor SM27, derived from the FGF-2-binding region of thrombospondin-1 (TSP-1), selectively bound the heparin-binding site of FGF-2 leading to reduced binding to FGFR1 and HS [
41]. As such, SM27 inhibited FGF-2-induced angiogenesis by interfering with a ternary complex formation between FGF, FGFR and HS. A computational and chemical strategy was used to investigate derivates of SM27, and several bi-naphtalenic small molecules were identified that bound FGF-2 and inhibited angiogenesis with increased potency over the original molecule [
42]. Recently, it was revealed that another TSP with anti-angiogenic activity, TSP-2, was also able to interact with FGF-2 and interfered with HS and FGFR1 binding. These findings indicated that TSP-2 could serve as a lead molecule to further develop FGF-2 inhibitors [
43]. In addition, a collagen Vα1-derived fragment with heparin-binding properties was shown to selectively bind to FGF-2, not VEGF-A, and inhibited only the FGF-2-mediated angiogenic processes [
44]. Our proposed CXCL9-derived peptide acted both as an HS- and GF-binding molecule. CXCL9(74-103) could potently inhibit VEGF165 binding to HS, which can account for the diminished phosphorylation of ERK by VEGF165 in the presence of CXCL9(74-103). Accordingly, we showed that CXCL9(74-103) interfered with VEGF165-induced endothelial cell proliferation, migration and adhesion. As such, it is plausible that CXCL9(74-103) prevents the formation of a functioning VEGF165:HS:VEGFR2 complex, resulting in inadequate signaling. Spontaneous proliferation was also significantly decreased by the high affinity GAG-binding peptide, which is probably due to inhibition of autocrine growth factors. Moreover, in a mouse model of vascular leakage, in which VEGF165 is thought to be a major contributor, CXCL9(74-103) ameliorated the diabetes-induced vascular permeability. The decrease in blood vessel outgrowth in the cornea after thermal cauterization in CXCL9(74-103)-treated mice, further underlines the effectiveness of the CXCL9-derived peptide to counteract VEGF in vivo.
In addition to binding to HS, CXCL9(74-103) also showed a direct association with FGF-2, with high nanomolar affinity. The obtained K
D value lies within the range of the concentrations that were applied in the different in vivo and in vitro assays reported here. FGF-2-mediated proliferation, chemotaxis and sprouting in vitro was diminished by CXCL9(74-103). Also in vivo, CXCL9(74-103) treatment attenuated FGF-2-mediated neovascularization in the Matrigel plug assay. The inhibitory mechanism responsible for countering FGF-2 could be that (1) by direct interaction with FGF-2, CXCL9(74-103) can trap the GF, preventing it from binding to HS and its FGFR or/and that (2) the concomitant binding of CXCL9(74-103) and FGF-2 to HS prevents effective signaling complex formation. Our observation that VEGF165 did not interact with CXCL9(74-103) could be explained by the observation that the structure and topology of the heparin-binding site of VEGF165 differ from that of other known heparin-binding proteins, such as FGF-2 [
45]. Furthermore, VEGF165 is also characterized by an 8-fold lower affinity for heparin compared to FGF-2 [
46].
Interestingly, in addition to functioning as a co-receptor initiating mitogen-activated protein kinase (MAPK) signaling, autonomous cell signaling through syndecans has also been reported [
14,
47]. Syndecan-4, the most abundant mammalian syndecan, has been described to function as an independent receptor for several heparin-binding growth factors such as FGFs, VEGFs and PDGFs [
47]. Upon ligand binding, the HSPGs oligomerize and initiate signaling components such as mammalian target of rapamycin (mTOR), RAC-α serine/threonine-protein kinase (AKT1) and the Rho family of GTPases, which are responsible for cell migration and adhesion [
47]. As such, syndecans are, unlike its family member glypicans, characterized by unique structural features. The extracellular domain bears the GAG chains which allows interaction with GAG-binding motifs of growth factors and with the extracellular matrix which enables adhesion. The transmembrane domain assists in efficient oligomerization and the intracellular domain initiates intracellular signaling cascades and recruits intracellular proteins to the cell surface. Therefore, it could also be possible that HS autonomous processes are targeted by CXCL9(74-103), but previous efforts have pinpointed the difficulty to dissect these events from those in cooperation with GFRs. As stated in the introduction, not EGF, but its receptor EGFR relies on interactions with syndecan-4, and integrins α
6β
4 and α
3β
1 to stimulate migration, survival and invasion [
16]. As such, a cooperative interplay between integrins and syndecans facilitates the translation of extracellular signals to the intracellular compartment. Moreover, a binding site in the extracellular domain of syndecan-4 spanning amino acids 87-131 captures the EGFR and α
3β
1 integrin and establishes a direct interaction between EGFR1 and syndecan-4. The cytoplasmic COOH-terminus of syndecan-4 engages with the COOH-terminal part of α
6β
4 integrin which in turn causes the cytoplasmic domain of α
6β
4 integrin to become phosphorylated and initiate downstream signaling. This led to the development of a peptide based on the EGFR interaction motifs, called synstatin (SSTN
EGFR) that displaces the RTK and α
3β
1 integrin from the syndecan and prevents cell migration [
48]. We showed that EGF-mediated endothelial cell proliferation, migration and sprouting were inhibited by CXCL9(74-103), including phosphorylation of ERK. Therefore, it is plausible that CXCL9(74-103) inhibits EGF-mediated processes by binding to the HS chains of endothelial syndecans, thereby interrupting syndecan:integrin:EGFR complex formation. This idea is strengthened by the observation that inhibition of EGF-induced endothelial sprouting by CXCL9(74-103) is abolished when the expression of cell surface HS is reduced. Also in vivo, CXCL9(74-103) attenuated angiogenesis in a breast tumor model wherein EGFR is upregulated. Similarly, in triple-negative breast cancer patients, an association between syndecan-1 and EGFR overexpression has been found [
49]. Upon syndecan-1 depletion, EGFR expression, as well as EGF-induced activation of Akt was reduced. In addition to EGF, a crosstalk between syndecans, integrin α
vβ
3 and VEGF in neovascularization has also been proposed. The used substratum gelatin in the adhesion assay also contains the RGD cell adhesive motif as a main receptor recognition pattern, which binds several integrins, but primarily α
5β
1 and α
vβ
3 integrins [
50]. Moreover, α
5β
1 and α
vβ
3 are two of the three integrins that mainly synergize with syndecans in the regulation of angiogenesis [
14]. Therefore, the disruption of an integrin:syndecan:VEGFR2 complex could also explain the CXCL9(74-103)-induced inhibition of endothelial cell adhesion in response to VEGF165.
Finally, we conclude that CXCL9(74-103) shows clear anti-angiogenic activity in vitro and in vivo by interfering with HSPG-mediated growth factor signaling. CXCL9(74-103) was able to counter various growth factor (EGF, VEGF165, FGF-2)-induced effects via different mechanisms. Considering the widespread involvement of HSPGs in different processes either autonomous or in conjunction with other players, difficulties arise to pinpoint the exact mechanism of action of CXCL9(74-103). However, more importantly, current anti-angiogenic cancer therapies targeting VEGF-A are confronted with resistance due to upregulation of other growth factors, such as FGF-2, EGF and PDGF by tumor cells. Therefore, strategies that target multiple growth factors can possibly subvert such compensatory mechanisms. In conclusion, CXCL9(74-103) forms an interesting lead molecule to further develop anti-angiogenics.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











