
Pancreatic Neuroendocrine Tumors: Molecular Mechanisms and Therapeutic Targets
 ,
,
Abstract
:Simple Summary
Abstract
1. pNET Introduction, Pathological Features and Classification
2. pNET-Associated Genes and Signaling Pathways
2.1. Menin
2.2. PI3K-Akt-mTOR Pathway
2.3. INK4A/ARF Locus and RB1 Pathway
2.4. ATRX/DAXX
2.5. p53 Pathway
2.6. VHL and Growth Factor Signaling
2.7. NF1 and RAS-RAF-MEK-ERK Pathway
2.8. Somatostatin Receptor Signaling
2.9. Miscellaneous Genes and Pathways
2.9.1. Myc
2.9.2. Src Family Kinases
2.9.3. RABL6A
2.9.4. HDACs
2.9.5. Heat Shock Protein (HSP) 90
2.9.6. Aurora Kinase
2.9.7. Developmental Pathways
3. In Vitro and Vivo pNET Models
3.1. Human pNET Cell Lines
3.2. 3D Cultures
3.3. Patient Derived Xenografts
3.4. Genetically Engineered Mouse Models
4. Concluding Thoughts
Author Contributions
Funding
Conflicts of Interest
References
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Oronsky, B.; Ma, P.C.; Morgensztern, D.; Carter, C.A. Nothing But NET: A Review of Neuroendocrine Tumors and Carcinomas. Neoplasia 2017, 19, 991–1002. [Google Scholar] [CrossRef]
- Sun, J. Pancreatic neuroendocrine tumors. Intractable Rare Dis. Res. 2017, 6, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halfdanarson, T.R.; Rabe, K.G.; Rubin, J.; Petersen, G.M. Pancreatic neuroendocrine tumors (PNETs): Incidence, prognosis and recent trend toward improved survival. Ann. Oncol. 2008, 19, 1727–1733. [Google Scholar] [CrossRef]
- Vortmeyer, A.O.; Huang, S.; Lubensky, I.; Zhuang, Z. Non-islet origin of pancreatic islet cell tumors. J. Clin. Endocrinol. Metab. 2004, 89, 1934–1938. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.T.; Berna, M.J.; Bingham, D.B.; Norton, J.A. Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer 2008, 113, 1807–1843. [Google Scholar] [CrossRef] [Green Version]
- Falconi, M.; Eriksson, B.; Kaltsas, G.; Bartsch, D.K.; Capdevila, J.; Caplin, M.; Kos-Kudla, B.; Kwekkeboom, D.; Rindi, G.; Klöppel, G.; et al. ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2016, 103, 153–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frizziero, M.; Chakrabarty, B.; Nagy, B.; Lamarca, A.; Hubner, R.A.; Valle, J.W.; McNamara, M.G. Mixed Neuroendocrine Non-Neuroendocrine Neoplasms: A Systematic Review of a Controversial and Underestimated Diagnosis. J. Clin. Med. 2020, 9, 273. [Google Scholar] [CrossRef] [Green Version]
- Choe, J.; Kim, K.W.; Kim, H.J.; Kim, D.W.; Kim, K.P.; Hong, S.-M.; Ryu, J.-S.; Tirumani, S.H.; Krajewski, K.; Ramaiya, N. What Is New in the 2017 World Health Organization Classification and 8th American Joint Committee on Cancer Staging System for Pancreatic Neuroendocrine Neoplasms? Korean J. Radiol. 2019, 20, 5–17. [Google Scholar] [CrossRef]
- Yachida, S.; Vakiani, E.; White, C.M.; Zhong, Y.; Saunders, T.; Morgan, R.; De Wilde, R.F.; Maitra, A.; Hicks, J.; DeMarzo, A.M.; et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am. J. Surg. Pathol. 2012, 36, 173–184. [Google Scholar] [CrossRef]
- Kawasaki, K.; Fujii, M.; Sato, T. Gastroenteropancreatic neuroendocrine neoplasms: Genes, therapies and models. Dis. Models Mech. 2018, 11, dmm029595. [Google Scholar] [CrossRef] [Green Version]
- Klöppel, G.; Rindi, G.; Perren, A.; Komminoth, P.; Klimstra, D.S. The ENETS and AJCC/UICC TNM classifications of the neuroendocrine tumors of the gastrointestinal tract and the pancreas: A statement. Virchows Arch. 2010, 456, 595–597. [Google Scholar] [CrossRef]
- Klöppel, G. Classification and pathology of gastroenteropancreatic neuroendocrine neoplasms. Endocr. Relat. Cancer 2011, 18, S1–S16. [Google Scholar] [CrossRef] [PubMed]
- Akirov, A.; Larouche, V.; Alshehri, S.; Asa, S.L.; Ezzat, S. Treatment Options for Pancreatic Neuroendocrine Tumors. Cancers 2019, 11, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscogiuri, G.; Altieri, B.; Albertelli, M.; Dotto, A.; Modica, R.; Barrea, L.; Fanciulli, G.; Feola, T.; Baldelli, R.; Ruggeri, R.M.; et al. Epidemiology of pancreatic neuroendocrine neoplasms: A gender perspective. Endocrine 2020, 69, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Zandee, W.T.; Brabander, T.; Blažević, A.; Kam, B.L.R.; Teunissen, J.J.M.; Feelders, R.A.; Hofland, J.; de Herder, W.W. Symptomatic and Radiological Response to 177Lu-DOTATATE for the Treatment of Functioning Pancreatic Neuroendocrine Tumors. J. Clin. Endocrinol. Metab. 2019, 104, 1336–1344. [Google Scholar] [CrossRef]
- Brabander, T.; van der Zwan, W.A.; Teunissen, J.J.M.; Kam, B.L.R.; Feelders, R.A.; de Herder, W.W.; van Eijck, C.H.J.; Franssen, G.J.H.; Krenning, E.P.; Kwekkeboom, D.J. Long-Term Efficacy, Survival, and Safety of [(177)Lu-DOTA(0),Tyr(3)]octreotate in Patients with Gastroenteropancreatic and Bronchial Neuroendocrine Tumors. Clin. Cancer Res. 2017, 23, 4617–4624. [Google Scholar] [CrossRef] [Green Version]
- Zandee, W.T.; van Adrichem, R.C.; Kamp, K.; Feelders, R.A.; van Velthuysen, M.-L.F.; de Herder, W.W. Incidence and prognostic value of serotonin secretion in pancreatic neuroendocrine tumours. Clin. Endocrinol. 2017, 87, 165–170. [Google Scholar] [CrossRef]
- Kulke, M.H.; O’Dorisio, T.; Phan, A.; Bergsland, E.; Law, L.; Banks, P.; Freiman, J.; Frazier, K.; Jackson, J.; Yao, J.C.; et al. Telotristat etiprate, a novel serotonin synthesis inhibitor, in patients with carcinoid syndrome and diarrhea not adequately controlled by octreotide. Endocr. Relat. Cancer 2014, 21, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Hruban, R.H.; van Mansfeld, A.D.; Offerhaus, G.J.; van Weering, D.H.; Allison, D.C.; Goodman, S.N.; Kensler, T.W.; Bose, K.K.; Cameron, J.L.; Bos, J.L. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am. J. Pathol. 1993, 143, 545–554. [Google Scholar]
- Redston, M.S.; Caldas, C.; Seymour, A.B.; Hruban, R.H.; da Costa, L.; Yeo, C.J.; Kern, S.E. p53 Mutations in Pancreatic Carcinoma and Evidence of Common Involvement of Homocopolymer Tracts in DNA Microdeletions. Cancer Res. 1994, 54, 3025–3033. [Google Scholar] [PubMed]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, C.; Skogseid, B.; Oberg, K.; Nakamura, Y.; Nordenskjold, M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 1988, 332, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Byström, C.; Larsson, C.; Blomberg, C.; Sandelin, K.; Falkmer, U.; Skogseid, B.; Oberg, K.; Werner, S.; Nordenskjöld, M. Localization of the MEN1 gene to a small region within chromosome 11q13 by deletion mapping in tumors. Proc. Natl. Acad. Sci. USA 1990, 87, 1968–1972. [Google Scholar] [CrossRef]
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelenko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, I.; Van de Ven, W.J.; Kas, K.; Zhang, C.X.; Giraud, S.; Wautot, V.; Buisson, N.; De Witte, K.; Salandre, J.; Lenoir, G.; et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum. Mol. Genet. 1997, 6, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Toliat, M.R.; Berger, W.; Ropers, H.H.; Neuhaus, P.; Wiedenmann, B. Mutations in the MEN I gene in sporadic neuroendocrine tumours of gastroenteropancreatic system. Lancet 1997, 350, 1223. [Google Scholar] [CrossRef] [Green Version]
- Corbo, V.; Dalai, I.; Scardoni, M.; Barbi, S.; Beghelli, S.; Bersani, S.; Albarello, L.; Doglioni, C.; Schott, C.; Capelli, P.; et al. MEN1 in pancreatic endocrine tumors: Analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr. Relat. Cancer 2010, 17, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Rigaud, G.; Missiaglia, E.; Moore, P.S.; Zamboni, G.; Falconi, M.; Talamini, G.; Pesci, A.; Baron, A.; Lissandrini, D.; Rindi, G.; et al. High Resolution Allelotype of Nonfunctional Pancreatic Endocrine Tumors: Identification of Two Molecular Subgroups with Clinical Implications. Cancer Res. 2001, 61, 285–292. [Google Scholar] [PubMed]
- Mailman, M.D.; Muscarella, P.; Schirmer, W.J.; Ellison, E.C.; O’Dorisio, T.M.; Prior, T.W. Identification of MEN1 Mutations in Sporadic Enteropancreatic Neuroendocrine Tumors by Analysis of Paraffin-embedded Tissue. Clin. Chem. 1999, 45, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Eubanks, P.J.; Sawicki, M.P.; Samara, G.J.; Gatti, R.; Nakamura, Y.; Tsao, D.; Johnson, C.; Hurwitz, M.; Wan, Y.J.; Passaro, E., Jr. Putative tumor-suppressor gene on chromosome 11 is important in sporadic endocrine tumor formation. Am. J. Surg. 1994, 167, 180–185. [Google Scholar] [CrossRef]
- Hessman, O.; Lindberg, D.; Skogseid, B.; Carling, T.; Hellman, P.; Rastad, J.; Akerstrom, G.; Westin, G. Mutation of the multiple endocrine neoplasia type 1 gene in nonfamilial, malignant tumors of the endocrine pancreas. Cancer Res. 1998, 58, 377–379. [Google Scholar] [PubMed]
- Keutgen, X.M.; Kumar, S.; Gara, S.K.; Boufraqech, M.; Agarwal, S.; Hruban, R.H.; Nilubol, N.; Quezado, M.; Finney, R.; Cam, M.; et al. Transcriptional alterations in hereditary and sporadic nonfunctioning pancreatic neuroendocrine tumors according to genotype. Cancer 2018, 124, 636–647. [Google Scholar] [CrossRef] [Green Version]
- Görtz, B.; Roth, J.; Krähenmann, A.; de Krijger, R.R.; Muletta-Feurer, S.; Rütimann, K.; Saremaslani, P.; Speel, E.J.; Heitz, P.U.; Komminoth, P. Mutations and allelic deletions of the MEN1 gene are associated with a subset of sporadic endocrine pancreatic and neuroendocrine tumors and not restricted to foregut neoplasms. Am. J. Pathol. 1999, 154, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Z.; Vortmeyer, A.O.; Pack, S.; Huang, S.; Pham, T.A.; Wang, C.; Park, W.S.; Agarwal, S.K.; Debelenko, L.V.; Kester, M.; et al. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res 1997, 57, 4682–4686. [Google Scholar]
- Shan, L.; Nakamura, Y.; Nakamura, M.; Yokoi, T.; Tsujimoto, M.; Arima, R.; Kameya, T.; Kakudo, K. Somatic mutations of multiple endocrine neoplasia type 1 gene in the sporadic endocrine tumors. Lab. Investig. A J. Tech. Methods Pathol. 1998, 78, 471–475. [Google Scholar]
- Lawrence, B.; Blenkiron, C.; Parker, K.; Tsai, P.; Fitzgerald, S.; Shields, P.; Robb, T.; Yeong, M.L.; Kramer, N.; James, S.; et al. Recurrent loss of heterozygosity correlates with clinical outcome in pancreatic neuroendocrine cancer. NPJ Genom. Med. 2018, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Garg, A.; Chen, D.; Capdevila, J.; Engstrom, P.; Pommier, R.; Van Cutsem, E.; Singh, S.; Fazio, N.; He, W.; et al. Genomic profiling of NETs: A comprehensive analysis of the RADIANT trials. Endocr.-Relat. Cancer 2019, 26, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Cavallari, I.; D’Agostino, D.M.; Ferro, T.; Rosato, A.; Barzon, L.; Pasquali, C.; Fogar, P.; Theodoropoulou, M.; Esposito, G.; Boscaro, M.; et al. In situ analysis of human menin in normal and neoplastic pancreatic tissues: Evidence for differential expression in exocrine and endocrine cells. J. Clin. Endocrinol. Metab. 2003, 88, 3893–3901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaji, H.; Canaff, L.; Lebrun, J.J.; Goltzman, D.; Hendy, G.N. Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 3837–3842. [Google Scholar] [CrossRef] [Green Version]
- Raj, N.; Shah, R.; Stadler, Z.; Mukherjee, S.; Chou, J.; Untch, B.; Li, J.; Kelly, V.; Saltz, L.B.; Mandelker, D.; et al. Real-Time Genomic Characterization of Metastatic Pancreatic Neuroendocrine Tumors Has Prognostic Implications and Identifies Potential Germline Actionability. JCO Precis Oncol. 2018, 2018, PO.17.00267. [Google Scholar] [CrossRef]
- Guru, S.C.; Goldsmith, P.K.; Burns, A.L.; Marx, S.J.; Spiegel, A.M.; Collins, F.S.; Chandrasekharappa, S.C. Menin, the product of the MEN1 gene, is a nuclear protein. Proc. Natl. Acad. Sci. USA 1998, 95, 1630–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wautot, V.; Khodaei, S.; Frappart, L.; Buisson, N.; Baro, E.; Lenoir, G.M.; Calender, A.; Zhang, C.X.; Weber, G. Expression analysis of endogenous menin, the product of the multiple endocrine neoplasia type 1 gene, in cell lines and human tissues. Int. J. Cancer 2000, 85, 877–881. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Guru, S.C.; Heppner, C.; Erdos, M.R.; Collins, R.M.; Park, S.Y.; Saggar, S.; Chandrasekharappa, S.C.; Collins, F.S.; Spiegel, A.M.; et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 1999, 96, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Heppner, C.; Bilimoria, K.Y.; Agarwal, S.K.; Kester, M.; Whitty, L.J.; Guru, S.C.; Chandrasekharappa, S.C.; Collins, F.S.; Spiegel, A.M.; Marx, S.J.; et al. The tumor suppressor protein menin interacts with NF-kappaB proteins and inhibits NF-kappaB-mediated transactivation. Oncogene 2001, 20, 4917–4925. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Mao, H.; Schnepp, R.W.; Sykes, S.M.; Silva, A.C.; D’Andrea, A.D.; Hua, X. Menin Associates with FANCD2, a Protein Involved in Repair of DNA Damage. Cancer Res. 2003, 63, 4204–4210. [Google Scholar] [PubMed]
- Hessman, O.; Skogseid, B.; Westin, G.; Akerström, G.R. Multiple Allelic Deletions and Intratumoral Genetic Heterogeneity in MEN1 Pancreatic Tumors1. J. Clin. Endocrinol. Metab. 2001, 86, 1355–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, A.; Wang, Z.; Wysocka, J.; Sanyal, M.; Aufiero, D.J.; Kitabayashi, I.; Herr, W.; Cleary, M.L. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol. Cell. Biol. 2004, 24, 5639–5649. [Google Scholar] [CrossRef] [Green Version]
- Karnik, S.K.; Hughes, C.M.; Gu, X.; Rozenblatt-Rosen, O.; McLean, G.W.; Xiong, Y.; Meyerson, M.; Kim, S.K. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc. Natl. Acad. Sci. USA 2005, 102, 14659–14664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, T.A.; Hughes, C.M.; Lloyd, R.; Yang, Z.; Rozenblatt-Rosen, O.; Dou, Y.; Schnepp, R.W.; Krankel, C.; LiVolsi, V.A.; Gibbs, D.; et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnepp, R.W.; Chen, Y.X.; Wang, H.; Cash, T.; Silva, A.; Diehl, J.A.; Brown, E.; Hua, X. Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 2006, 66, 5707–5715. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, A.; Mukherjee, S.; Lack, J.; Gara, S.K.; Wang, S.; Quezado, M.M.; Keutgen, X.M.; Wu, X.; Cam, M.; Kumar, S.; et al. Distinct genome-wide methylation patterns in sporadic and hereditary nonfunctioning pancreatic neuroendocrine tumors. Cancer 2019, 125, 1247–1257. [Google Scholar] [CrossRef]
- Feng, Z.; Wang, L.; Sun, Y.; Jiang, Z.; Domsic, J.; An, C.; Xing, B.; Tian, J.; Liu, X.; Metz, D.C.; et al. Menin and Daxx Interact to Suppress Neuroendocrine Tumors through Epigenetic Control of the Membrane Metallo-Endopeptidase. Cancer Res. 2017, 77, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.; Tang, L.H.; Davidson, C.; Vosburgh, E.; Chen, W.; Foran, D.J.; Notterman, D.A.; Levine, A.J.; Xu, E.Y. Two well-differentiated pancreatic neuroendocrine tumor mouse models. Cell Death Differ. 2019, 27, 269–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Heritable Formation OF Pancreatic Beta-Cell Tumors in Transgenic Mice Expressing Recombinant Insulin Simian Virus-40 Oncogenes. Nature 1985, 315, 115–122. [Google Scholar] [CrossRef]
- Kobayashi, S.; Contractor, T.; Vosburgh, E.; Du, Y.N.; Tang, L.H.; Clausen, R.; Harris, C.R. Alleles of Insm1 determine whether RIP1-Tag2 mice produce insulinomas or nonfunctioning pancreatic neuroendocrine tumors. Oncogenesis 2019, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Alliouachene, S.; Tuttle, R.L.; Boumard, S.; Lapointe, T.; Berissi, S.; Germain, S.; Jaubert, F.; Tosh, D.; Birnbaum, M.J.; Pende, M. Constitutively active Akt1 expression in mouse pancreas requires S6 kinase 1 for insulinoma formation. J. Clin. Investig. 2008, 118, 3629–3638. [Google Scholar] [CrossRef]
- Murphy, D.; Bishop, A.; Rindi, G.; Murphy, M.N.; Stamp, G.W.; Hanson, J.; Polak, J.M.; Hogan, B. Mice transgenic for a vasopressin-SV40 hybrid oncogene develop tumors of the endocrine pancreas and the anterior pituitary. A possible model for human multiple endocrine neoplasia type 1. Am. J. Pathol. 1987, 129, 552–566. [Google Scholar]
- Crabtree, J.S.; Scacheri, P.C.; Ward, J.M.; Garrett-Beal, L.; Emmert-Buck, M.R.; Edgemon, K.A.; Lorang, D.; Libutti, S.K.; Chandrasekharappa, S.C.; Marx, S.J.; et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 1118–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolino, P.; Tong, W.M.; Galendo, D.; Wang, Z.Q.; Zhang, C.X. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol. Endocrinol. 2003, 17, 1880–1892. [Google Scholar] [CrossRef] [Green Version]
- Biondi, C.A.; Gartside, M.G.; Waring, P.; Loffler, K.A.; Stark, M.S.; Magnuson, M.A.; Kay, G.F.; Hayward, N.K. Conditional Inactivation of the Men1 Gene Leads to Pancreatic and Pituitary Tumorigenesis but Does Not Affect Normal Development of These Tissues. Mol. Cell. Biol. 2004, 24, 3125–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.C.; Ylaya, K.; Pechhold, K.; Wilson, A.; Adem, A.; Hewitt, S.M.; Libutti, S.K. Multiple endocrine neoplasia type 1 deletion in pancreatic alpha-cells leads to development of insulinomas in mice. Endocrinology 2010, 151, 4024–4030. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.C.J.; He, M.; Powell, A.; Adem, A.; Lorang, D.; Heller, C.; Grover, A.C.; Ylaya, K.; Hewitt, S.M.; Marx, S.J.; et al. Recapitulation of Pancreatic Neuroendocrine Tumors in Human Multiple Endocrine Neoplasia Type I Syndrome via Pdx1-Directed Inactivation of Men1. Cancer Res. 2009, 69, 1858–1866. [Google Scholar] [CrossRef] [Green Version]
- Lines, K.E.; Vas Nunes, R.P.; Frost, M.; Yates, C.J.; Stevenson, M.; Thakker, R.V. A MEN1 pancreatic neuroendocrine tumour mouse model under temporal control. Endocr. Connect. 2017, 6, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Crabtree, J.S.; Scacheri, P.C.; Ward, J.M.; McNally, S.R.; Swain, G.P.; Montagna, C.; Hager, J.H.; Hanahan, D.; Edlund, H.; Magnuson, M.A.; et al. Of mice and MEN1: Insulinomas in a conditional mouse knockout. Mol. Cell. Biol. 2003, 23, 6075–6085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolino, P.; Tong, W.M.; Herrera, P.L.; Casse, H.; Zhang, C.X.; Wang, Z.Q. Pancreatic beta-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer Res. 2003, 63, 4836–4841. [Google Scholar]
- Lu, J.; Herrera, P.L.; Carreira, C.; Bonnavion, R.; Seigne, C.; Calender, A.; Bertolino, P.; Zhang, C.X. Alpha cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology 2010, 138, 1954–1965. [Google Scholar] [CrossRef]
- Efrat, S.; Teitelman, G.; Anwar, M.; Ruggiero, D.; Hanahan, D. Glucagon gene regulatory region directs oncoprotein expression to neurons and pancreatic a cells. Neuron 1988, 1, 605–613. [Google Scholar] [CrossRef]
- Yu, R.; Dhall, D.; Nissen, N.N.; Zhou, C.; Ren, S.G. Pancreatic neuroendocrine tumors in glucagon receptor-deficient mice. PLoS ONE 2011, 6, e23397. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.B.; Reens, J.; Brocklehurst, S.R.; Betts, C.J.; Bickerton, S.; Bigley, A.L.; Jenkins, R.P.; Whalley, N.M.; Morgan, D.; Smith, D.M. Islets of Langerhans from prohormone convertase-2 knockout mice show alpha-cell hyperplasia and tumorigenesis with elevated alpha-cell neogenesis. Int. J. Exp. Pathol. 2014, 95, 29–48. [Google Scholar] [CrossRef]
- Azzopardi, S.; Pang, S.; Klimstra, D.S.; Du, Y.N. p53 and p16(Ink4a)/p19(Arf) Loss Promotes Different Pancreatic Tumor Types from PyMT-Expressing Progenitor Cells. Neoplasia 2016, 18, 610–617. [Google Scholar] [CrossRef] [Green Version]
- Pelengaris, S.; Khan, M.; Evan, G.I. Suppression of Myc-Induced Apoptosis in β Cells Exposes Multiple Oncogenic Properties of Myc and Triggers Carcinogenic Progression. Cell 2002, 109, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, Y.; Kodama, Y.; Shiokawa, M.; Kakiuchi, N.; Marui, S.; Kuwada, T.; Sogabe, Y.; Tomono, T.; Mima, A.; Morita, T.; et al. Rb and p53 Execute Distinct Roles in the Development of Pancreatic Neuroendocrine Tumors. Cancer Res. 2020, 80, 3620–3630. [Google Scholar] [CrossRef]
- Xu, E.Y.; Vosburgh, E.; Wong, C.; Tang, L.H.; Notterman, D.A. Genetic analysis of the cooperative tumorigenic effects of targeted deletions of tumor suppressors Rb1, Trp53, Men1, and Pten in neuroendocrine tumors in mice. Oncotarget 2020, 11, 2718–2739. [Google Scholar] [CrossRef]
- Maharjan, C.K.; Umesalma, S.; Kaemmer, C.A.; Muniz, V.P.; Bauchle, C.; Mott, S.L.; Zamba, K.D.; Breheny, P.; Leidinger, M.R.; Darbro, B.W.; et al. RABL6A Promotes Pancreatic Neuroendocrine Tumor Angiogenesis and Progression In Vivo. Biomedicines 2021, 9, 633. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Gardiner, J.C.; Maggi, E.C.; Huang, S.; Adem, A.; Bagdasarov, S.; Li, G.; Lee, S.; Slegowski, D.; Exarchakis, A.; et al. B7 immune-checkpoints as targets for the treatment of neuroendocrine tumors. Endocr. Relat. Cancer 2021, 28, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Neychev, V.; Sadowski, S.M.; Zhu, J.; Allgaeuer, M.; Kilian, K.; Meltzer, P.; Kebebew, E. Neuroendocrine Tumor of the Pancreas as a Manifestation of Cowden Syndrome: A Case Report. J. Clin. Endocrinol. Metab. 2016, 101, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, A.F.; Rebhun, J.F.; Clark, G.J.; Quilliam, L.A. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J. Biol. Chem. 2003, 278, 32493–32496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francalanci, P.; Diomedi-Camassei, F.; Purificato, C.; Santorelli, F.M.; Giannotti, A.; Dominici, C.; Inserra, A.; Boldrini, R. Malignant pancreatic endocrine tumor in a child with tuberous sclerosis. Am. J. Surg. Pathol. 2003, 27, 1386–1389. [Google Scholar] [CrossRef]
- Merritt, J.L., 2nd; Davis, D.M.; Pittelkow, M.R.; Babovic-Vuksanovic, D. Extensive acrochordons and pancreatic islet-cell tumors in tuberous sclerosis associated with TSC2 mutations. Am. J. Med Genet. Part A 2006, 140, 1669–1672. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, S.; van Diemen-Steenvoorde, R.; Akkersdijk, W.L.; Bax, N.M.; Ariyurek, Y.; Hermans, C.J.; van Nieuwenhuizen, O.; Nikkels, P.G.; Lindhout, D.; Halley, D.J.; et al. Malignant pancreatic tumour within the spectrum of tuberous sclerosis complex in childhood. Eur. J. Pediatrics 1999, 158, 284–287. [Google Scholar] [CrossRef] [Green Version]
- Simpson, L.; Parsons, R. PTEN: Life as a tumor suppressor. Exp. Cell Res. 2001, 264, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef]
- Chung, D.C.; Brown, S.B.; Graeme-Cook, F.; Tillotson, L.G.; Warshaw, A.L.; Jensen, R.T.; Arnold, A. Localization of Putative Tumor Suppressor Loci by Genome-wide Allelotyping in Human Pancreatic Endocrine Tumors. Cancer Res. 1998, 58, 3706–3711. [Google Scholar] [PubMed]
- Perren, A.; Komminoth, P.; Saremaslani, P.; Matter, C.; Feurer, S.; Lees, J.A.; Heitz, P.U.; Eng, C. Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. Am. J. Pathol. 2000, 157, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Missiaglia, E.; Dalai, I.; Barbi, S.; Beghelli, S.; Falconi, M.; della Peruta, M.; Piemonti, L.; Capurso, G.; Di Florio, A.; delle Fave, G.; et al. Pancreatic Endocrine Tumors: Expression Profiling Evidences a Role for AKT-mTOR Pathway. J. Clin. Oncol. 2010, 28, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Ghayouri, M.; Boulware, D.; Nasir, A.; Strosberg, J.; Kvols, L.; Coppola, D. Activation of the serine/theronine protein kinase Akt in enteropancreatic neuroendocrine tumors. Anticancer Res. 2010, 30, 5063–5067. [Google Scholar]
- Shida, T.; Kishimoto, T.; Furuya, M.; Nikaido, T.; Koda, K.; Takano, S.; Kimura, F.; Shimizu, H.; Yoshidome, H.; Ohtsuka, M.; et al. Expression of an activated mammalian target of rapamycin (mTOR) in gastroenteropancreatic neuroendocrine tumors. Cancer Chemother. Pharm. 2010, 65, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Djukom, C.; Porro, L.J.; Mrazek, A.; Townsend, C.M., Jr.; Hellmich, M.R.; Chao, C. Dual inhibition of PI3K and mTOR signaling pathways decreases human pancreatic neuroendocrine tumor metastatic progression. Pancreas 2014, 43, 88–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umesalma, S.; Kaemmer, C.A.; Kohlmeyer, J.L.; Letney, B.; Schab, A.M.; Reilly, J.A.; Sheehy, R.M.; Hagen, J.; Tiwari, N.; Zhan, F.; et al. RABL6A inhibits tumor-suppressive PP2A/AKT signaling to drive pancreatic neuroendocrine tumor growth. J. Clin. Investig. 2019, 130, 1641–1653. [Google Scholar] [CrossRef]
- Soler, A.; Figueiredo, A.M.; Castel, P.; Martin, L.; Monelli, E.; Angulo-Urarte, A.; Mila-Guasch, M.; Vinals, F.; Baselga, J.; Casanovas, O.; et al. Therapeutic Benefit of Selective Inhibition of p110alpha PI3-Kinase in Pancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2016, 22, 5805–5817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolin, E.M. PI3K/Akt/mTOR pathway inhibitors in the therapy of pancreatic neuroendocrine tumors. Cancer Lett. 2013, 335, 1–8. [Google Scholar] [CrossRef]
- Chiu, C.W.; Nozawa, H.; Hanahan, D. Survival Benefit With Proapoptotic Molecular and Pathologic Responses From Dual Targeting of Mammalian Target of Rapamycin and Epidermal Growth Factor Receptor in a Preclinical Model of Pancreatic Neuroendocrine Carcinogenesis. J. Clin. Oncol. 2010, 28, 4425–4433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grozinsky-Glasberg, S.; Franchi, G.; Teng, M.; Leontiou, C.A.; de Oliveira, A.R., Jr.; Dalino, P.; Salahuddin, N.; Korbonits, M.; Grossman, A.B. Octreotide and the mTOR inhibitor RAD001 (everolimus) block proliferation and interact with the Akt-mTOR-p70S6K pathway in a neuro-endocrine tumour cell Line. Neuroendocrinology 2008, 87, 168–181. [Google Scholar] [CrossRef]
- Moreno, A.; Akcakanat, A.; Munsell, M.F.; Soni, A.; Yao, J.C.; Meric-Bernstam, F. Antitumor activity of rapamycin and octreotide as single agents or in combination in neuroendocrine tumors. Endocr. Relat. Cancer 2008, 15, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Zitzmann, K.; De Toni, E.N.; Brand, S.; Goke, B.; Meinecke, J.; Spottl, G.; Meyer, H.H.; Auernhammer, C.J. The novel mTOR inhibitor RAD001 (everolimus) induces antiproliferative effects in human pancreatic neuroendocrine tumor cells. Neuroendocrinology 2007, 85, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.C.; Lombard-Bohas, C.; Baudin, E.; Kvols, L.K.; Rougier, P.; Ruszniewski, P.; Hoosen, S.; St Peter, J.; Haas, T.; Lebwohl, D.; et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: A phase II trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 69–76. [Google Scholar] [CrossRef]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.E.; et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Gallo, M.; Malandrino, P.; Fanciulli, G.; Rota, F.; Faggiano, A.; Colao, A. Everolimus as first line therapy for pancreatic neuroendocrine tumours: Current knowledge and future perspectives. J. Cancer Res. Clin. Oncol. 2017, 143, 1209–1224. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, C.E.; German, M.S.; Yang, K.; Wang, J.; VanBrocklin, H.; Regan, M.; Shokat, K.M.; Ducker, G.S.; Kim, G.E.; Hann, B.; et al. A Patient-derived Xenograft Model of Pancreatic Neuroendocrine Tumors Identifies Sapanisertib as a Possible New Treatment for Everolimus-resistant Tumors. Mol. Cancer 2018, 17, 2702–2709. [Google Scholar] [CrossRef] [Green Version]
- Serrano, J.; Goebel, S.U.; Peghini, P.L.; Lubensky, I.A.; Gibril, F.; Jensen, R.T. Alterations in the p16INK4a/CDKN2A tumor suppressor gene in gastrinomas. J. Clin. Endocrinol. Metab. 2000, 85, 4146–4156. [Google Scholar] [CrossRef]
- Lubomierski, N.; Kersting, M.; Bert, T.; Muench, K.; Wulbrand, U.; Schuermann, M.; Bartsch, D.; Simon, B. Tumor suppressor genes in the 9p21 gene cluster are selective targets of inactivation in neuroendocrine gastroenteropancreatic tumors. Cancer Res. 2001, 61, 5905–5910. [Google Scholar]
- Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt. Cancer Res. 2006, 66, 1500. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.T.; Weitz, M.; Breheny, P.J.; Ear, P.H.; Darbro, B.; Brown, B.J.; Braun, T.A.; Li, G.; Umesalma, S.; Kaemmer, C.A.; et al. Gene Expression Signatures Identify Novel Therapeutics for Metastatic Pancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2020, 26, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-M.; Shan, Y.-S.; Chu, P.-Y.; Jiang, S.S.; Hung, W.-C.; Chen, Y.-L.; Tu, H.-C.; Lin, H.-Y.; Tsai, H.-J.; Chen, L.-T. The regulatory role of aberrant Phosphatase and Tensin Homologue and Liver Kinase B1 on AKT/mTOR/c-Myc axis in pancreatic neuroendocrine tumors. Oncotarget 2017, 8, 98068–98083. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Quelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [CrossRef] [Green Version]
- Quelle, D.E.; Nteeba, J.; Darbro, B.W. The INK4a/ARF Locus. In Encyclopedia of Cell Biology; Stahl, P., Bradshaw, R., Eds.; Academic Press: Waltham, MA, USA, 2016; Volume 3, pp. 447–457. [Google Scholar]
- Sherr, C.J. Divorcing ARF and p53: An unsettled case. Nat. Rev. Cancer 2006, 6, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Muscarella, P.; Melvin, W.S.; Fisher, W.E.; Foor, J.; Ellison, E.C.; Herman, J.G.; Schirmer, W.J.; Hitchcock, C.L.; DeYoung, B.R.; Weghorst, C.M. Genetic alterations in gastrinomas and nonfunctioning pancreatic neuroendocrine tumors: An analysis of p16/MTS1 tumor suppressor gene inactivation. Cancer Res. 1998, 58, 237–240. [Google Scholar]
- Simon, B.; Lubomierski, N. Implication of the INK4a/ARF Locus in Gastroenteropancreatic Neuroendocrine Tumorigenesis. Ann. N. Y. Acad. Sci. 2004, 1014, 284–299. [Google Scholar] [CrossRef]
- Chan, A.O.; Kim, S.G.; Bedeir, A.; Issa, J.P.; Hamilton, S.R.; Rashid, A. CpG island methylation in carcinoid and pancreatic endocrine tumors. Oncogene 2003, 22, 924–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zindy, F.; Quelle, D.E.; Roussel, M.F.; Sherr, C.J. Expression of the p16(INK4a) tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dammann, R.; Schagdarsurengin, U.; Liu, L.; Otto, N.; Gimm, O.; Dralle, H.; Boehm, B.O.; Pfeifer, G.P.; Hoang-Vu, C. Frequent RASSF1A promoter hypermethylation and K-ras mutations in pancreatic carcinoma. Oncogene 2003, 22, 3806–3812. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Chang, Y.; Ma, J.; Li, X.; Li, X.; Fan, J.; Huang, R.; Duan, G.; Sun, X. Prognostic impact of p16 and p21 on gastroenteropancreatic neuroendocrine tumors. Oncol. Lett. 2013, 6, 1641–1645. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lee, H.; Chin, L.; Cordon-Cardo, C.; Beach, D.; DePinho, R.A. Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996, 85, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R.A. Loss of p16(Ink4a) with retention of p19(Arf) predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar] [CrossRef]
- Krimpenfort, P.; Quon, K.C.; Mooi, W.J.; Loonstra, A.; Berns, A. Loss of p16(Ink4a) confers susceptibility to metastatic melanoma in mice. Nature 2001, 413, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Ulanet, D.B.; Hanahan, D. Loss of p19Arf Facilitates the Angiogenic Switch and Tumor Initiation in a Multi-Stage Cancer Model via p53-Dependent and Independent Mechanisms. PLoS ONE 2010, 5, e12454. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.H.; Contractor, T.; Clausen, R.; Klimstra, D.S.; Du, Y.-C.N.; Allen, P.J.; Brennan, M.F.; Levine, A.J.; Harris, C.R. Attenuation of the Retinoblastoma Pathway in Pancreatic Neuroendocrine Tumors Due to Increased Cdk4/Cdk6. Clin. Cancer Res. 2012, 18, 4612–4620. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Qian, Z.R.; Zhang, S.; Li, W.; Masugi, Y.; Li, T.; Chan, J.A.; Yang, J.; Da Silva, A.; Gu, M.; et al. Cell Cycle Protein Expression in Neuroendocrine Tumors: Association of CDK4/CDK6, CCND1, and Phosphorylated Retinoblastoma Protein With Proliferative Index. Pancreas 2017, 46, 1347–1353. [Google Scholar] [CrossRef]
- Scholz, A.; Wagner, K.; Welzel, M.; Remlinger, F.; Wiedenmann, B.; Siemeister, G.; Rosewicz, S.; Detjen, K.M. The oral multitarget tumour growth inhibitor, ZK 304709, inhibits growth of pancreatic neuroendocrine tumours in an orthotopic mouse model. Gut 2009, 58, 261. [Google Scholar] [CrossRef] [PubMed]
- Aristizabal Prada, E.T.; Nölting, S.; Spoettl, G.; Maurer, J.; Auernhammer, C.J. The Novel Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) Alone and in Dual-Targeting Approaches Demonstrates Antitumoral Efficacy in Neuroendocrine Tumors in vitro. Neuroendocrinology 2018, 106, 58–73. [Google Scholar] [CrossRef]
- Bartsch, D.K.; Kersting, M.; Wild, A.; Ramaswamy, A.; Gerdes, B.; Schuermann, M.; Simon, B.; Rothmund, M. Low Frequency of p16INK4a Alterations in Insulinomas. Digestion 2000, 62, 171–177. [Google Scholar] [CrossRef]
- Grande, E.; Teule, A.; Alonso-Gordoa, T.; Jimenez-Fonseca, P.; Benavent, M.; Capdevila, J.; Custodio, A.; Vera, R.; Munarriz, J.; La Casta, A.; et al. The PALBONET Trial: A Phase II Study of Palbociclib in Metastatic Grade 1 and 2 Pancreatic Neuroendocrine Tumors (GETNE-1407). Oncologist 2020, 25, 745.e1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, N.; Zheng, Y.; Hauser, H.; Chou, J.; Rafailov, J.; Bou-Ayache, J.; Sawan, P.; Chaft, J.; Chan, J.; Perez, K.; et al. Ribociclib and everolimus in well-differentiated foregut neuroendocrine tumors. Endocr. Relat. Cancer 2021, 28, 237–246. [Google Scholar] [CrossRef]
- Kohlmeyer, J.L.; Kaemmer, C.A.; Pulliam, C.; Maharjan, C.K.; Samayoa, A.M.; Major, H.J.; Cornick, K.E.; Knepper-Adrian, V.; Khanna, R.; Sieren, J.C.; et al. RABL6A Is an Essential Driver of MPNSTs that Negatively Regulates the RB1 Pathway and Sensitizes Tumor Cells to CDK4/6 Inhibitors. Clin. Cancer Res. 2020, 26, 2997. [Google Scholar] [CrossRef] [Green Version]
- Hagen, J.; Muniz, V.P.; Falls, K.C.; Reed, S.M.; Taghiyev, A.F.; Quelle, F.W.; Gourronc, F.A.; Klingelhutz, A.J.; Major, H.J.; Askeland, R.W.; et al. RABL6A promotes G1-S phase progression and pancreatic neuroendocrine tumor cell proliferation in an Rb1-dependent manner. Cancer Res. 2014, 74, 6661–6670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Ji, F.; Sun, J.; Xie, Y.; Xu, Y.; Yue, H. RBEL1 is required for osteosarcoma cell proliferation via inhibiting retinoblastoma 1. Mol. Med. Rep. 2016, 13, 1275–1280. [Google Scholar] [CrossRef] [Green Version]
- Kohlmeyer, J.L.; Gordon, D.J.; Tanas, M.R.; Dodd, R.D.; Monga, V.; Darbro, B.W.; Quelle, D.E. Combination therapies for MPNSTs targeting RABL6A-RB1 signaling. Oncotarget 2021, 12, 10–14. [Google Scholar] [CrossRef]
- Nikitin, A.Y.; Juárez-Pérez, M.I.; Li, S.; Huang, L.; Lee, W.H. RB-mediated suppression of spontaneous multiple neuroendocrine neoplasia and lung metastases in Rb+/− mice. Proc. Natl. Acad. Sci. USA 1999, 96, 3916–3921. [Google Scholar] [CrossRef] [Green Version]
- Harvey, M.; Vogel, H.; Lee, E.Y.; Bradley, A.; Donehower, L.A. Mice deficient in both p53 and Rb develop tumors primarily of endocrine origin. Cancer Res. 1995, 55, 1146–1151. [Google Scholar]
- Tang, J.; Wu, S.; Liu, H.; Stratt, R.; Barak, O.G.; Shiekhattar, R.; Picketts, D.J.; Yang, X. A novel transcription regulatory complex containing death domain-associated protein and the ATR-X syndrome protein. J. Biol. Chem. 2004, 279, 20369–20377. [Google Scholar] [CrossRef] [Green Version]
- Salomoni, P.; Khelifi, A.F. Daxx: Death or survival protein? Trends Cell Biol. 2006, 16, 97–104. [Google Scholar] [CrossRef]
- Gibbons, R.J.; Picketts, D.J.; Villard, L.; Higgs, D.R. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell 1995, 80, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, R.J.; Bachoo, S.; Picketts, D.J.; Aftimos, S.; Asenbauer, B.; Bergoffen, J.; Berry, S.A.; Dahl, N.; Fryer, A.; Keppler, K.; et al. Mutations in transcriptional regulator ATRX establish the functional significance of a PHD-like domain. Nat. Genet. 1997, 17, 146–148. [Google Scholar] [CrossRef]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [Green Version]
- Khelifi, A.F.; D’Alcontres, M.S.; Salomoni, P. Daxx is required for stress-induced cell death and JNK activation. Cell Death Differ. 2005, 12, 724–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Gibbons, R.; Yan, Z.; Yang, D.; McDowell, T.L.; Sechi, S.; Qin, J.; Zhou, S.; Higgs, D.; Wang, W. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc. Natl. Acad. Sci. USA 2003, 100, 10635–10640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drane, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wilde, R.F.; Heaphy, C.M.; Maitra, A.; Meeker, A.K.; Edil, B.H.; Wolfgang, C.L.; Ellison, T.A.; Schulick, R.D.; Molenaar, I.Q.; Valk, G.D.; et al. Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod. Pathol. 2012, 25, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Ronai, Z. Balancing Mdm2—A Daxx-HAUSP matter. Nat. Cell Biol. 2006, 8, 790–791. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.Y.; Liu, J.; Sidhu, G.S.; Niu, Y.; Liu, Y.; Wang, R.; Liao, D. Negative regulation of p53 functions by Daxx and the involvement of MDM2. J. Biol. Chem. 2004, 279, 50566–50579. [Google Scholar] [CrossRef] [Green Version]
- Wasylishen, A.R.; Estrella, J.S.; Pant, V.; Chau, G.P.; Lozano, G. Daxx Functions Are p53-Independent In Vivo. Mol. Cancer Res. 2018, 16, 1523–1529. [Google Scholar] [CrossRef] [Green Version]
- Wasylishen, A.R.; Sun, C.; Moyer, S.M.; Qi, Y.; Chau, G.P.; Aryal, N.K.; McAllister, F.; Kim, M.P.; Barton, M.C.; Estrella, J.S.; et al. Daxx maintains endogenous retroviral silencing and restricts cellular plasticity in vivo. Sci. Adv. 2020, 6, eaba8415. [Google Scholar] [CrossRef]
- Chan, C.S.; Laddha, S.V.; Lewis, P.W.; Koletsky, M.S.; Robzyk, K.; Da Silva, E.; Torres, P.J.; Untch, B.R.; Li, J.; Bose, P.; et al. ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat. Commun. 2018, 9, 4158. [Google Scholar] [CrossRef] [Green Version]
- Cejas, P.; Drier, Y.; Dreijerink, K.M.A.; Brosens, L.A.A.; Deshpande, V.; Epstein, C.B.; Conemans, E.B.; Morsink, F.H.M.; Graham, M.K.; Valk, G.D.; et al. Enhancer signatures stratify and predict outcomes of non-functional pancreatic neuroendocrine tumors. Nat. Med. 2019, 25, 1260–1265. [Google Scholar] [CrossRef]
- Marinoni, I.; Kurrer, A.S.; Vassella, E.; Dettmer, M.; Rudolph, T.; Banz, V.; Hunger, F.; Pasquinelli, S.; Speel, E.J.; Perren, A. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology 2014, 146, 453–460.e455. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Meek, D.W. Regulation of the p53 response and its relationship to cancer. Biochem. J. 2015, 469, 325–346. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1476–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aristizabal Prada, E.T.; Auernhammer, C.J. Targeted therapy of gastroenteropancreatic neuroendocrine tumours: Preclinical strategies and future targets. Endocr. Connect. 2018, 7, R1–R25. [Google Scholar] [CrossRef] [Green Version]
- Le Guezennec, X.; Bulavin, D.V. WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem. Sci. 2010, 35, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Feng, Z.; Modica, I.; Klimstra, D.S.; Song, L.; Allen, P.J.; Brennan, M.F.; Levine, A.J.; Tang, L.H. Gene Amplifications in Well-Differentiated Pancreatic Neuroendocrine Tumors Inactivate the p53 Pathway. Genes Cancer 2010, 1, 360–368. [Google Scholar] [CrossRef]
- Ohki, R.; Saito, K.; Chen, Y.; Kawase, T.; Hiraoka, N.; Saigawa, R.; Minegishi, M.; Aita, Y.; Yanai, G.; Shimizu, H.; et al. PHLDA3 is a novel tumor suppressor of pancreatic neuroendocrine tumors. Proc. Natl. Acad. Sci. USA 2014, 111, E2404–E2413. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, P.; Schrader, J.; Wagner, J.; Hörsch, D.; Arnold, R.; Arnold, C.N.; Georgieva, I.; Stein, H.; Zeitz, M.; Daniel, P.T.; et al. Loss of Nuclear p27 Expression and Its Prognostic Role in Relation to Cyclin E and p53 Mutation in Gastroenteropancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2008, 14, 7378. [Google Scholar] [CrossRef] [Green Version]
- Furlan, D.; Bernasconi, B.; Uccella, S.; Cerutti, R.; Carnevali, I.; Capella, C. Allelotypes and fluorescence in situ hybridization profiles of poorly differentiated endocrine carcinomas of different sites. Clin. Cancer Res. 2005, 11, 1765–1775. [Google Scholar] [CrossRef] [Green Version]
- Pizzi, S.; Azzoni, C.; Bassi, D.; Bottarelli, L.; Milione, M.; Bordi, C. Genetic alterations in poorly differentiated endocrine carcinomas of the gastrointestinal tract. Cancer 2003, 98, 1273–1282. [Google Scholar] [CrossRef]
- Dilworth, S.M. Polyoma virus middle T antigen and its role in identifying cancer-related molecules. Nat. Rev. Cancer 2002, 2, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Shanzer, M.; Ricardo-Lax, I.; Keshet, R.; Reuven, N.; Shaul, Y. The polyomavirus middle T-antigen oncogene activates the Hippo pathway tumor suppressor Lats in a Src-dependent manner. Oncogene 2015, 34, 4190–4198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Lubensky, I.A.; Pack, S.; Ault, D.; Vortmeyer, A.O.; Libutti, S.K.; Choyke, P.L.; Walther, M.M.; Linehan, W.M.; Zhuang, Z. Multiple neuroendocrine tumors of the pancreas in von Hippel-Lindau disease patients: Histopathological and molecular genetic analysis. Am. J. Pathol. 1998, 153, 223–231. [Google Scholar] [CrossRef]
- Neumann, H.P.; Dinkel, E.; Brambs, H.; Wimmer, B.; Friedburg, H.; Volk, B.; Sigmund, G.; Riegler, P.; Haag, K.; Schollmeyer, P.; et al. Pancreatic lesions in the von Hippel-Lindau syndrome. Gastroenterology 1991, 101, 465–471. [Google Scholar] [CrossRef]
- Libutti, S.K.; Choyke, P.L.; Bartlett, D.L.; Vargas, H.; Walther, M.; Lubensky, I.; Glenn, G.; Linehan, W.M.; Alexander, H.R. Pancreatic neuroendocrine tumors associated with von Hippel Lindau disease: Diagnostic and management recommendations. Surgery 1998, 124, 1153–1159. [Google Scholar] [CrossRef]
- Charlesworth, M.; Verbeke, C.S.; Falk, G.A.; Walsh, M.; Smith, A.M.; Morris-Stiff, G. Pancreatic lesions in von Hippel-Lindau disease? A systematic review and meta-synthesis of the literature. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 2012, 16, 1422–1428. [Google Scholar] [CrossRef]
- Binkovitz, L.A.; Johnson, C.D.; Stephens, D.H. Islet cell tumors in von Hippel-Lindau disease: Increased prevalence and relationship to the multiple endocrine neoplasias. AJR Am. J. Roentgenol. 1990, 155, 501–505. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Ebert, B.L.; Bunn, H.F. Regulation of the erythropoietin gene. Blood 1999, 94, 1864–1877. [Google Scholar] [CrossRef]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Perigny, M.; Hammel, P.; Corcos, O.; Larochelle, O.; Giraud, S.; Richard, S.; Sauvanet, A.; Belghiti, J.; Ruszniewski, P.; Bedossa, P.; et al. Pancreatic endocrine microadenomatosis in patients with von Hippel-Lindau disease: Characterization by VHL/HIF pathway proteins expression. Am. J. Surg. Pathol. 2009, 33, 739–748. [Google Scholar] [CrossRef]
- Speisky, D.; Duces, A.; Bieche, I.; Rebours, V.; Hammel, P.; Sauvanet, A.; Richard, S.; Bedossa, P.; Vidaud, M.; Murat, A.; et al. Molecular profiling of pancreatic neuroendocrine tumors in sporadic and Von Hippel-Lindau patients. Clin. Cancer Res. 2012, 18, 2838–2849. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jia, Z.; Li, Q.; Wang, L.; Rashid, A.; Zhu, Z.; Evans, D.B.; Vauthey, J.N.; Xie, K.; Yao, J.C. Elevated expression of vascular endothelial growth factor correlates with increased angiogenesis and decreased progression-free survival among patients with low-grade neuroendocrine tumors. Cancer 2007, 109, 1478–1486. [Google Scholar] [CrossRef] [PubMed]
- Terris, B.; Scoazec, J.Y.; Rubbia, L.; Bregeaud, L.; Pepper, M.S.; Ruszniewski, P.; Belghiti, J.; Flejou, J.; Degott, C. Expression of vascular endothelial growth factor in digestive neuroendocrine tumours. Histopathology 1998, 32, 133–138. [Google Scholar] [CrossRef]
- Couvelard, A.; O’Toole, D.; Turley, H.; Leek, R.; Sauvanet, A.; Degott, C.; Ruszniewski, P.; Belghiti, J.; Harris, A.L.; Gatter, K.; et al. Microvascular density and hypoxia-inducible factor pathway in pancreatic endocrine tumours: Negative correlation of microvascular density and VEGF expression with tumour progression. Br. J. Cancer 2005, 92, 94–101. [Google Scholar] [CrossRef]
- Hansel, D.E.; Rahman, A.; Hermans, J.; de Krijger, R.R.; Ashfaq, R.; Yeo, C.J.; Cameron, J.L.; Maitra, A. Liver metastases arising from well-differentiated pancreatic endocrine neoplasms demonstrate increased VEGF-C expression. Mod. Pathol. 2003, 16, 652–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, A.M.; Bouchier-Hayes, D.J.; Harmey, J.H. Vascular endothelial growth factor (VEGF) and its role in non-endothelial cells: Autocrine signalling by VEGF. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Christofori, G.; Naik, P.; Hanahan, D. Vascular endothelial growth factor and its receptors, flt-1 and flk-1, are expressed in normal pancreatic islets and throughout islet cell tumorigenesis. Mol. Endocrinol. 1995, 9, 1760–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Hager, J.H.; Ferrara, N.; Gerber, H.P.; Hanahan, D. VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell 2002, 1, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Adhikari, L.J.; Lloyd, R.V.; Halfdanarson, T.R.; Muders, M.H.; Ames, M.M. Molecular Markers for Novel Therapeutic Strategies in Pancreatic Endocrine Tumors. Pancreas 2013, 42, 411–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelescu, R.; Burada, F.; Angelescu, C.; Gheonea, D.I.; Iordache, S.; Mixich, F.; Ioana, M.; Săftoiu, A. Expression of vascular endothelial growth factor and epidermal growth factor receptor in pancreatic ductal adenocarcinomas, neuroendocrine tumours and chronic pancreatitis. Endosc. Ultrasound 2013, 2, 86–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fjallskog, M.L.; Lejonklou, M.H.; Oberg, K.E.; Eriksson, B.K.; Janson, E.T. Expression of molecular targets for tyrosine kinase receptor antagonists in malignant endocrine pancreatic tumors. Clin. Cancer Res. 2003, 9, 1469–1473. [Google Scholar] [PubMed]
- Papouchado, B.; Erickson, L.A.; Rohlinger, A.L.; Hobday, T.J.; Erlichman, C.; Ames, M.M.; Lloyd, R.V. Epidermal growth factor receptor and activated epidermal growth factor receptor expression in gastrointestinal carcinoids and pancreatic endocrine carcinomas. Mod. Pathol. 2005, 18, 1329–1335. [Google Scholar] [CrossRef] [Green Version]
- Fjallskog, M.L.; Hessman, O.; Eriksson, B.; Janson, E.T. Upregulated expression of PDGF receptor beta in endocrine pancreatic tumors and metastases compared to normal endocrine pancreas. Acta Oncol. 2007, 46, 741–746. [Google Scholar] [CrossRef]
- Pavel, M.E.; Hassler, G.; Baum, U.; Hahn, E.G.; Lohmann, T.; Schuppan, D. Circulating levels of angiogenic cytokines can predict tumour progression and prognosis in neuroendocrine carcinomas. Clin. Endocrinol. 2005, 62, 434–443. [Google Scholar] [CrossRef]
- Capdevila, J.; Fazio, N.; Lopez, C.; Teulé, A.; Valle, J.W.; Tafuto, S.; Custodio, A.; Reed, N.; Raderer, M.; Grande, E.; et al. Lenvatinib in Patients With Advanced Grade 1/2 Pancreatic and Gastrointestinal Neuroendocrine Tumors: Results of the Phase II TALENT Trial (GETNE1509). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 2304–2312. [Google Scholar] [CrossRef]
- Koumarianou, A.; Kaltsas, G. Surufatinib—A novel oral agent for neuroendocrine tumours. Nat. Rev. Endocrinol. 2021, 17, 9–10. [Google Scholar] [CrossRef]
- Xu, J.; Shen, L.; Zhou, Z.; Li, J.; Bai, C.; Chi, Y.; Li, Z.; Xu, N.; Li, E.; Liu, T.; et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET-ep): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 1500–1512. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S.; Meyer-Morse, N.; Bergsland, E.; Hanahan, D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J. Clin. Investig. 2003, 111, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Raymond, E.; Dahan, L.; Raoul, J.-L.; Bang, Y.-J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib Malate for the Treatment of Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011, 364, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Pietras, K.; Hanahan, D. A multitargeted, metronomic, and maximum-tolerated dose “chemo-switch” regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 939–952. [Google Scholar] [CrossRef]
- Chandrasekharan, C. Medical Management of Gastroenteropancreatic Neuroendocrine Tumors. Surg. Oncol. Clin. N. Am. 2020, 29, 293–316. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shen, L.; Bai, C.; Wang, W.; Li, J.; Yu, X.; Li, Z.; Li, E.; Yuan, X.; Chi, Y.; et al. Surufatinib in advanced pancreatic neuroendocrine tumours (SANET-p): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 1489–1499. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Cives, M.; Hwang, J.; Weber, T.; Nickerson, M.; Atreya, C.E.; Venook, A.; Kelley, R.K.; Valone, T.; Morse, B.; et al. A phase II study of axitinib in advanced neuroendocrine tumors. Endocr. Relat. Cancer 2016, 23, 411–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.K.; Chapman, H.A.; Christensen, J.G.; et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sennino, B.; Ishiguro-Oonuma, T.; Schriver, B.J.; Christensen, J.G.; McDonald, D.M. Inhibition of c-Met reduces lymphatic metastasis in RIP-Tag2 transgenic mice. Cancer Res. 2013, 73, 3692–3703. [Google Scholar] [CrossRef] [Green Version]
- Krampitz, G.W.; George, B.M.; Willingham, S.B.; Volkmer, J.-P.; Weiskopf, K.; Jahchan, N.; Newman, A.M.; Sahoo, D.; Zemek, A.J.; Yanovsky, R.L.; et al. Identification of tumorigenic cells and therapeutic targets in pancreatic neuroendocrine tumors. Proc. Natl. Acad. Sci. USA 2016, 113, 4464–4469. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.A.; Faris, J.E.; Murphy, J.E.; Blaszkowsky, L.S.; Kwak, E.L.; McCleary, N.J.; Fuchs, C.S.; Meyerhardt, J.A.; Ng, K.; Zhu, A.X.; et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J. Clin. Oncol. 2017, 35, 228. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Lloyd, R.V.; Ames, M.M. Lack of mutations in EGFR in gastroenteropancreatic neuroendocrine tumors. N. Engl. J. Med. 2005, 353, 209–210. [Google Scholar] [CrossRef]
- Von Wichert, G.; Jehle, P.M.; Hoeflich, A.; Koschnick, S.; Dralle, H.; Wolf, E.; Wiedenmann, B.; Boehm, B.O.; Adler, G.; Seufferlein, T. Insulin-like Growth Factor-I Is an Autocrine Regulator of Chromogranin A Secretion and Growth in Human Neuroendocrine Tumor Cells. Cancer Res. 2000, 60, 4573–4581. [Google Scholar] [PubMed]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef]
- Tchernev, G.; Chokoeva, A.A.; Patterson, J.W.; Bakardzhiev, I.; Wollina, U.; Tana, C. Plexiform Neurofibroma: A Case Report. Medicine 2016, 95, e2663. [Google Scholar] [CrossRef]
- Cawthon, R.M.; Weiss, R.; Xu, G.F.; Viskochil, D.; Culver, M.; Stevens, J.; Robertson, M.; Dunn, D.; Gesteland, R.; O’Connell, P.; et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 1990, 62, 193–201. [Google Scholar] [CrossRef]
- Viskochil, D.; Buchberg, A.M.; Xu, G.; Cawthon, R.M.; Stevens, J.; Wolff, R.K.; Culver, M.; Carey, J.C.; Copeland, N.G.; Jenkins, N.A.; et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 1990, 62, 187–192. [Google Scholar] [CrossRef]
- Wallace, M.R.; Marchuk, D.A.; Andersen, L.B.; Letcher, R.; Odeh, H.M.; Saulino, A.M.; Fountain, J.W.; Brereton, A.; Nicholson, J.; Mitchell, A.L.; et al. Type 1 neurofibromatosis gene: Identification of a large transcript disrupted in three NF1 patients. Science 1990, 249, 181–186. [Google Scholar] [CrossRef]
- Zakka, K.; Nagy, R.; Drusbosky, L.; Akce, M.; Wu, C.; Alese, O.B.; El-Rayes, B.F.; Kasi, P.M.; Mody, K.; Starr, J.; et al. Blood-based next-generation sequencing analysis of neuroendocrine neoplasms. Oncotarget 2020, 11, 1749–1757. [Google Scholar] [CrossRef]
- Pipinikas, C.; Gutteridge, A.; Forshew, T.; Childs, A.; Meyer, T.; Oukrif, D.; Luong, T.; Thirlwell, C. Abstracts of the 13th Annual ENETS Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease. Neuroendocrinology 2016, 103 (Suppl. S1), 1–128. [Google Scholar] [CrossRef]
- Beltran, H.; Romanel, A.; Casiraghi, N.; Sigouros, M.; Benelli, M.; Xiang, J.; Demichelis, F. Whole exome sequencing (WES) of circulating tumor DNA (ctDNA) in patients with neuroendocrine prostate cancer (NEPC) informs tumor heterogeneity. J. Clin. Oncol. 2017, 35, 5011. [Google Scholar] [CrossRef]
- Zitzmann, K.; Ruden, J.; Brand, S.; Goke, B.; Lichtl, J.; Spottl, G.; Auernhammer, C.J. Compensatory activation of Akt in response to mTOR and Raf inhibitors—A rationale for dual-targeted therapy approaches in neuroendocrine tumor disease. Cancer Lett. 2010, 295, 100–109. [Google Scholar] [CrossRef]
- Zitzmann, K.; de Toni, E.; von Ruden, J.; Brand, S.; Goke, B.; Laubender, R.P.; Auernhammer, C.J. The novel Raf inhibitor Raf265 decreases Bcl-2 levels and confers TRAIL-sensitivity to neuroendocrine tumour cells. Endocr. Relat. Cancer 2011, 18, 277–285. [Google Scholar] [CrossRef]
- Iida, S.; Miki, Y.; Ono, K.; Akahira, J.; Nakamura, Y.; Suzuki, T.; Sasano, H. Synergistic anti-tumor effects of RAD001 with MEK inhibitors in neuroendocrine tumors: A potential mechanism of therapeutic limitation of mTOR inhibitor. Mol. Cell. Endocrinol. 2012, 350, 99–106. [Google Scholar] [CrossRef]
- Valentino, J.D.; Li, J.; Zaytseva, Y.Y.; Mustain, W.C.; Elliott, V.A.; Kim, J.T.; Harris, J.W.; Campbell, K.; Weiss, H.; Wang, C.; et al. Cotargeting the PI3K and RAS Pathways for the Treatment of Neuroendocrine Tumors. Clin. Cancer Res. 2014, 20, 1212–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyronnet, S.; Bousquet, C.; Najib, S.; Azar, R.; Laklai, H.; Susini, C. Antitumor effects of somatostatin. Mol. Cell. Endocrinol. 2008, 286, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, Y.C.; Srikant, C.B. Subtype selectivity of peptide analogs for all five cloned human somatostatin receptors (hsstr 1–5). Endocrinology 1994, 135, 2814–2817. [Google Scholar] [CrossRef]
- Lopez, F.; Ferjoux, G.; Cordelier, P.; Saint-Laurent, N.; Estève, J.P.; Vaysse, N.; Buscail, L.; Susini, C. Neuronal nitric oxide synthase: A substrate for SHP-1 involved in sst2 somatostatin receptor growth inhibitory signaling. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 2300–2302. [Google Scholar] [CrossRef]
- Pagès, P.; Benali, N.; Saint-Laurent, N.; Estève, J.P.; Schally, A.V.; Tkaczuk, J.; Vaysse, N.; Susini, C.; Buscail, L. sst2 somatostatin receptor mediates cell cycle arrest and induction of p27(Kip1). Evidence for the role of SHP-1. J. Biol. Chem. 1999, 274, 15186–15193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillermet, J.; Saint-Laurent, N.; Rochaix, P.; Cuvillier, O.; Levade, T.; Schally, A.V.; Pradayrol, L.; Buscail, L.; Susini, C.; Bousquet, C. Somatostatin receptor subtype 2 sensitizes human pancreatic cancer cells to death ligand-induced apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Van Gompel, J.J.; Kunnimalaiyaan, M.; Holen, K.; Chen, H. ZM336372, a Raf-1 activator, suppresses growth and neuroendocrine hormone levels in carcinoid tumor cells. Mol. Cancer Ther. 2005, 4, 910–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunnimalaiyaan, M.; Ndiaye, M.; Chen, H. Neuroendocrine tumor cell growth inhibition by ZM336372 through alterations in multiple signaling pathways. Surgery 2007, 142, 959–964. [Google Scholar] [CrossRef] [Green Version]
- Theodoropoulou, M.; Stalla, G.K. Somatostatin receptors: From signaling to clinical practice. Front. Neuroendocrinol. 2013, 34, 228–252. [Google Scholar] [CrossRef] [PubMed]
- Stueven, A.K.; Kayser, A.; Wetz, C.; Amthauer, H.; Wree, A.; Tacke, F.; Wiedenmann, B.; Roderburg, C.; Jann, H. Somatostatin Analogues in the Treatment of Neuroendocrine Tumors: Past, Present and Future. Int. J. Mol. Sci. 2019, 20, 3049. [Google Scholar] [CrossRef] [Green Version]
- Lopez, F.; Estève, J.P.; Buscail, L.; Delesque, N.; Saint-Laurent, N.; Vaysse, N.; Susini, C. Molecular mechanisms of antiproliferative effect of somatostatin: Involvement of a tyrosine phosphatase. Metab. Clin. Exp. 1996, 45, 14–16. [Google Scholar] [CrossRef]
- Mehta, S.; de Reuver, P.R.; Gill, P.; Andrici, J.; D’Urso, L.; Mittal, A.; Pavlakis, N.; Clarke, S.; Samra, J.S.; Gill, A.J. Somatostatin Receptor SSTR-2a Expression Is a Stronger Predictor for Survival Than Ki-67 in Pancreatic Neuroendocrine Tumors. Medicine (Baltimore) 2015, 94, e1281. [Google Scholar] [CrossRef]
- Papotti, M.; Bongiovanni, M.; Volante, M.; Allìa, E.; Landolfi, S.; Helboe, L.; Schindler, M.; Cole, S.L.; Bussolati, G. Expression of somatostatin receptor types 1-5 in 81 cases of gastrointestinal and pancreatic endocrine tumors. A correlative immunohistochemical and reverse-transcriptase polymerase chain reaction analysis. Virchows Arch. Int. J. Pathol. 2002, 440, 461–475. [Google Scholar] [CrossRef]
- Leu, F.P.; Nandi, M.; Niu, C. The Effect of Transforming Growth Factor β on Human Neuroendocrine Tumor BON Cell Proliferation and Differentiation Is Mediated through Somatostatin Signaling. Mol. Cancer Res. 2008, 6, 1029–1042. [Google Scholar] [CrossRef] [Green Version]
- Guenter, R.; Aweda, T.; Carmona Matos, D.M.; Jang, S.; Whitt, J.; Cheng, Y.-Q.; Liu, X.M.; Chen, H.; Lapi, S.E.; Jaskula-Sztul, R. Overexpression of somatostatin receptor type 2 in neuroendocrine tumors for improved Ga68-DOTATATE imaging and treatment. Surgery 2020, 167, 189–196. [Google Scholar] [CrossRef]
- Wanek, J.; Gaisberger, M.; Beyreis, M.; Mayr, C.; Helm, K.; Primavesi, F.; Jäger, T.; Di Fazio, P.; Jakab, M.; Wagner, A.; et al. Pharmacological Inhibition of Class IIA HDACs by LMK-235 in Pancreatic Neuroendocrine Tumor Cells. Int. J. Mol. Sci. 2018, 19, 3128. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Qian, Q.; Sun, G.; Mackey, L.V.; Fuselier, J.A.; Coy, D.H.; Yu, C.Y. Valproic acid induces NET cell growth arrest and enhances tumor suppression of the receptor-targeted peptide-drug conjugate via activating somatostatin receptor type II. J. Drug Target 2016, 24, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.G.; Scott, A.T.; Li, G.; Sherman, S.K.; Ear, P.H.; Howe, J.R. Metastatic pancreatic neuroendocrine tumors have decreased somatostatin expression and increased Akt signaling. Surgery 2021, 169, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.R.; Li, T.; Ter-Minassian, M.; Yang, J.; Chan, J.A.; Brais, L.K.; Masugi, Y.; Thiaglingam, A.; Brooks, N.; Nishihara, R.; et al. Association Between Somatostatin Receptor Expression and Clinical Outcomes in Neuroendocrine Tumors. Pancreas 2016, 45, 1386–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, W.; Jin, K.; Fang, C.; Lin, Y.; Xue, L.; Feng, S.; Zhou, Z.; Shao, C.; Chen, M.; et al. Somatostatin receptor expression indicates improved prognosis in gastroenteropancreatic neuroendocrine neoplasm, and octreotide long-acting release is effective and safe in Chinese patients with advanced gastroenteropancreatic neuroendocrine tumors. Oncol. Lett. 2017, 13, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Okuwaki, K.; Kida, M.; Mikami, T.; Yamauchi, H.; Imaizumi, H.; Miyazawa, S.; Iwai, T.; Takezawa, M.; Saegusa, M.; Watanabe, M.; et al. Clinicopathologic characteristics of pancreatic neuroendocrine tumors and relation of somatostatin receptor type 2A to outcomes. Cancer 2013, 119, 4094–4102. [Google Scholar] [CrossRef]
- Kimura, N.; Pilichowska, M.; Date, F.; Kimura, I.; Schindler, M. Immunohistochemical Expression of Somatostatin Type 2A Receptor in Neuroendocrine Tumors. Clin. Cancer Res. 1999, 5, 3483. [Google Scholar] [PubMed]
- Ono, K.; Suzuki, T.; Miki, Y.; Taniyama, Y.; Nakamura, Y.; Noda, Y.; Watanabe, M.; Sasano, H. Somatostatin receptor subtypes in human non-functioning neuroendocrine tumors and effects of somatostatin analogue SOM230 on cell proliferation in cell line NCI-H727. Anticancer Res. 2007, 27, 2231–2239. [Google Scholar] [PubMed]
- Mizutani, G.; Nakanishi, Y.; Watanabe, N.; Honma, T.; Obana, Y.; Seki, T.; Ohni, S.; Nemoto, N. Expression of Somatostatin Receptor (SSTR) Subtypes (SSTR-1, 2A, 3, 4 and 5) in Neuroendocrine Tumors Using Real-time RT-PCR Method and Immunohistochemistry. Acta Histochem. Cytochem. 2012, 45, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Patel, Y.C.; Wheatley, T. In vivo and in vitro plasma disappearance and metabolism of somatostatin-28 and somatostatin-14 in the rat. Endocrinology 1983, 112, 220–225. [Google Scholar] [CrossRef]
- Rinke, A.; Mueller, H.-H.; Schade-Brittinger, C.; Klose, K.-J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.-F.; Blaeker, M.; et al. Placebo-Controlled, Double-Blind, Prospective, Randomized Study on the Effect of Octreotide LAR in the Control of Tumor Growth in Patients With Metastatic Neuroendocrine Midgut Tumors: A Report From the PROMID Study Group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef]
- Rinke, A.; Wittenberg, M.; Schade-Brittinger, C.; Aminossadati, B.; Ronicke, E.; Gress, T.M.; Muller, H.H.; Arnold, R. Placebo-Controlled, Double-Blind, Prospective, Randomized Study on the Effect of Octreotide LAR in the Control of Tumor Growth in Patients with Metastatic Neuroendocrine Midgut Tumors (PROMID): Results of Long-Term Survival. Neuroendocrinology 2017, 104, 26–32. [Google Scholar] [CrossRef]
- Caplin, M.E.; Pavel, M.; Ruszniewski, P. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 2014, 371, 1556–1557. [Google Scholar] [CrossRef]
- Tirosh, A.; Kebebew, E. The utility of (68)Ga-DOTATATE positron-emission tomography/computed tomography in the diagnosis, management, follow-up and prognosis of neuroendocrine tumors. Future Oncol. 2018, 14, 111–122. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Blanchard, M.P.; Albertelli, M.; Barbieri, F.; Brue, T.; Niccoli, P.; Delpero, J.R.; Monges, G.; Garcia, S.; Ferone, D.; et al. Pasireotide and octreotide antiproliferative effects and sst2 trafficking in human pancreatic neuroendocrine tumor cultures. Endocr. Relat. Cancer 2014, 21, 691–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, G.V.; Stevenson, M.; Soukup, B.S.; Lines, K.E.; Grossman, A.B.; Schmid, H.A.; Thakker, R.V. Pasireotide Therapy of Multiple Endocrine Neoplasia Type 1-Associated Neuroendocrine Tumors in Female Mice Deleted for an Men1 Allele Improves Survival and Reduces Tumor Progression. Endocrinology 2016, 157, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T.J.; Yuan, Z.; Adem, A.; Geha, R.; Vrikshajanani, C.; Koba, W.; Fine, E.; Hughes, D.T.; Schmid, H.A.; Libutti, S.K. Pasireotide (SOM230) is effective for the treatment of pancreatic neuroendocrine tumors (PNETs) in a multiple endocrine neoplasia type 1 (MEN1) conditional knockout mouse model. Surgery 2012, 152, 1068–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cives, M.; Kunz, P.L.; Morse, B.; Coppola, D.; Schell, M.J.; Campos, T.; Nguyen, P.T.; Nandoskar, P.; Khandelwal, V.; Strosberg, J.R. Phase II clinical trial of pasireotide long-acting repeatable in patients with metastatic neuroendocrine tumors. Endocr. Relat. Cancer 2015, 22, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wolin, E.M.; Jarzab, B.; Eriksson, B.; Walter, T.; Toumpanakis, C.; Morse, M.A.; Tomassetti, P.; Weber, M.M.; Fogelman, D.R.; Ramage, J.; et al. Phase III study of pasireotide long-acting release in patients with metastatic neuroendocrine tumors and carcinoid symptoms refractory to available somatostatin analogues. Drug Des. Dev. Ther. 2015, 9, 5075–5086. [Google Scholar] [CrossRef] [Green Version]
- Kulke, M.H.; Ruszniewski, P.; Van Cutsem, E.; Lombard-Bohas, C.; Valle, J.W.; De Herder, W.W.; Pavel, M.; Degtyarev, E.; Brase, J.C.; Bubuteishvili-Pacaud, L.; et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-differentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann. Oncol. 2017, 28, 1309–1315. [Google Scholar] [CrossRef]
- Wang, L.; Tang, K.; Zhang, Q.; Li, H.; Wen, Z.; Zhang, H.; Zhang, H. Somatostatin Receptor-Based Molecular Imaging and Therapy for Neuroendocrine Tumors. BioMed Res. Int. 2013, 2013, 102819. [Google Scholar] [CrossRef]
- Kwekkeboom, D.J.; Kam, B.L.; van Essen, M.; Teunissen, J.J.M.; van Eijck, C.H.J.; Valkema, R.; de Jong, M.; de Herder, W.W.; Krenning, E.P. Somatostatin receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr.-Relat. Cancer 2010, 17, R53–R73. [Google Scholar] [CrossRef] [Green Version]
- Carlsen, E.A.; Fazio, N.; Granberg, D.; Grozinsky-Glasberg, S.; Ahmadzadehfar, H.; Grana, C.M.; Zandee, W.T.; Cwikla, J.; Walter, M.A.; Oturai, P.S.; et al. Peptide receptor radionuclide therapy in gastroenteropancreatic NEN G3: A multicenter cohort study. Endocr.-Relat. Cancer 2019, 26, 227–239. [Google Scholar] [CrossRef]
- Levens, D. Disentangling the MYC web. Proc. Natl. Acad. Sci. USA 2002, 99, 5757. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Kress, T.R.; Sabò, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-G.; Johnston, C.F.; Buchanan, K.D. Oncogene expression in gastroenteropancreatic neuroendocrine tumors. Cancer 1997, 80, 668–675. [Google Scholar] [CrossRef]
- Alvarez, M.J.; Subramaniam, P.S.; Tang, L.H.; Grunn, A.; Aburi, M.; Rieckhof, G.; Komissarova, E.V.; Hagan, E.A.; Bodei, L.; Clemons, P.A.; et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 2018, 50, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Fraedrich, K.; Schrader, J.; Ittrich, H.; Keller, G.; Gontarewicz, A.; Matzat, V.; Kromminga, A.; Pace, A.; Moll, J.; Bläker, M.; et al. Targeting Aurora Kinases with Danusertib (PHA-739358) Inhibits Growth of Liver Metastases from Gastroenteropancreatic Neuroendocrine Tumors in an Orthotopic Xenograft Model. Clin. Cancer Res. 2012, 18, 4621–4632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, B.C.; Klimstra, D.S.; Varmus, H.E. The c-myc and PyMT oncogenes induce different tumor types in a somatic mouse model for pancreatic cancer. Genes Dev. 2003, 17, 3127–3138. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.W.; Kutok, J.L.; Lee, N.H.; Piao, H.Y.; Fletcher, C.D.M.; Kanki, J.P.; Look, A.T. Targeted Expression of Human MYCN Selectively Causes Pancreatic Neuroendocrine Tumors in Transgenic Zebrafish. Cancer Res. 2004, 64, 7256–7262. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.V.; Ravitz, M.J.; Chen, L.; Lynch, M. Growth controls connect: Interactions between c-myc and the tuberous sclerosis complex-mTOR pathway. Cell Cycle 2009, 8, 1344–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.; Laddha, S.V.; Tang, L.; Vosburgh, E.; Levine, A.J.; Normant, E.; Sandy, P.; Harris, C.R.; Chan, C.S.; Xu, E.Y. The bromodomain and extra-terminal inhibitor CPI203 enhances the antiproliferative effects of rapamycin on human neuroendocrine tumors. Cell Death Dis. 2014, 5, e1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef] [Green Version]
- Mizukami, Y.; Fujiki, K.; Duerr, E.M.; Gala, M.; Jo, W.S.; Zhang, X.; Chung, D.C. Hypoxic regulation of vascular endothelial growth factor through the induction of phosphatidylinositol 3-kinase/Rho/ROCK and c-Myc. J. Biol. Chem. 2006, 281, 13957–13963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodir, N.M.; Swigart, L.B.; Karnezis, A.N.; Hanahan, D.; Evan, G.I.; Soucek, L. Endogenous Myc maintains the tumor microenvironment. Genes Dev. 2011, 25, 907–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capurso, G.; Lattimore, S.; Crnogorac-Jurcevic, T.; Panzuto, F.; Milione, M.; Bhakta, V.; Campanini, N.; Swift, S.M.; Bordi, C.; Fave, G.D.; et al. Gene expression profiles of progressive pancreatic endocrine tumours and their liver metastases reveal potential novel markers and therapeutic targets. Endocr.-Relat. Cancer 2006, 13, 541–558. [Google Scholar] [CrossRef]
- Klieser, E.; Urbas, R.; Stättner, S.; Primavesi, F.; Jäger, T.; Dinnewitzer, A.; Mayr, C.; Kiesslich, T.; Holzmann, K.; Di Fazio, P.; et al. Comprehensive immunohistochemical analysis of histone deacetylases in pancreatic neuroendocrine tumors: HDAC5 as a predictor of poor clinical outcome. Hum. Pathol. 2017, 65, 41–52. [Google Scholar] [CrossRef]
- Gurung, B.; Hua, X.; Runske, M.; Bennett, B.; LiVolsi, V.; Roses, R.; Fraker, D.A.; Metz, D.C. PTCH 1 staining of pancreatic neuroendocrine tumor (PNET) samples from patients with and without multiple endocrine neoplasia (MEN-1) syndrome reveals a potential therapeutic target. Cancer Biol. 2015, 16, 219–224. [Google Scholar] [CrossRef]
- Weiss, V.; Dueber, J.; Wright, J.P.; Cates, J.; Revetta, F.; Parikh, A.A.; Merchant, N.B.; Shi, C. Immunohistochemical analysis of the Wnt/β-catenin signaling pathway in pancreatic neuroendocrine neoplasms. World J. Gastrointest. Oncol. 2016, 8, 615–622. [Google Scholar] [CrossRef]
- Wimmel, A.; Wiedenmann, B.; Rosewicz, S. Autocrine growth inhibition by transforming growth factor beta-1 (TGFbeta-1) in human neuroendocrine tumour cells. Gut 2003, 52, 1308–1316. [Google Scholar] [CrossRef] [Green Version]
- Shattuck, T.M.; Costa, J.; Bernstein, M.; Jensen, R.T.; Chung, D.C.; Arnold, A. Mutational analysis of Smad3, a candidate tumor suppressor implicated in TGF-beta and menin pathways, in parathyroid adenomas and enteropancreatic endocrine tumors. J. Clin. Endocrinol. Metab. 2002, 87, 3911–3914. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, D.; Hahn, S.A.; Danichevski, K.D.; Ramaswamy, A.; Bastian, D.; Galehdari, H.; Barth, P.; Schmiegel, W.; Simon, B.; Rothmund, M. Mutations of the DPC4/Smad4 gene in neuroendocrine pancreatic tumors. Oncogene 1999, 18, 2367–2371. [Google Scholar] [CrossRef] [Green Version]
- Capurso, G.; Di Florio, A.; Sette, C.; Delle Fave, G. Signalling pathways passing Src in pancreatic endocrine tumours: Relevance for possible combined targeted therapies. Neuroendocrinology 2013, 97, 67–73. [Google Scholar] [CrossRef]
- Alessia Di, F.; Laura, A.; Simona, P.; Gabriele, C.; Emanuela, P.; Vincenzo, C.; Aldo, S.; Raffaele, G.; Gianfranco Delle, F.; Claudio, S. Src kinase activity coordinates cell adhesion and spreading with activation of mammalian target of rapamycin in pancreatic endocrine tumour cells. Endocr.-Relat. Cancer 2011, 18, 541–554. [Google Scholar] [CrossRef] [Green Version]
- Tompkins, V.; Hagen, J.; Zediak, V.P.; Quelle, D.E. Identification of novel ARF binding proteins by two-hybrid screening. Cell Cycle 2006, 5, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Montalbano, J.; Jin, W.; Sheikh, M.S.; Huang, Y. RBEL1 Is a Novel Gene That Encodes a Nucleocytoplasmic Ras Superfamily GTP-binding Protein and Is Overexpressed in Breast Cancer. J. Biol. Chem. 2007, 282, 37640–37649. [Google Scholar] [CrossRef] [Green Version]
- Montalbano, J.; Lui, K.; Sheikh, M.S.; Huang, Y. Identification and Characterization of RBEL1 Subfamily of GTPases in the Ras Superfamily Involved in Cell Growth Regulation. J. Biol. Chem. 2009, 284, 18129–18142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-Y.; Fu, S.; Wang, X.-P.; Wang, H.-Y.; Zeng, M.-S.; Shao, J.-Y. Down-Regulation of C9orf86 in Human Breast Cancer Cells Inhibits Cell Proliferation, Invasion and Tumor Growth and Correlates with Survival of Breast Cancer Patients. PLoS ONE 2013, 8, e71764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muniz, V.P.; Askeland, R.W.; Zhang, X.; Reed, S.M.; Tompkins, V.S.; Hagen, J.; McDowell, B.D.; Button, A.; Smith, B.J.; Weydert, J.A.; et al. RABL6A Promotes Oxaliplatin Resistance in Tumor Cells and Is a New Marker of Survival for Resected Pancreatic Ductal Adenocarcinoma Patients. Genes Cancer 2013, 4, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, K.; Osman, M.; Inoue, Y.; Suda, T.; Sugimura, H. A novel prognostic marker of non-small cell lung cancer: Chromosome 9 open reading frame 86 (C9orf86). J. Thorac. Dis. 2016, 8, 2284–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, G.L.; Tao, Y.L.; Wu, Q.N.; Zhang, Y.; He, J.X. Positive expression of protein chromosome 9 open reading frame 86 (C9orf86) correlated with poor prognosis in non-small cell lung cancer patients. J. Thorac. Dis. 2016, 8, 1449–1459. [Google Scholar] [CrossRef] [Green Version]
- Klieser, E.; Swierczynski, S.; Mayr, C.; Schmidt, J.; Neureiter, D.; Kiesslich, T.; Illig, R. Role of histone deacetylases in pancreas: Implications for pathogenesis and therapy. World J. Gastrointest. Oncol. 2015, 7, 473–483. [Google Scholar] [CrossRef]
- Klieser, E.; Urbas, R.; Swierczynski, S.; Stättner, S.; Primavesi, F.; Jäger, T.; Mayr, C.; Kiesslich, T.; Fazio, P.D.; Helm, K.; et al. HDAC-Linked “Proliferative” miRNA Expression Pattern in Pancreatic Neuroendocrine Tumors. Int. J. Mol. Sci. 2018, 19, 2781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, P.L.; Chao, Y.L.; Tsai, Y.-H.; Ghosh, S.K.; Porrello, A.; Van Swearingen, A.E.D.; Harrison, E.B.; Cooley, B.C.; Parker, J.S.; Carey, L.A.; et al. Histone deacetylase 11 inhibition promotes breast cancer metastasis from lymph nodes. Nat. Commun. 2019, 10, 4192. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.T.; Wang, Y.W.; Chen, C.T.; Ho, C.M.; Su, W.H.; Jou, Y.S. HDAC inhibitors augmented cell migration and metastasis through induction of PKCs leading to identification of low toxicity modalities for combination cancer therapy. Clin. Cancer Res. 2012, 18, 4691–4701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenblatt, D.Y.; Vaccaro, A.M.; Jaskula-Sztul, R.; Ning, L.; Haymart, M.; Kunnimalaiyaan, M.; Chen, H. Valproic acid activates notch-1 signaling and regulates the neuroendocrine phenotype in carcinoid cancer cells. Oncologist 2007, 12, 942–951. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, T.A.; Holen, K.D.; Jaskula-Sztul, R.; Mulkerin, D.; Lubner, S.J.; Schelman, W.R.; Eickhoff, J.; Chen, H.; LoConte, N.K. A Pilot Phase II Study of Valproic Acid for Treatment of Low-Grade Neuroendocrine Carcinoma. Oncologist 2011, 16, 835–843. [Google Scholar] [CrossRef]
- Jin, N.; Lubner, S.J.; Mulkerin, D.L.; Rajguru, S.; Carmichael, L.; Chen, H.; Holen, K.D.; LoConte, N.K. A Phase II Trial of a Histone Deacetylase Inhibitor Panobinostat in Patients With Low-Grade Neuroendocrine Tumors. Oncologist 2016, 21, 785–786. [Google Scholar] [CrossRef] [Green Version]
- Butler, L.M.; Ferraldeschi, R.; Armstrong, H.K.; Centenera, M.M.; Workman, P. Maximizing the Therapeutic Potential of HSP90 Inhibitors. Mol. Cancer Res. 2015, 13, 1445–1451. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Adhikari, L.J.; Lloyd, R.V.; Rubin, J.; Haluska, P.; Carboni, J.M.; Gottardis, M.M.; Ames, M.M. Molecular markers for novel therapies in neuroendocrine (carcinoid) tumors. Endocr. Relat. Cancer 2010, 17, 623. [Google Scholar] [CrossRef]
- Zitzmann, K.; Ailer, G.; Vlotides, G.; Spoettl, G.; Maurer, J.; Göke, B.; Beuschlein, F.; Auernhammer, C.J. Potent antitumor activity of the novel HSP90 inhibitors AUY922 and HSP990 in neuroendocrine carcinoid cells. Int. J. Oncol. 2013, 43, 1824–1832. [Google Scholar] [CrossRef] [Green Version]
- Gloesenkamp, C.; Nitzsche, B.; Lim, A.R.; Normant, E.; Vosburgh, E.; Schrader, M.; Ocker, M.; Scherübl, H.; Höpfner, M. Heat shock protein 90 is a promising target for effective growth inhibition of gastrointestinal neuroendocrine tumors. Int. J. Oncol. 2012, 40, 1659–1667. [Google Scholar] [CrossRef] [Green Version]
- Keen, N.; Taylor, S. Aurora-kinase inhibitors as anticancer agents. Nat. Rev. Cancer 2004, 4, 927–936. [Google Scholar] [CrossRef]
- Katayama, H.; Sasai, K.; Kawai, H.; Yuan, Z.M.; Bondaruk, J.; Suzuki, F.; Fujii, S.; Arlinghaus, R.B.; Czerniak, B.A.; Sen, S. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat. Genet. 2004, 36, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsha, A.; Belkhiri, A.; Goff, L.; El-Rifai, W. Aurora kinase A in gastrointestinal cancers: Time to target. Mol. Cancer 2015, 14, 106. [Google Scholar] [CrossRef] [Green Version]
- Georgieva, I.; Koychev, D.; Wang, Y.; Holstein, J.; Hopfenmüller, W.; Zeitz, M.; Grabowski, P. ZM447439, a novel promising aurora kinase inhibitor, provokes antiproliferative and proapoptotic effects alone and in combination with bio- and chemotherapeutic agents in gastroenteropancreatic neuroendocrine tumor cell lines. Neuroendocrinology 2010, 91, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Hofving, T.; Arvidsson, Y.; Almobarak, B.; Inge, L.; Pfragner, R.; Persson, M.; Stenman, G.; Kristiansson, E.; Johanson, V.; Nilsson, O. The neuroendocrine phenotype, genomic profile and therapeutic sensitivity of GEPNET cell lines. Endocr.-Relat. Cancer 2018, 25, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Briest, F.; Wang, Y.; Arsenic, R.; Elezkurtaj, S.; Berg, E.; Greshake, S.; Lock, A.C.; Hörsch, D.; Arnold, C.N.; Hummel, M.; et al. Immunohistochemical Study of Mitosis-regulatory Proteins in Gastroenteropancreatic Neuroendocrine Neoplasms. Anticancer Res. 2018, 38, 3863–3870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, E.; Capdevila, J.; Barriuso, J.; Anton-Aparicio, L.; Castellano, D. Gastroenteropancreatic neuroendocrine tumor cancer stem cells: Do they exist? Cancer Metastasis Rev. 2012, 31, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, M.; Tian, H.; de Sauvage, F.J. The Hedgehog Signaling Pathway in Cancer. Clin. Cancer Res. 2006, 12, 5924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shida, T.; Furuya, M.; Nikaido, T.; Hasegawa, M.; Koda, K.; Oda, K.; Miyazaki, M.; Kishimoto, T.; Nakatani, Y.; Ishikura, H. Sonic Hedgehog-Gli1 signaling pathway might become an effective therapeutic target in gastrointestinal neuroendocrine carcinomas. Cancer Biol. Ther. 2006, 5, 1530–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fendrich, V.; Waldmann, J.; Esni, F.; Ramaswamy, A.; Mullendore, M.; Buchholz, M.; Maitra, A.; Feldmann, G. Snail and Sonic Hedgehog activation in neuroendocrine tumors of the ileum. Endocr.-Relat. Cancer 2007, 14, 865. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [Green Version]
- Lau, J.; Hebrok, M. Hedgehog signaling in pancreas epithelium regulates embryonic organ formation and adult beta-cell function. Diabetes 2010, 59, 1211–1221. [Google Scholar] [CrossRef] [Green Version]
- Fendrich, V.; Ramerth, R.; Waldmann, J.; Maschuw, K.; Langer, P.; Bartsch, D.K.; Slater, E.P.; Ramaswamy, A.; Rothmund, M. Sonic hedgehog and pancreatic-duodenal homeobox 1 expression distinguish between duodenal and pancreatic gastrinomas. Endocr. Relat. Cancer 2009, 16, 613–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fendrich, V.; Rehm, J.; Waldmann, J.; Buchholz, M.; Christofori, G.; Lauth, M.; Slater, E.P.; Bartsch, D.K. Hedgehog Inhibition With Cyclopamine Represses Tumor Growth and Prolongs Survival in a Transgenic Mouse Model of Islet Cell Tumors. Ann. Surg. 2011, 253, 546–552. [Google Scholar] [CrossRef]
- Fendrich, V.; Wiese, D.; Waldmann, J.; Lauth, M.; Heverhagen, A.E.; Rehm, J.; Bartsch, D.K. Hedgehog Inhibition With the Orally Bioavailable Smo Antagonist LDE225 Represses Tumor Growth and Prolongs Survival in a Transgenic Mouse Model of Islet Cell Neoplasms. Ann. Surg. 2011, 254, 818–823. [Google Scholar] [CrossRef]
- Gurung, B.; Hua, X. Menin/PRMT5/hedgehog signaling: A potential target for the treatment of multiple endocrine neoplasia type 1 tumors. Epigenomics 2013, 5, 469–471. [Google Scholar] [CrossRef] [Green Version]
- Gurung, B.; Feng, Z.; Iwamoto, D.V.; Thiel, A.; Jin, G.; Fan, C.-M.; Ng, J.M.Y.; Curran, T.; Hua, X. Menin epigenetically represses Hedgehog signaling in MEN1 tumor syndrome. Cancer Res. 2013, 73, 2650–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, H.; Hong, X.; Wang, X.; Lin, C.; Wu, W.; Zhao, Y. Pancreatic neuroendocrine tumor cancer stem cells: Potential novel therapeutic targets? Transl. Cancer Res. 2016, 5, 860–870. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Zarebczan, B.; Chen, H. Signaling mechanisms in neuroendocrine tumors as targets for therapy. Endocrinol. Metab. Clin. N. Am. 2010, 39, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, Y.; Fernandez-Del Castillo, C.; Yilmaz, O.; Deshpande, V. Heterogeneity in signaling pathways of gastroenteropancreatic neuroendocrine tumors: A critical look at notch signaling pathway. Mod. Pathol. 2013, 26, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Nakakura, E.K.; Sriuranpong, V.R.; Kunnimalaiyaan, M.; Hsiao, E.C.; Schuebel, K.E.; Borges, M.W.; Jin, N.; Collins, B.J.; Nelkin, B.D.; Chen, H.; et al. Regulation of Neuroendocrine Differentiation in Gastrointestinal Carcinoid Tumor Cells by Notch Signaling. J. Clin. Endocrinol. Metab. 2005, 90, 4350–4356. [Google Scholar] [CrossRef] [Green Version]
- Sriuranpong, V.; Borges, M.W.; Strock, C.L.; Nakakura, E.K.; Watkins, D.N.; Blaumueller, C.M.; Nelkin, B.D.; Ball, D.W. Notch Signaling Induces Rapid Degradation of Achaete-Scute Homolog 1. Mol. Cell. Biol. 2002, 22, 3129–3139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunnimalaiyaan, M.; Traeger, K.; Chen, H. Conservation of the Notch1 signaling pathway in gastrointestinal carcinoid cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G636–G642. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.-X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Sehrawat, A.; Shiota, C.; Mohamed, N.; DiNicola, J.; Saleh, M.; Kalsi, R.; Zhang, T.; Wang, Y.; Prasadan, K.; Gittes, G.K. SMAD7 enhances adult β-cell proliferation without significantly affecting β-cell function in mice. J. Biol. Chem. 2020, 295, 4858–4869. [Google Scholar] [CrossRef] [PubMed]
- Adler, J.T.; Hottinger, D.G.; Kunnimalaiyaan, M.; Chen, H. Histone deacetylase inhibitors upregulate Notch-1 and inhibit growth in pheochromocytoma cells. Surgery 2008, 144, 956–962. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulou, S.; Edlund, H. Attenuated Wnt Signaling Perturbs Pancreatic Growth but Not Pancreatic Function. Diabetes 2005, 54, 2844–2851. [Google Scholar] [CrossRef]
- Kim, J.T.; Li, J.; Jang, E.R.; Gulhati, P.; Rychahou, P.G.; Napier, D.L.; Wang, C.; Weiss, H.L.; Lee, E.Y.; Anthony, L.; et al. Deregulation of Wnt/β-catenin signaling through genetic or epigenetic alterations in human neuroendocrine tumors. Carcinogenesis 2013, 34, 953–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.-F.; Spoettl, G.; Maurer, J.; Nölting, S.; Auernhammer, C.J. Inhibition of Wnt/β-Catenin Signaling in Neuroendocrine Tumors in vitro: Antitumoral Effects. Cancers 2020, 12, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saupe, F.; Schwenzer, A.; Jia, Y.; Gasser, I.; Spenlé, C.; Langlois, B.; Kammerer, M.; Lefebvre, O.; Hlushchuk, R.; Rupp, T.; et al. Tenascin-C Downregulates Wnt Inhibitor Dickkopf-1, Promoting Tumorigenesis in a Neuroendocrine Tumor Model. Cell Rep. 2013, 5, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.T.; Liu, C.; Zaytseva, Y.Y.; Weiss, H.L.; Townsend, C.M., Jr.; Evers, B.M. Neurotensin, a novel target of Wnt/β-catenin pathway, promotes growth of neuroendocrine tumor cells. Int. J. Cancer 2015, 136, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Cao, Y.; Li, F.; Su, Y.; Li, Y.; Peng, Y.; Cheng, Y.; Zhang, C.; Wang, W.; Ning, G. Targeting β-catenin signaling for therapeutic intervention in MEN1-deficient pancreatic neuroendocrine tumours. Nat. Commun. 2014, 5, 5809. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Jingbo, A.; Wang, M.; Farley, S.; Lee, L.Y.; Lee, L.C.; Sawicki, M.P. Menin promotes the Wnt signaling pathway in pancreatic endocrine cells. Mol. Cancer Res. 2008, 6, 1894–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidd, M.; Schimmack, S.; Lawrence, B.; Alaimo, D.; Modlin, I.M. EGFR/TGFα and TGFβ/CTGF Signaling in Neuroendocrine Neoplasia: Theoretical Therapeutic Targets. Neuroendocrinology 2013, 97, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Panarelli, N.; Tyryshkin, K.; Wong, J.J.M.; Majewski, A.; Yang, X.; Scognamiglio, T.; Kim, M.K.; Bogardus, K.; Tuschl, T.; Chen, Y.-T.; et al. Evaluating gastroenteropancreatic neuroendocrine tumors through microRNA sequencing. Endocr.-Relat. Cancer 2019, 26, 47–57. [Google Scholar] [CrossRef]
- Hellberg, T.; Mohr, R.; Geisler, L.; Knorr, J.; Wree, A.; Demir, M.; Benz, F.; Lambrecht, J.; Loosen, S.H.; Tacke, F.; et al. Serum levels of miR-223 but not miR-21 are decreased in patients with neuroendocrine tumors. PLoS ONE 2020, 15, e0244504. [Google Scholar] [CrossRef]
- Özdirik, B.; Stueven, A.K.; Mohr, R.; Geisler, L.; Wree, A.; Knorr, J.; Demir, M.; Vucur, M.; Loosen, S.H.; Benz, F.; et al. Analysis of miR-29 Serum Levels in Patients with Neuroendocrine Tumors-Results from an Exploratory Study. J. Clin. Med. 2020, 9, 2881. [Google Scholar] [CrossRef] [PubMed]
- Moschovis, D.; Gazouli, M.; Tzouvala, M.; Vezakis, A.; Karamanolis, G. Long non-coding RNA in pancreatic adenocarcinoma and pancreatic neuroendocrine tumors. Ann. Gastroenterol. 2017, 30, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Modali, S.D.; Parekh, V.I.; Kebebew, E.; Agarwal, S.K. Epigenetic regulation of the lncRNA MEG3 and its target c-MET in pancreatic neuroendocrine tumors. Mol. Endocrinol. 2015, 29, 224–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, M.; Tang, L.; Ding, R.; Shi, L.; Liu, A.; Chen, D.; Shao, C. Long noncoding RNA-mRNA expression profiles and validation in pancreatic neuroendocrine neoplasms. Clin. Endocrinol. 2020, 92, 312–322. [Google Scholar] [CrossRef]
- Kaku, M.; Nishiyama, T.; Yagawa, K.; Abe, M. Establishment of a carcinoembryonic antigen-producing cell line from human pancreatic carcinoma. Gan 1980, 71, 596–601. [Google Scholar]
- Evers, B.M.; Townsend, C.M., Jr.; Upp, J.R.; Allen, E.; Hurlbut, S.C.; Kim, S.W.; Rajaraman, S.; Singh, P.; Reubi, J.C.; Thompson, J.C. Establishment and characterization of a human carcinoid in nude mice and effect of various agents on tumor growth. Gastroenterology 1991, 101, 303–311. [Google Scholar] [CrossRef]
- Benten, D.; Behrang, Y.; Unrau, L.; Weissmann, V.; Wolters-Eisfeld, G.; Burdak-Rothkamm, S.; Stahl, F.R.; Anlauf, M.; Grabowski, P.; Möbs, M.; et al. Establishment of the First Well-differentiated Human Pancreatic Neuroendocrine Tumor Model. Mol. Cancer Res. 2018, 16, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, T.; Peeters, M.; Dogan, F.; Pauwels, P.; Van Assche, E.; Beyens, M.; Mortier, G.; Vandeweyer, G.; de Herder, W.; Van Camp, G.; et al. Whole-exome characterization of pancreatic neuroendocrine tumor cell lines BON-1 and QGP-1. J. Mol. Endocrinol. 2015, 54, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Boora, G.K.; Kanwar, R.; Kulkarni, A.A.; Pleticha, J.; Ames, M.; Schroth, G.; Beutler, A.S.; Banck, M.S. Exome-level comparison of primary well-differentiated neuroendocrine tumors and their cell lines. Cancer Genet. 2015, 208, 374–381. [Google Scholar] [CrossRef]
- Kaemmer, C.A.; Umesalma, S.; Maharjan, C.K.; Moose, D.L.; Narla, G.; Mott, S.L.; Zamba, G.K.D.; Breheny, P.; Darbro, B.W.; Bellizzi, A.M.; et al. Development and comparison of novel bioluminescent mouse models of pancreatic neuroendocrine neoplasm metastasis. Sci. Rep. 2021, 11, 10252. [Google Scholar] [CrossRef]
- Wong, C.; Vosburgh, E.; Levine, A.J.; Cong, L.; Xu, E.Y. Human neuroendocrine tumor cell lines as a three-dimensional model for the study of human neuroendocrine tumor therapy. J. Vis. Exp. 2012, 14, e4218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ear, P.H.; Li, G.; Wu, M.; Abusada, E.; Bellizzi, A.M.; Howe, J.R. Establishment and Characterization of Small Bowel Neuroendocrine Tumor Spheroids. J. Vis. Exp. 2019, 14, e60303. [Google Scholar] [CrossRef]
- April-Monn, S.L.; Wiedmer, T.; Skowronska, M.; Maire, R.; Schiavo Lena, M.; Trippel, M.; Di Domenico, A.; Muffatti, F.; Andreasi, V.; Capurso, G.; et al. Three-Dimensional Primary Cell Culture: A Novel Preclinical Model for Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2021, 111, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Siolas, D.; Hannon, G.J. Patient-derived tumor xenografts: Transforming clinical samples into mouse models. Cancer Res. 2013, 73, 5315–5319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, K.; Teng, L.; Shen, Y.; He, K.; Xu, Z.; Li, G. Patient-derived human tumour tissue xenografts in immunodeficient mice: A systematic review. Clin. Transl. Oncol. 2010, 12, 473–480. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- DeRose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef]
- Tanaka, N.; Onda, M.; Seya, T.; Kanazawa, Y.; Naito, Z.; Asano, G.; Oguro, T. Establishment and characterization of a human rectal neuroendocrine carcinoma xenograft into nude mice. Digestion 1999, 60, 117–124. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, D.D.; Yang, M.; Chen, D.; Pang, L.; Guo, S.; Cai, J.; Wery, J.P.; Li, L.; Li, H.Q.; et al. Comprehensive characterization of chemotherapeutic efficacy on metastases in the established gastric neuroendocrine cancer patient derived xenograft model. Oncotarget 2015, 6, 15639–15651. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Zhang, L.; Serra, S.; Law, C.; Wei, A.; Stockley, T.L.; Ezzat, S.; Asa, S.L. Establishment and Characterization of a Human Neuroendocrine Tumor Xenograft. Endocr. Pathol. 2016, 27, 97–103. [Google Scholar] [CrossRef]
- Gaudenzi, G.; Vitale, G. Transplantable zebrafish models of neuroendocrine tumors. Ann. Endocrinol. 2019, 80, 149–152. [Google Scholar] [CrossRef]
- Gaudenzi, G.; Carra, S.; Dicitore, A.; Cantone, M.C.; Persani, L.; Vitale, G. Fishing for neuroendocrine tumors. Endocr.-Relat. Cancer 2020, 27, R163–R176. [Google Scholar] [CrossRef] [PubMed]
- Peverelli, E.; Giardino, E.; Treppiedi, D.; Meregalli, M.; Belicchi, M.; Vaira, V.; Corbetta, S.; Verdelli, C.; Verrua, E.; Serban, A. Dopamine receptor type 2 (DRD2) and somatostatin receptor type 2 (SSTR2) agonists are effective in inhibiting proliferation of progenitor/stem-like cells isolated from nonfunctioning pituitary tumors. Int. J. Cancer 2017, 140, 1870–1880. [Google Scholar] [CrossRef]
- Wurth, R.; Barbieri, F.; Pattarozzi, A.; Gaudenzi, G.; Gatto, F.; Fiaschi, P.; Ravetti, J.; Zona, G.; Daga, A.; Persani, L. Phenotypical and pharmacological characterization of stem-like cells in human pituitary adenomas. Mol. Neurobiol. 2017, 54, 4879–4895. [Google Scholar] [CrossRef]
- Parangi, S.; Dietrich, W.; Christofori, G.; Lander, E.S.; Hanahan, D. Tumor Suppressor Loci on Mouse Chromosomes 9 and 16 Are Lost at Distinct Stages of Tumorigenesis in a Transgenic Model of Islet Cell Carcinoma. Cancer Res. 1995, 55, 6071–6076. [Google Scholar] [PubMed]
- Hunter, K.E.; Quick, M.L.; Sadanandam, A.; Hanahan, D.; Joyce, J.A. Identification and characterization of poorly differentiated invasive carcinomas in a mouse model of pancreatic neuroendocrine tumorigenesis. PLoS ONE 2013, 8, e64472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, P.; Chu, G.C.; Perry, S.R.; Nolan-Stevaux, O.; Hanahan, D. Imaging guided trials of the angiogenesis inhibitor sunitinib in mouse models predict efficacy in pancreatic neuroendocrine but not ductal carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, E1275–E1284. [Google Scholar] [CrossRef] [Green Version]
- Carter, A.M.; Kumar, N.; Herring, B.; Tan, C.; Guenter, R.; Telange, R.; Howse, W.; Viol, F.; Graham, C.; McCaw, T.R.; et al. Cdk5 drives formation of heterogeneous pancreatic neuroendocrine tumors. bioRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Pozo, K.; Castro-Rivera, E.; Tan, C.; Plattner, F.; Schwach, G.; Siegl, V.; Meyer, D.; Guo, A.; Gundara, J.; Mettlach, G.; et al. The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell 2013, 24, 499–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, A.M.; Tan, C.; Pozo, K.; Telange, R.; Molinaro, R.; Guo, A.; De Rosa, E.; Martinez, J.O.; Zhang, S.; Kumar, N.; et al. Phosphoprotein-based biomarkers as predictors for cancer therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 18401–18411. [Google Scholar] [CrossRef] [PubMed]



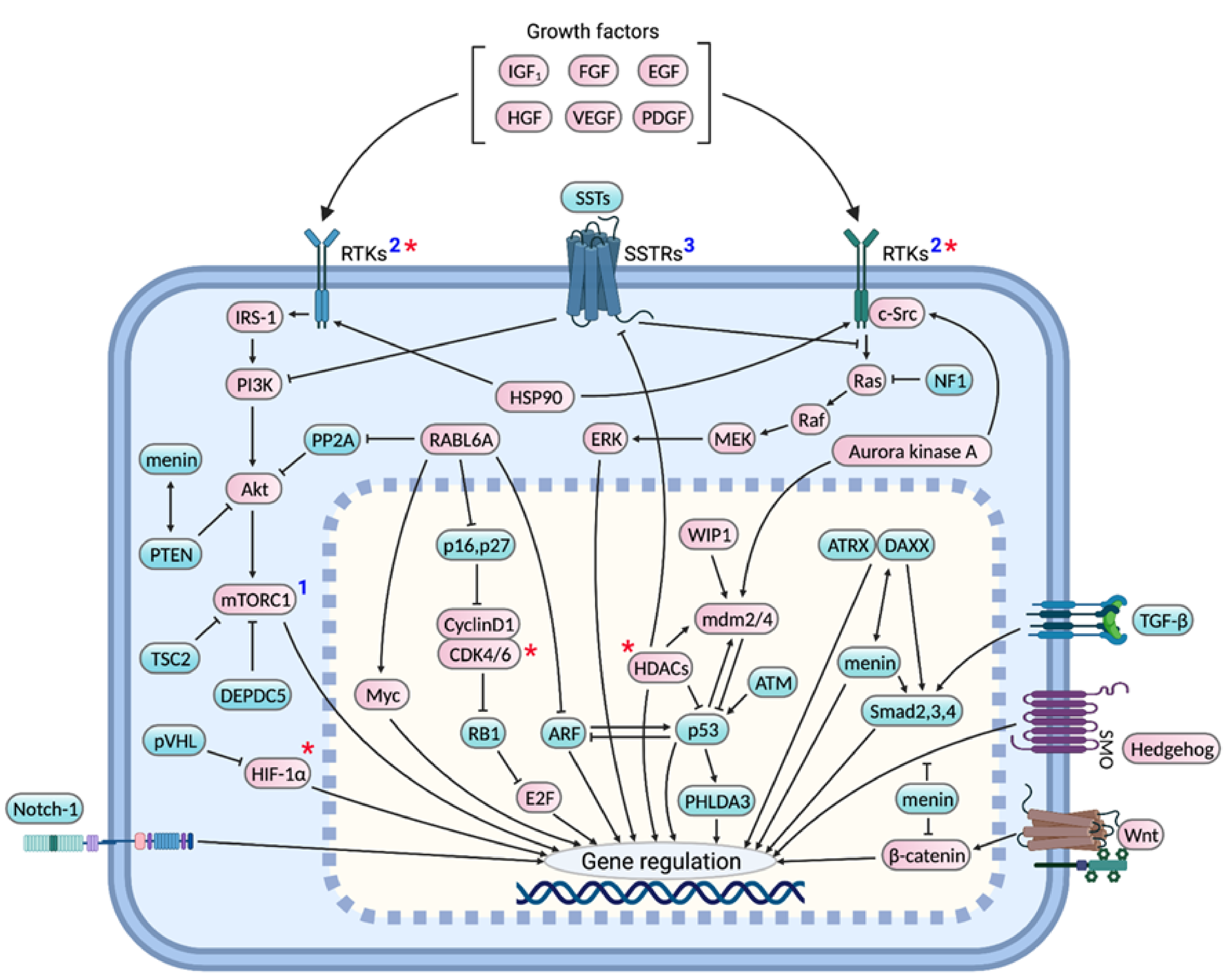{kind=link}
{kind=link}
| Technique | Reference | Key Findings |
|---|---|---|
| Exome/genome sequencing | [24] | Somatic MEN1 mutations in 44.1% of 68 sporadic pNETs. MEN1 mutations correlated with poor patient survival. |
| [23] | Somatic MEN1 mutations in 41% of 102 primary pNETs. Abnormal telomere length observed in MEN1-mutated tumors. | |
| [39] | In total, 26% of 57 sporadic well differentiated pNETs had recurrent LOH of 10 specific chromosomes and biallelic MEN1 inactivation. Another 40% had chromosome 11 LOH and biallelic MEN1 inactivation. The first patient group had worse clinical outcomes compared to the second. | |
| [40] | MEN1 mutations in 43% of 65 pNETs. | |
| [43] | Somatic MEN1 mutations in 56% of 80 patient pNETs. In total, 1 of 17 patients carried a germline MEN1 mutation. | |
| Allelotyping/LOH analysis | [31] | Loss of chromosomal segment 11q (where MEN1 is located) in >60% cases. |
| [34] | LOH of 11q13 in 70% and MEN1 mutations in 27% of 11 advanced pNETs (9 NF and 2 glucagonomas). | |
| [29] | MEN1 mutatons in 6 (mostly pNETs) of 43 sporadic GEPNETs. | |
| [37] | MEN1 allelic deletions in 93% of gastrinomas and 50% of 12 insulinomas; mutations in 33% gastrinomas and 17% insulinomas. | |
| Microarray | [35] | Consensus cluster analysis of microarray results showed clustering of 5 out of 9 sporadic NF-pNETs with MEN1-associated familial pNETs. In total, 4 of those 5 sporadic pNETs had MEN1 LOH. |
| Model | Strain | pNET Type | Key Findings |
|---|---|---|---|
| RIP-Tag2 | C57BL/6 | Insulinoma | SV40 large T-antigen inactivates p53 and Rb in islet-β cells and promotes insulinoma development in a multi-stage, synchronized fashion [57] |
| Men1f/f Ptenf/f; MIP-Cre or RIP-Cre (MPM or MPR) | C57BL/6J | Insulinoma | Loss of Pten co-operates with that of Men1 to develop well differentiated G1/G2 pNETs [56] |
| RIP-Tag2 AB6F1 | AB6F1 (hybrids from A/J dam and RT2 C57BL/6 sire) | Non-functional (NF) pNETs | RT2 mice develop NF pNETs with higher rate of liver metastases on AB6F1 genetic background, which is attributed to low expression of Insm1, a β-cell specific differentiation factor required for insulin secretion [58] |
| RIP-MyrAkt1 | C57/BL6 | Insulinoma | β-cell specific expression of constitutively active Myr-Akt leads to formation of insulinomas in S6K1 (a mTOR downstream target) dependent manner [59] |
| Avp-Tag | C57B1/10; CBA/J | Insulinoma | Mice bearing vasopressin promoter (1.2 kb 5′ sequence)-SV40 hybrid transgene uncharacteristically transformed pancreatic β-cells and anterior pituitary cells with no effect in hypothalamus and other organs where vasopressin is normally expressed [60] |
| Men1TSM/+ | NIH Black Swiss; 129/Sv | Insulinoma | Mutation of one Men1 allele by homologous recombination leads to insulinoma development by 9 months and other tumors involving parathyroid, thyroid, adrenal cortex, and pituitary by 16 months mimicking human MEN1 syndrome [61] |
| Men1+/T | 129 | Insulinoma and glucagonoma that dedifferentiated into advanced NF-pNETs | Disruption of one Men1 allele by gene targeting resulted in tumors of pancreatic, parathyroid, thyroid, pituitary, and adrenal glands exhibiting multistage progression and metastatic potential [62] |
| Men1f/f; RIP-Cre | C57BL/6 | Insulinoma | RIP-Cre mediated conditional knockout of Men1 gene leads to insulinomas and pituitary prolactinomas by 9 months [63] |
| Men1f/f; Glu-Cre | Not specified | Insulinoma | Surprisingly, loss of Men1 in α-cells resulted in β-cell insulinomas rather than glucagonomas suggesting the role of intercellular talk between islet cells [64] |
| Men1f/f; Pdx1-Cre | FVB; 129Sv | Insulinoma | Although Men1 is lost in both pancreatic exocrine and endocrine cells, only endocrine cells developed into highly angiogenic tumors suggesting the role of tissue-specific menin modulators and surrounding microenvironment during tumorigenesis [65] |
| Men1f/f; RIP2-CreER | 129; (C57BL/6 X CBA) | Insulinoma | Temporally controlled β-cell specific loss of Men1 led to insulinomas. Moreover, the model helps elucidate early stage events such as β-cell hyperproliferation [66]. |
| Men1f/f; RIP-Cre | B6; FVB; 129Sv | Insulinoma | Conditional knockout of both Men1 alleles promoted islet cell tumor development much faster than that of one allele [67] |
| Men1f/f; RIP-Cre | 129 | Insulinoma | Disruption of Men1 gene directly in β-cells led to insulinoma development by 6 months in a multistage fashion, exhibiting angiogenesis and altered E-cadherin and β-catenin expression [68] |
| Men1f/f; Glu-Cre | 129; B6/CBAJ-F1 | Insulinoma, glucagonoma, and mixed islet cell tumor | α-cell specific loss of Men1 leads to α-cell hyperplasia that grow into glucagonomas, however, majority of the hyperplastic α-cells transdifferentiate into insulinomas and mixed islet tumors [69] |
| Glu2-Tag | C57BL/6 | Glucagonoma | Expression of Tag under preproglucagon promoter drives hyperproliferation of alpha cells and formation of glucagonomas by 9–12 months. Promiscuous expression of T-antigen in hind brain neurons is not sufficient for their hyperplasia or tumorigenesis [70]. |
| Gcgr−/− | DBA/1 | Glucagonoma | Inhibition of glucagon signaling by glucagon receptor mutation causes α-cell hyperplasia that progress into islet dysplasia and solid tumors. A few animals develop mixed tumors or NF-pNETs [71]. |
| Pc2−/− | C57BL/6 | Glucagonoma | Loss of prohormone convertase 2 required for glucagon synthesis leads to α-cell hyperplasia that develop glucagonomas and mixed islet tumors by 6–8 months [72] |
| RIP7-rtTA; tet-o-MT; p48-Cre; Ink4a/Arf f/f | C57BL6; FVB; ICR | Not determined | Loss of Ink4a/Arf tumor suppressor locus cooperates with overexpression of PyMT in pancreatic progenitor cells to induce pNET formation however at low incidence rate of 20% [73] |
| RIP7-rtTA; tet-o-MT; p48-Cre; Trp53f/f; Ink4a/Arf f/f | C57BL6; FVB; ICR | Not determined | Overexpression of oncogenic PyMT in β-cells together with deletion of P53 and Ink4a/Arf loci results in pNET incidence in 40% mutant mice [73] |
| pIns-c-MycERTAM/RIP-Bcl-xL | CBAxC57BL/6 | Insulinoma | Conditional expression of transgenic Myc and Bcl-xL to suppress Myc-induced apoptosis in islet β cells causes islet tumor development in a reversible fashion [74] |
| Pdx1-Cre; Trp53R172H;Rbf/f | FVB/N; J1; | Well differentiated, metastatic insulinoma and glucagonoma | Pancreas-specific p53 mutation and Rb deletion caused islet dysplasia that progressed to indolent and metastatic pNET in stepwise fashion [75] |
| Men1f/f Rb1f/f RIP-Cre; Men1f/f Ptenf/f RIP-Cre; Trp53f/f Rb1f/f RIP-Cre; | Men1f/f (129S, FVB) Rb1f/f(FVB;129) Ptenf/f(C;129S4) Trp53f/f(B6.129P2) RIP-Cre (C57BL/6) | Well differentiated G1, G2, and G3 pNETs (insulinoma) | Demonstrated the cooperative role of tumor suppressor genes, Men1, Rb1, Pten, and Trp53 in pNET suppression [76] |
| RIP-Tag2; Rabl6m/m | C57BL/6N | Insulinoma | Loss of oncogenic RABL6A attenuates pNET progression and angiogenesis in RIP-Tag2 mice [77] |
| Pdx1-Cre; Men1f/f; B7x KO | C57BL/6 | Insulinoma | Loss of B7x, an immune-checkpoint ligand, reduces islet β-cell proliferation and pNET formation consistent with increased T-cell infiltration [78] |
| Technique | Reference | Key Findings |
|---|---|---|
| Exome sequencing | [24] | Somatic PTEN, TSC2, and PIK3CA mutations in 7.3%, 8.8%, and 1.4% of 68 sporadic pNETs, respectively. |
| [23] | Somatic mutations in mTOR pathway genes observed in 102 primary pNETs: PTEN (7%), DEPDC (2%), TSC1 (2%), and TSC2 (2%). mTOR pathway gene mutations associated with poor survival. | |
| [40] | 11% of 65 pNETs had mTOR pathway gene mutations: TSC2 (6%) and PTEN (5%). | |
| [43] | TSC2 mutations in 25% of 80 patient pNETs. In total, 1 of 17 patients carried a germline TSC2 mutation. | |
| Allelotyping/LOH analysis | [87] | LOH of 10q23 (where PTEN is located) in >50% of 22 pNETs. PTEN mutations rarely observed. |
| [86] | Allelic deletions of 16p13 (where TSC2 is located) in 36% of 28 pNETs | |
| Microarray | [88] | TSC2, PTEN or both downregulated in 85% of primary PNETs, subsequently validated by qRT-PCR and IHC studies. Reduced expression of TSC2 and PTEN correlated with poor patient prognosis. |
| RNA sequencing | [106] | Ingenuity pathway analysis (IPA) and Connectivity Map (CMap) analysis of 626 metastatic gene signatures obtained from 39 primary tumors, 21 lymph node metastases and 17 liver metastases predicted mTOR and PI3K as top pNET pharmacological targets. |
| [92] | Alteration of Akt signaling genes revealed by sequencing of 20 primary pNETs. | |
| IHC | [107] | Low PTEN expression in 48% of 21 pNETs. |
| Technique | Reference | Key Findings |
|---|---|---|
| Sequencing and mutational analysis | [24] | No INK4A/ARF mutation observed in 68 pNETs. |
| [11] | Inactivating mutations in the RB1 gene identified in 75% (3/4) small cell pNECs and 66.67% (2 of 3) large cell pNECs, however, absent in 11 well-differentiated pNETs analyzed. No CDKN2A mutations observed in 7 pNECs and 11 pNETs. | |
| Methylation Specific PCR (MSP) | [112] | In total, 91.7% of 12 gastrinomas and non-functioning pNETs demonstrated INK4a homozygous gene deletions (41.7%) or 5′ CpG island hypermethylation. However, no mutations were found by single-strand conformation polymorphism (SSCP) analyses. |
| [103] | INK4a 5′-CpG island hypermethylation in 52% of 44 gastrinomas, along with homozygous gene deletions. | |
| [126] | In total, 17% of 17 insulinomas exhibited INK4a gene alterations: homozygous deletion in 5.9% and promoter hypermethylation in 11.8%. No INK4a mutations were identified by SSCP. IHC confirmed loss of the p16 protein expression in samples harboring gene alterations. | |
| [116] | INK4a CpG island hypermethylation in 17% of 12 pNETs, more frequently in malignant (29%) than in benign (0%) tumors. | |
| PCR (MSP or gene specific and LOH) | [114] | INK4a and ARF CpG island hypermethylation in 9% (1 out of 11) pNETs each vs. 44% and 31%, respectively, in carcinoid NETs. Chromosome 9p loss identified in 18% (2 of 11) pNETs. |
| RT-PCR | [104] | Absent expression of INK4a, INK4b, and ARF in 28% (2/7), 57% (4/7), and 43% (3/7) NF-pNETs. Loss of INK4b observed in 26% insulinomas and gastrinomas (N = 19), however, INK4a and ARF found to be expressed. |
| [35] | Overexpression of CDK4 and CDK6 in MEN1 NF-pNETs (N = 10) compared to VHL (N = 9) and sporadic (N = 9) NF-pNETs and normal islets (N = 4). | |
| Tissue microarray, qPCR, and FISH | [122] | IHC revealed high CDK4, cyclin D1 and phospho-RB1 levels in 58–68% of total pNETs (N = 92) in contrast to negative staining in the normal pancreas. qRT-PCR revealed marked upregulation of CDK4 in 19% of 26 pNETs, which were found to have amplified CDK4 or CDK6 genes by qPCR and FISH. |
| IHC | [11] | RB1 protein expression intact in well-differentiated pNETs, however, lost in 88.9% of 9 small cell and 60% of large cell pNECs. Loss of p16 expression observed in pNECs with intact RB1 suggest p16/Rb pathway is disrupted in virtually all pNECs. |
| Technique | Reference | Key Findings |
|---|---|---|
| Exome sequencing and mutational analysis | [24] | Somatic ATRX and DAXX inactivating-to-missense mutations in 25% and 17.6% of 68 sporadic pNETs, respectively. IHC showed complete loss of ATRX or DAXX in pNETs harboring the corresponding gene mutations. ATRX/DAXX mutations with or without MEN1 mutations correlated with improved patient survival. |
| [23] | Somatic mutations in ATRX (10%) and DAXX (22%) of 102 primary pNETs. ATRX/DAXX mutations linked with longer telomere length and poor patient survival. | |
| [40] | DAXX and ATRX mutations in 28% and 11% of 65 pNETs, respectively. These mutations were mutually exclusive. | |
| [43] | Somatic alterations in DAXX and ATRX observed in 40% and 25% of 80 patient pNETs. | |
| IHC | [11] | ATRX or DAXX immunolabeling lost in 45% of 11 pNETs, but intact in all of 19 pNECs. |
| Technique | Reference | Key Findings |
|---|---|---|
| Sequencing and mutational analysis | [11] | Inactivating TP53 mutations found in 4 of 7 pNECs, however, in none of 11 well-differentiated pNETs. |
| [24] | TP53 gene mutations identified in 3% of 68 pNETs. | |
| PCR and IHC | [157] | Amplification of the MDM2 gene in 22% (38 of 169), MDM4 in 30% (45 of 150), and WIP1 in 76% (34 of 45) well differential pNETs, which correlated with corresponding changes in mRNA and protein expression. |
| IHC | [11] | 11 well differentiated pNETs exhibited normal p53 labeling, however, abnormal labeling observed in 9 (100%) small cell pNECs and 9 of 10 (90%) large cell pNECs. |
| Technique | Reference | Key Findings |
|---|---|---|
| FISH | [185] | EGFR copy number found to be elevated in 38% of 44 pNET cases. |
| qRT-PCR | [186] | Positive VEGF expression in 5 of 8 pNETs and EGFR in 4 pNETs of which 2 had overexpressed EGFR compared to the normal pancreatic tissue. |
| IHC | [178] | Positive expression of VEGF in 16 (11 mild, 3 moderate, and 2 strong) out of 20 pNETs. |
| [187] | Of 38 malignant pNETs, 100% expressed PDGFRα tumor cells, 57% in stromal cells; 74% expressed PDGFRβ in tumor cells, 97% in stromal cells; 92% of tumors expressed c-kit and 55% expressed EGFR. | |
| [180] | Positive VEGF-A expression in all 19 primary well-differentiated pNETs and 7 liver metastases. Mild to moderate VEGF-C immunostaining in the majority of primary pNETs with significantly increased expression in liver metastases. High immunoreactivity for VEGFR2 observed in all 8 primary pNETs and 3 liver metastases examined. | |
| [179] | Positive expression of VEGF in pNETs 73% (33/45) pNETs which correlated negatively with WHO disease stage. In total, 91% (41/45) pNETs showed high HIF-1α staining. | |
| [188] | Positive expression of EGFR or p-EGFR in 25–50% of 48 primary pNETs or their metastases. Higher percentage of carcinoid NETs were found to be EGFR- or p-EGFR positive. Activated p-EGFR in primary NETs correlated with poor prognosis. | |
| [189] | Positive PDGFRβ expression observed in 18 of 21 (86%) primary tumors and all of 19 pNET metastases. PDGFRβ shown to be more frequently expressed in primary pNETs and metastases as compared to normal endocrine pancreas. | |
| [177] | In total, 80% of 15 pNETs showed mild to moderate VEGF staining. | |
| [185] | High IHC staining (score 3) of VEGFR1, TGFBR1, PDGFRA, SSTR5, SSTR2A, and IGF1R in 80%, 69% 65%, 55%, 55%, and 47% of 44 pNETs, respectively. |
| Technique | Reference | Key Findings |
|---|---|---|
| Sequencing and mutational analysis | [24] | KRAS mutation observed in none of the 68 pNETs studied. |
| [155] | HRAS, NRAS, or BRAF mutations in less than 1% pNETs. | |
| ctDNA sequencing | [212] | ctDNA NGS of 280 NET/NEC patient plasma samples revealed KRAS mutations in 22% (n = 61) and NF1 mutations in 7% (n = 19) samples. |
| Mutational analysis | [185] | No pNETs (n = 35) showed KRAS exon 2 mutations. |
| Technique | Reference | Key Findings |
|---|---|---|
| RT PCR and IHC | [230] | mRNA amplification of SSTR1 in 90.1%, SSTR2 in 84.8%, SSTR3 in 78.8%, SSTR4 in 24.2% and SSTR5 in 42.4% of pNET cases. Positive SSTR2 immunoreactivity in 15 of 22 tumors (68.2%), SSTR3 in 8 of 22 (36.4%), and SSTR5 in 14 of 22 (63.6%). |
| Microarray and IHC | [88] | SSTR2 expression is significantly upregulated in NF-pNETs compared to insulinomas. |
| IHC | [239] | Mild to strong immunoreactivity for SSTR2A observed in 15 out of 16 (94%) pNETs. |
| [238] | Positive (IHC score 1 to 4) SSTR2A immunostaining in 63% of 79 pNETs. Negative staining correlated with poor outcomes. | |
| [185] | Positive SSTR2A and SSTR5 staining in 68% and 58% of 44 pNETs, respectively. | |
| [240] | Immunoreactivity for SSTR1 in 40%, SSTR2A in 90%, SSTR2B in 39%, SSTR3 in 51% and SSTR5 in 76% of 71 NETS of the GI tract and lungs. | |
| [241] | Positive immunostaining of SSTR1 and SSTR2 in 100% of 11 G1 and G2 NETs of the GI tract and lungs. | |
| [229] | Positive SSTR2A and SSTR5 staining in 86% and 35%, respectively, of 99 pNETs. Positive SSTR2A expression correlated with better overall survival. |
| Technique | Reference | Key Findings |
|---|---|---|
| IHC | [107] | In total, 17 out of 21 patients (81%) pNETs showed strong immunoreactivity for Myc. |
| IHC | [260] | Positive Myc expression in all 39 benign or metastatic pNETs. |
| Microarray | [270] | Upregulation of LCK, a Src family kinase, in primary (n = 8) and metastatic pNETs (n = 5), and pNET cell lines compared to normal islets. Microarray results were validated by qRT-PCR and IHC. |
| qPCR and IHC | [130] | RABL6A amplification in 6 of 11 primary pNETs and their matched metastases. High RABL6A protein expression observed by IHC in all pNETs (n = 5). |
| IHC | [271] | Significant upregulation of all HDACs (I, IIa, IIIb, III and IV) in pNETs whose expression was found correlate with tumor grading and predict disease outcomes. |
| RNA seq | [261] | Transcriptome analysis of 212 patient GEP-NETs complemented by systematic drug perturbation assays identified HDAC class I inhibitor, entinostat, as a potent agent to treat 42% GEP-NET patients. |
| RNA Seq | [106] | IPA and cMAP analysis of differentially expressed genes in 43 primary pNETs vs. their matched metastases predicted HDAC as one of the top pharmacological targets to treat metastatic pNETs. |
| IHC | [185] | Positive immunoreactivity for HSP90 and TGF-βRI in 75% of 67 primary and metastatic pNETs. |
| IHC | [262] | Positive aurora kinase A expression in 8 of 10 insulinomas, in all of 13 nonfunctional pNETs and 20 SBNETs. |
| IHC | [272] | Ptch1, the sonic hedgehog receptor, expressed in 12 of 22 sporadic pNETs and 4 of 5 MEN-1 pNETs with no significant correlation with clinical outcomes. |
| IHC | [273] | Nuclear β-catenin immunostaining in higher percentage of stage III/IV pNETs (2/13, 15%) vs. stage I/II pNETs (0/74). Negative APC expression in 70% (57/81) of the cases. |
| IHC | [274] | Positive expression of TGF-β, TGF-βRI, and TGF-βRII in 75–100% patient pNETs. |
| LOH and sequencing | [275] | LOH of Smad3 in 20% of 20 pNETs; no inactivating Smad3 mutations observed. |
| PCR and SSCP mutational analysis | [276] | DPC4 mutation or deletion detected in 55% (5 of 9) non-functional pNETs in contrast to none of the 16 functional tumors-insulinomas, gastrinomas, and VIPnomas. |
| Cell Type | Source | Key Features |
|---|---|---|
| QGP-1 | Metastatic human islet cell carcinoma | Production of carcinoembryonic antigen and absence of hormonal secretion recapitulating the parent tumor [338]. |
| BON-1 | Peripancreatic lymph node metastasis of pancreatic carcinoid tumor | Express gastrin and somatostatin receptors; synthesize serotonin and chromogranin [339]. |
| NT-3 | Lymph node metastasis of an insulinoma | Model of well-differentiated insulinoma, highly expresses somatostatin receptors and neuroendocrine markers, exhibit slow growth in contrast to BON-1 and QGP-1 cells. Gene sequencing revealed a homozygous missense mutation in MEN1 gene with other pNET-associated genes intact [340]. |
| Type | Source Cell/Tissue | Key Features |
|---|---|---|
| pNET spheroids | pNET cell lines: BON-1 and QGP-1 Human lung neuroendocrine cell line: H727 | Three-dimensional spheroids start forming by day 3 of cell plating and contain highly proliferative cells at the periphery and a necrotic center, proposed to be useful models for in vitro drug screening [344]. |
| SBNET spheroids | Patient SBNETs | Cultured with the help of extracellular matrix (Matrigel); doubling time was 14 days; expressed synaptophysin, chromogranin and SSTR2; undergo apoptosis with rapamycin treatment [345]. |
| pNET spheroids | Patient pNETs | Primary tumor cells isolated from pNETs and cultured in vitro to form islet-like tumoroids that retain a neuroendocrine phenotype and are viable for at least 2 weeks in culture for drug testing [346]. |
| Reference | Source Tissue | Key Features |
|---|---|---|
| [203] | Primary tumor fragments from patients’ lymph node metastases used for s.c. transplantion into NOD scid mouse (NSG) | Successful engraftment required MET proto-oncogene activation by HGF or its analogue. Critical comments: validity, utility unclear, no studies utilizing the model. |
| [102] | pNET liver metastases | pNETs were well-differentiated, pNET gene signatures were retained, tumor growth inhibition observed with everolimus and sapanisertib. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maharjan, C.K.; Ear, P.H.; Tran, C.G.; Howe, J.R.; Chandrasekharan, C.; Quelle, D.E. Pancreatic Neuroendocrine Tumors: Molecular Mechanisms and Therapeutic Targets. Cancers 2021, 13, 5117. https://doi.org/10.3390/cancers13205117
Maharjan CK, Ear PH, Tran CG, Howe JR, Chandrasekharan C, Quelle DE. Pancreatic Neuroendocrine Tumors: Molecular Mechanisms and Therapeutic Targets. Cancers. 2021; 13(20):5117. https://doi.org/10.3390/cancers13205117
Chicago/Turabian StyleMaharjan, Chandra K., Po Hien Ear, Catherine G. Tran, James R. Howe, Chandrikha Chandrasekharan, and Dawn E. Quelle. 2021. "Pancreatic Neuroendocrine Tumors: Molecular Mechanisms and Therapeutic Targets" Cancers 13, no. 20: 5117. https://doi.org/10.3390/cancers13205117
APA StyleMaharjan, C. K., Ear, P. H., Tran, C. G., Howe, J. R., Chandrasekharan, C., & Quelle, D. E. (2021). Pancreatic Neuroendocrine Tumors: Molecular Mechanisms and Therapeutic Targets. Cancers, 13(20), 5117. https://doi.org/10.3390/cancers13205117