
The Current Landscape of NKT Cell Immunotherapy and the Hills Ahead


Abstract
:Simple Summary
Abstract
1. Introduction
2. NKT Cells
3. iNKT Cells in Cancer
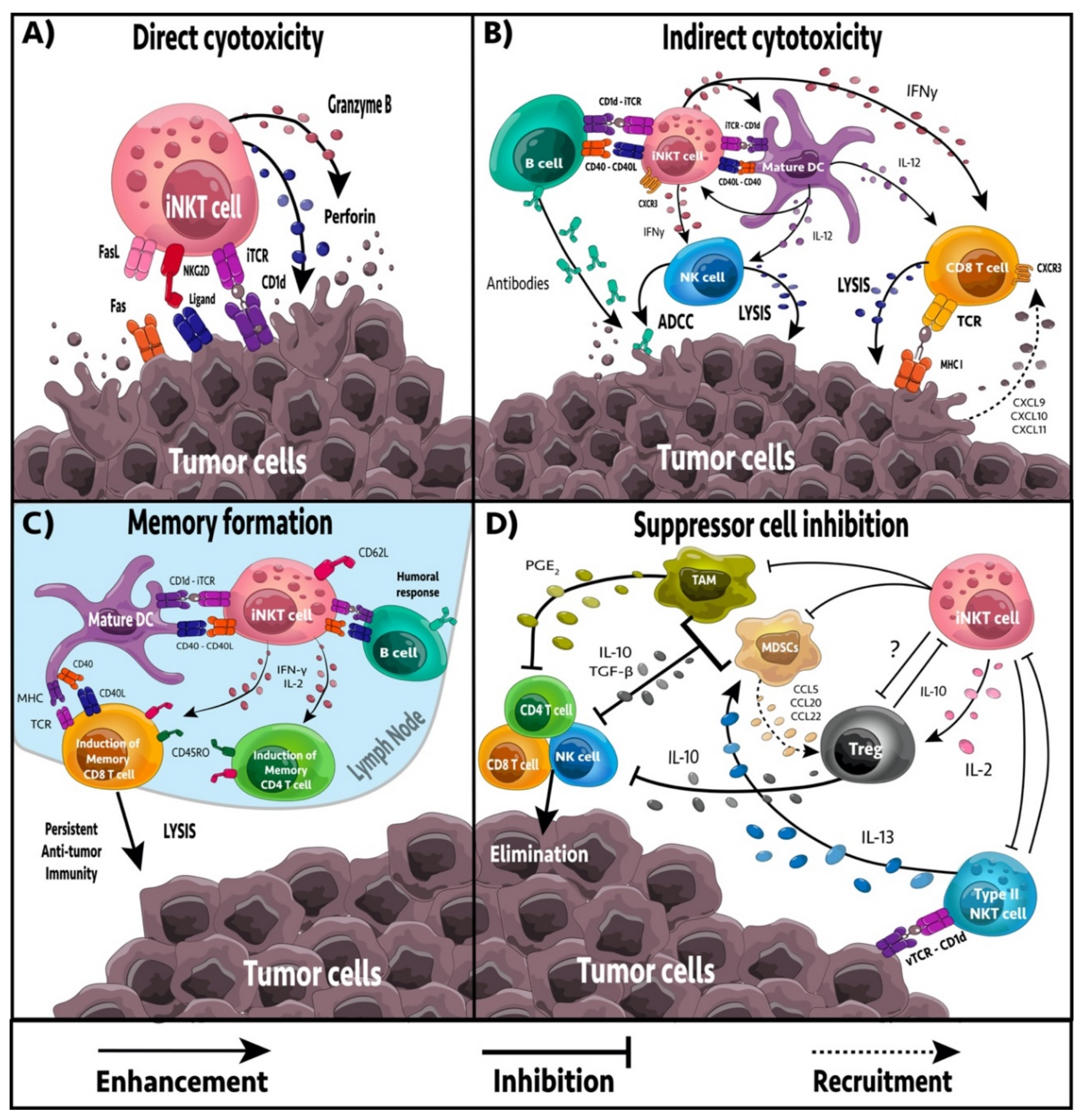
4. iNKT Cell-Mediated Anti-Tumor Effector Responses
5. Regulation of Immune Suppression
6. Formation of Tumor Immune Memory
7. iNKT Cell Immunotherapy
7.1. Free α-GalCer Administration
7.2. Adoptive Transfer of DCs Presenting α-GalCer
7.3. Adoptive Transfer of Activated iNKT Cells
7.4. iNKT Cell Ligands as Cancer Vaccine Adjuvants
7.5. CD1d-Antibody Fusion Proteins
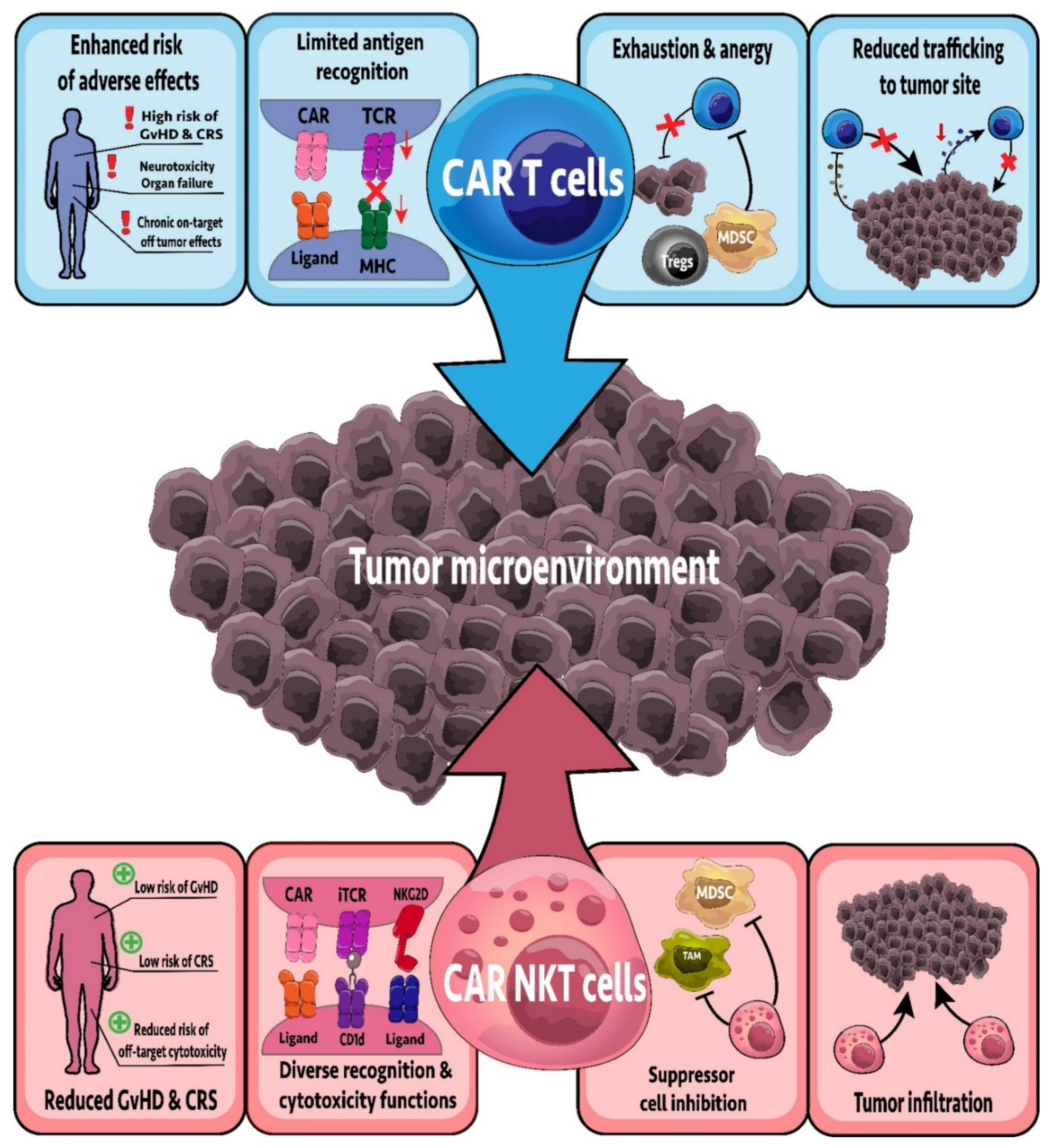
7.6. CAR-NKT Cells
7.7. Combination Therapies
7.7.1. Combination with Chemotherapy
7.7.2. Combination with Oncolytic Viruses
7.7.3. Combination with Immunotherapy
8. Next Steps and Challenges
8.1. Alternatative Glycolipid Delivery
8.2. Alternatative Glycolipids
8.3. Induced Pluripotent Stem Cell-Derived iNKT Cells
8.4. Inhibition of Type II NKT Cells
8.5. CAR-NKT Cells: Lessons from CAR-T Cells
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Mohan, G.; Hama, A.; Jijo, A.J.; Saradha Devi, K.M.; Narayanasamy, A.; Vellingiri, B. Recent advances in radiotherapy and its associated side effects in cancer—A review. J. Basic Appl. Zool. 2019, 80, 14. [Google Scholar] [CrossRef]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse effects of cancer chemotherapy: Anything new to improve tolerance and reduce sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Rueff, J.; Rodrigues, A.S. Cancer drug resistance: A brief overview from a genetic viewpoint. Methods Mol. Biol. 2016, 1395, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Borst, P. Cancer drug pan-resistance: Pumps, cancer stem cells, quiescence, epithelial to mesenchymal transition, blocked cell death pathways, persisters or what? Open Biol. 2012, 2, 120066. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Dotti, G.; Yee, C.; Goff, S.L. An update on adoptive T-cell therapy and neoantigen vaccines. Am. Soc. Clin. Oncol. Educ. B. 2019, 39, e70–e78. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T cells for solid tumors: New dtrategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Fujii, S.-I.; Shimizu, K.; Okamoto, Y.; Kunii, N.; Nakayama, T.; Motohashi, S.; Taniguchi, M. NKT cells as an ideal anti-tumor immunotherapeutic. Front. Immunol. 2013, 4, 409. [Google Scholar] [CrossRef] [Green Version]
- Swann, J.B.; Uldrich, A.P.; van Dommelen, S.; Sharkey, J.; Murray, W.K.; Godfrey, D.I.; Smyth, M.J. Type I natural killer T cells suppress tumors caused by p53 loss in mice. Blood 2009, 113, 6382–6385. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Springfield, R.; Chen, S.; Li, X.; Feng, X.; Moshirian, R.; Yang, R.; Yuan, W. α-GalCer and iNKTcell-based cancer immunotherapy: Realizing the therapeutic potentials. Front. Immunol. 2019, 10, 1126. [Google Scholar] [CrossRef] [Green Version]
- Makino, Y.; Kanno, R.; Ito, T.; Higashino, K.; Taniguchi, M. Predominant expression of invariant Vα14+ TCR α chain in NK1.1+ T cell populations. Int. Immunol. 1995, 7, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of NKT cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hix, L.M.; Shi, Y.H.; Brutkiewicz, R.R.; Stein, P.L.; Wang, C.-R.; Zhang, M. CD1d-expressing breast cancer cells modulate NKT cell-mediated antitumor immunity in a murine model of breast cancer metastasis. PLoS ONE 2011, 6, e20702. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Ugolini, S.; Blaise, D.; Chabannon, C.; Brossay, L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol. 2012, 12, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Horiguchi, S.; Kurosaki, M.; Kunii, N.; Nagato, K.; Hanaoka, H.; Shimizu, N.; Ueno, N.; Yamamoto, S.; Taniguchi, M.; et al. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin. Immunol. 2011, 138, 255–265. [Google Scholar] [CrossRef]
- Kawano, T.; Cui, J.; Koezuka, Y.; Toura, I.; Kaneko, Y.; Motoki, K.; Ueno, H.; Nakagawa, R.; Sato, H.; Kondo, E.; et al. CD1d-restricted and TCR-mediated activation of Vα14 NKT cells by glycosylceramides. Science 1997, 278, 1626–1629. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.I.; MacDonald, H.R.; Kronenberg, M.; Smyth, M.J.; Kaer, L. Van NKT cells: What’s in a name? Nat. Rev. Immunol. 2004, 4, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Lantz, O.; Bendelac, A. An invariant T cell receptor α chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4-8- T cells in mice and humans. J. Exp. Med. 1994, 180, 1097–1106. [Google Scholar] [CrossRef] [Green Version]
- Coquet, J.M.; Chakravarti, S.; Kyparissoudis, K.; McNab, F.W.; Pitt, L.A.; McKenzie, B.S.; Berzins, S.P.; Smyth, M.J.; Godfrey, D.I. Diverse cytokine production by NKT cell subsets and identification of an IL-17–producing CD4- NK1.1− NKT cell population. Proc. Natl. Acad. Sci. USA 2008, 105, 11287–11292. [Google Scholar] [CrossRef] [Green Version]
- Sag, D.; Özkan, M.; Kronenberg, M.; Wingender, G. Improved detection of cytokines produced by invariant NKT cells. Sci. Rep. 2017, 7, 16607. [Google Scholar] [CrossRef]
- Bendelac, A.; Killeen, N.; Littman, D.R.; Schwartz, R.H. A subset of CD4+ thymocytes selected by MHC class I molecules. Science 1994, 263, 1774–1778. [Google Scholar] [CrossRef]
- Gadola, S.D.; Dulphy, N.; Salio, M.; Cerundolo, V. Vα24-JαQ-independent, CD1d-restricted recognition of α-galactosylceramide by human CD4+ and CD8αβ+ T lymphocytes. J. Immunol. 2002, 168, 5514–5520. [Google Scholar] [CrossRef] [Green Version]
- Gumperz, J.E.; Miyake, S.; Yamamura, T.; Brenner, M.B. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J. Exp. Med. 2002, 195, 625–636. [Google Scholar] [CrossRef]
- Engel, I.; Hammond, K.; Sullivan, B.A.; He, X.; Taniuchi, I.; Kappes, D.; Kronenberg, M. Co-receptor choice by Vα14i NKT cells is driven by Th-POK expression rather than avoidance of CD8-mediated negative selection. J. Exp. Med. 2010, 207, 1015–1029. [Google Scholar] [CrossRef] [Green Version]
- Macho-Fernandez, E.; Brigl, M. The extended family of CD1d-restricted NKT cells: Sifting through a mixed bag of TCRs, antigens, and functions. Front. Immunol. 2015, 6, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardell, S.; Tangri, S.; Chan, S.; Kronenberg, M.; Benoist, C.; Mathis, D. CD1-restricted CD4+ T cells in major histocompatibility complex class II-deficient mice. J. Exp. Med. 1995, 182, 993–1004. [Google Scholar] [CrossRef]
- Stax, A.M.; Tuengel, J.; Girardi, E.; Kitano, N.; Allan, L.L.; Liu, V.; Zheng, D.; Panenka, W.J.; Guillaume, J.; Wong, C.-H.; et al. Autoreactivity to sulfatide by human invariant NKT cells. J. Immunol. 2017, 199, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maricic, I.; Girardi, E.; Zajonc, D.M.; Kumar, V. Recognition of lysophosphatidylcholine by Type II NKT cells and protection from an inflammatory liver disease. J. Immunol. 2014, 193, 4580–4589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, F.C.; Berzofsky, J.A.; Terabe, M. NKT cell networks in the regulation of tumor immunity. Front. Immunol. 2014, 5, 543. [Google Scholar] [CrossRef] [Green Version]
- Renukaradhya, G.J.; Khan, M.A.; Vieira, M.; Du, W.; Gervay-Hague, J.; Brutkiewicz, R.R. Type I NKT cells protect (and type II NKT cells suppress) the host’s innate antitumor immune response to a B-cell lymphoma. Blood 2008, 111, 5637–5645. [Google Scholar] [CrossRef] [Green Version]
- Crowe, N.Y.; Smyth, M.J.; Godfrey, D.I. A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas. J. Exp. Med. 2002, 196, 119–127. [Google Scholar] [CrossRef]
- Bellone, M.; Ceccon, M.; Grioni, M.; Jachetti, E.; Calcinotto, A.; Napolitano, A.; Freschi, M.; Casorati, G.; Dellabona, P. iNKT cells control mouse spontaneous carcinoma independently of tumor-specific cytotoxic T cells. PLoS ONE 2010, 5, e8646. [Google Scholar] [CrossRef] [Green Version]
- Muhammad Ali Tahir, S.; Cheng, O.; Shaulov, A.; Koezuka, Y.; Bubley, G.J.; Wilson, S.B.; Balk, S.P.; Exley, M.A. Loss of IFN-γ production byinvariant NKT cells in advanced cancer. J. Immunol. 2001, 167, 4046–4050. [Google Scholar] [CrossRef] [Green Version]
- Molling, J.W.; Kölgen, W.; van der Vliet, H.J.J.; Boomsma, M.F.; Kruizenga, H.; Smorenburg, C.H.; Molenkamp, B.G.; Langendijk, J.A.; Leemans, C.R.; von Blomberg, B.M.E.; et al. Peripheral blood IFN-γ-secreting Vα24+Vβ11+ NKT cell numbers are decreased in cancer patients independent of tumor type or tumor load. Int. J. Cancer 2005, 116, 87–93. [Google Scholar] [CrossRef]
- Yoneda, K.; Morii, T.; Nieda, M.; Tsukaguchi, N.; Amano, I.; Tanaka, H.; Yagi, H.; Narita, N.; Kimura, H. The peripheral blood Vα24+NKT cell numbers decrease in patients with haematopoietic malignancy. Leuk. Res. 2005, 29, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, K.; Seino, K.; Ishikawa, Y.; Nozue, M.; Todoroki, T.; Fukao, K. Impaired proliferative response of Vα24 NKT cells from cancer patients against α-galactosylceramide. J. Immunol. 2002, 168, 6494–6499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metelitsa, L.S.; Wu, H.-W.; Wang, H.; Yang, Y.; Warsi, Z.; Asgharzadeh, S.; Groshen, S.; Wilson, S.B.; Seeger, R.C. Natural Killer T Cells Infiltrate neuroblastomas expressing the chemokine CCL2. J. Exp. Med. 2004, 199, 1213–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, T. Increased intratumor Vα24-positive natural killer T cells: A prognostic factor for primary colorectal carcinomas. Clin. Cancer Res. 2005, 11, 7322–7327. [Google Scholar] [CrossRef] [Green Version]
- Lundgren, S.; Warfvinge, C.F.; Elebro, J.; Heby, M.; Nodin, B.; Krzyzanowska, A.; Bjartell, A.; Leandersson, K.; Eberhard, J.; Jirström, K. The prognostic impact of NK/NKT cell density in periampullary adenocarcinoma differs by morphological yype and adjuvant treatment. PLoS ONE 2016, 11, e0156497. [Google Scholar] [CrossRef]
- Tang, R.; Liu, X.; Liang, C.; Hua, J.; Xu, J.; Wang, W.; Meng, Q.; Liu, J.; Zhang, B.; Yu, X.; et al. Deciphering the prognostic implications of the components and signatures in the immune microenvironment of pancreatic ductal adenocarcinoma. Front. Immunol. 2021, 12, 575. [Google Scholar] [CrossRef]
- Gorini, F.; Azzimonti, L.; Delfanti, G.; Scarfò, L.; Scielzo, C.; Bertilaccio, M.T.; Ranghetti, P.; Gulino, A.; Doglioni, C.; Di Napoli, A.; et al. Invariant NKT cells contribute to chronic lymphocytic leukemia surveillance and prognosis. Blood 2017, 129, 3440–3451. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.H.; Osman, K.; Connolly, J.; Kukreja, A.; Krasovsky, J.; Pack, M.; Hutchinson, A.; Geller, M.; Liu, N.; Annable, R.; et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of α-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J. Exp. Med. 2005, 201, 1503–1517. [Google Scholar] [CrossRef]
- Baxevanis, C.N.; Gritzapis, A.D.; Papamichail, M. In vivo antitumor activity of NKT cells activated by the combination of IL-12 and IL-18. J. Immunol. 2003, 171, 2953–2959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite-de-Moraes, M.C.; Hameg, A.; Arnould, A.; Machavoine, F.; Koezuka, Y.; Schneider, E.; Herbelin, A.; Dy, M. A distinct IL-18-induced pathway to fully activate NK T lymphocytes independently from TCR engagement. J. Immunol. 1999, 163, 5871–5876. [Google Scholar] [PubMed]
- Wu, D.Y.; Segal, N.H.; Sidobre, S.; Kronenberg, M.; Chapman, P.B. Cross-presentation of disialoganglioside GD3 to natural killer T cells. J. Exp. Med. 2003, 198, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metelitsa, L.S. Anti-tumor potential of type-I NKT cells against CD1d-positive and CD1d-negative tumors in humans. Clin. Immunol. 2011, 140, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, S.; Kawana, K.; Schust, D.J.; Fujii, T.; Yokoyama, T.; Iwasawa, Y.; Nagamatsu, T.; Adachi, K.; Tomio, A.; Tomio, K.; et al. CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: A possible mechanism for immune evasion by HPV. J. Virol. 2010, 84, 11614–11623. [Google Scholar] [CrossRef] [Green Version]
- Dockry, É.; O’Leary, S.; Gleeson, L.E.; Lyons, J.; Keane, J.; Gray, S.G.; Doherty, D.G. Epigenetic induction of CD1d expression primes lung cancer cells for killing by invariant natural killer T cells. Oncoimmunology 2018, 7, e1428156. [Google Scholar] [CrossRef]
- Fujii, S.-I.; Shimizu, K.; Smith, C.; Bonifaz, L.; Steinman, R.M. Activation of natural killer T cells by α-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J. Exp. Med. 2003, 198, 267–279. [Google Scholar] [CrossRef]
- Gebremeskel, S.; Clattenburg, D.R.; Slauenwhite, D.; Lobert, L.; Johnston, B. Natural killer T cell activation overcomes immunosuppression to enhance clearance of postsurgical breast cancer metastasis in mice. Oncoimmunology 2015, 4, e995562. [Google Scholar] [CrossRef] [Green Version]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; Cretney, E.; Trapani, J.A.; Taniguchi, M.; Kawano, T.; Pelikan, S.B.; Crowe, N.Y.; Godfrey, D.I. Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med. 2000, 191, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Kawano, T.; Nakayama, T.; Kamada, N.; Kaneko, Y.; Harada, M.; Ogura, N.; Akutsu, Y.; Motohashi, S.; Iizasa, T.; Endo, H.; et al. Antitumor cytotoxicity mediated by ligand-activated human Vα24 NKT Cells. Cancer Res. 1999, 59, 5102–5105. [Google Scholar]
- Metelitsa, L.S.; Naidenko, O.V.; Kant, A.; Wu, H.; Loza, M.J.; Perussia, B.; Seeger, R.C. Human NKT cells mediate antitumor cytotoxicity directly by recognizing target cell CD1d with bound ligand or indirectly by producing IL-2 to activate NK cells. J. Immunol. 2001, 167, 3114–3122. [Google Scholar] [CrossRef]
- Kuylenstierna, C.; Björkström, N.K.; Andersson, S.K.; Sahlström, P.; Bosnjak, L.; Paquin-Proulx, D.; Malmberg, K.-J.; Ljunggren, H.-G.; Moll, M.; Sandberg, J.K. NKG2D performs two functions in invariant NKT cells: Direct TCR-independent activation of NK-like cytolysis and co-stimulation of activation by CD1d. Eur. J. Immunol. 2011, 41, 1913–1923. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.H.; Liu, N.; Klimek, V.; Hassoun, H.; Mazumder, A.; Nimer, S.D.; Jagannath, S.; Dhodapkar, M.V. Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: Therapeutic implications. Blood 2006, 108, 618–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Asgharzadeh, S.; Salo, J.; Engell, K.; Wu, H.; Sposto, R.; Ara, T.; Silverman, A.M.; DeClerck, Y.A.; Seeger, R.C.; et al. Vα24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J. Clin. Invest. 2009, 119, 1524–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santo, C.; Salio, M.; Masri, S.H.; Lee, L.Y.H.; Dong, T.; Speak, A.O.; Porubsky, S.; Booth, S.; Veerapen, N.; Besra, G.S.; et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J. Clin. Invest. 2008, 118, 4036–4048. [Google Scholar] [CrossRef] [Green Version]
- Bezbradica, J.S.; Stanic, A.K.; Matsuki, N.; Bour-Jordan, H.; Bluestone, J.A.; Thomas, J.W.; Unutmaz, D.; Van Kaer, L.; Joyce, S. Distinct roles of dendritic cells and B cells in Va14Ja18 natural T cell activation in vivo. J. Immunol. 2005, 174, 4696–4705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, S.; Shimizu, K.; Kronenberg, M.; Steinman, R.M. Prolonged IFN-γ-producing NKT response induced with α-galactosylceramide-loaded DCs. Nat. Immunol. 2002, 3, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Constantinides, M.G.; Thomas, S.Y.; Reboulet, R.; Meng, F.; Koentgen, F.; Teyton, L.; Savage, P.B.; Bendelac, A. Distinct APCs explain the cytokine bias of α-galactosylceramide variants in vivo. J. Immunol. 2012, 188, 3053–3061. [Google Scholar] [CrossRef]
- Cullen, R.; Germanov, E.; Shimaoka, T.; Johnston, B. Enhanced tumor metastasis in response to blockade of the chemokine receptor CXCR6 is overcome by NKT cell activation. J. Immunol. 2009, 183, 5807–5815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, M.H.; Hood, B.L.; Beck, H.C.; Conrads, T.P.; Ditzel, H.J.; Leth-Larsen, R. Downregulation of antigen presentation-associated pathway proteins is linked to poor outcome in triple-negative breast cancer patient tumors. Oncoimmunology 2017, 6, e1305531. [Google Scholar] [CrossRef]
- Keller, C.W.; Freigang, S.; Lünemann, J.D. Reciprocal crosstalk between dendritic cells and natural killer T cells: Mechanisms and therapeutic potential. Front. Immunol. 2017, 8, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harimoto, H.; Shimizu, M.; Nakagawa, Y.; Nakatsuka, K.; Wakabayashi, A.; Sakamoto, C.; Takahashi, H. Inactivation of tumor-specific CD8+ CTLs by tumor-infiltrating tolerogenic dendritic cells. Immunol. Cell Biol. 2013, 91, 545–555. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor cells convert immature myeloid dendritic cells into TGF-β-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med. 2005, 202, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Joyee, A.G.; Uzonna, J.; Yang, X. Invariant NKT Cells Preferentially modulate the function of CD8α+ dendritic cell subset in inducing type 1 immunity against infection. J. Immunol. 2010, 184, 2095–2106. [Google Scholar] [CrossRef] [Green Version]
- Veinotte, L.; Gebremeskel, S.; Johnston, B. CXCL16-positive dendritic cells enhance invariant natural killer T cell-dependent IFNγ production and tumor control. Oncoimmunology 2016, 5, e1160979. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, H.; Iwakabe, K.; Yahata, T.; Nishimura, S.; Ohta, A.; Ohmi, Y.; Sato, M.; Takeda, K.; Okumura, K.; Van Kaer, L.; et al. The natural killer T (NKT) cell ligand α-galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J. Exp. Med. 1999, 189, 1121–1128. [Google Scholar] [CrossRef] [Green Version]
- Yanaba, K.; Bouaziz, J.-D.; Matsushita, T.; Magro, C.M.; St.Clair, E.W.; Tedder, T.F. B-lymphocyte contributions to human autoimmune disease. Immunol. Rev. 2008, 223, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.P.; Haynes, L.; Sayles, P.C.; Duso, D.K.; Eaton, S.M.; Lepak, N.M.; Johnson, L.L.; Swain, S.L.; Lund, F.E. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat. Immunol. 2000, 1, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.H. CD20+ B Cells: The other tumor-infiltrating lymphocytes. J. Immunol. 2010, 185, 4977–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-associated macrophages: Rcent insights and therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Courtney, A.N.; Tian, G.; Marinova, E.; Wei, J.; Guo, L.; Jin, J.; Gao, X.; Ghaghada, K.B.; Heczey, A.; Metelitsa, L.S. NKT cells control tumor associated macrophages and metastatic growth in neuroblastoma. J. Immunol. 2017, 198, 204–224. [Google Scholar]
- Paul, S.; Chhatar, S.; Mishra, A.; Lal, G. Natural killer T cell activation increases iNOS+CD206- M1 macrophage and controls the growth of solid tumor. J. Immunother. Cancer 2019, 7, 208. [Google Scholar] [CrossRef] [Green Version]
- Cortesi, F.; Delfanti, G.; Grilli, A.; Calcinotto, A.; Gorini, F.; Pucci, F.; Lucianò, R.; Grioni, M.; Recchia, A.; Benigni, F.; et al. Bimodal CD40/Fas-dependent crosstalk between iNKT cells and tumor-associated macrophages impairs prostate cancer progression. Cell Rep. 2018, 22, 3006–3020. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Massarotti, M. Myeloid suppressor cells in cancer and autoimmunity. J. Autoimmun. 2017, 85, 117–125. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Joshi, N.; Choi, H.; Ryu, S.; Hahn, M.; Catena, R.; Sadik, H.; Argani, P.; Wagner, P.; Vahdat, L.T.; et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 2012, 72, 1384–1394. [Google Scholar] [CrossRef] [Green Version]
- Ko, H.-J.; Lee, J.-M.; Kim, Y.-J.; Kim, Y.-S.; Lee, K.-A.; Kang, C.-Y. Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of sctivated NKT cells: An alternative cell-based antitumor vaccine. J. Immunol. 2009, 182, 1818–1828. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, E.; Terabe, M.; Halder, R.C.; Peng, J.; Takaku, S.; Miyake, S.; Yamamura, T.; Kumar, V.; Berzofsky, J.A. Cross-regulation between type I and tpe II NKT cells in regulating Tumor immunity: A new immunoregulatory axis. J. Immunol. 2007, 179, 5126–5136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terabe, M.; Berzofsky, J.A. Tissue-specific roles of NKT cells in tumor immunity. Front. Immunol. 2018, 9, 1838. [Google Scholar] [CrossRef]
- Kanamori, M.; Tasumi, Y.; Iyoda, T.; Ushida, M.; Inaba, K. Sulfatide inhibits α-galactosylceramide presentation by dendritic cells. Int. Immunol. 2012, 24, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, V.; Cho, S.; Li, P.; O’Donnell, P.W.; Dunn, C.; Hayakawa, K.; Blum, J.S.; Brutkiewicz, R.R. Inhibition of glycolipid shedding rescues recognition of a CD1+ T cell lymphoma by natural killer T (NKT) cells. Proc. Natl. Acad. Sci. USA 2002, 99, 8197–8202. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-E.; Wu, D.Y.; Prendes, M.; Lu, S.X.; Ragupathi, G.; Schrantz, N.; Chapman, P.B. Fine specificity of natural killer T cells against GD3 ganglioside and identification of GM3 as an inhibitory natural killer T-cell ligand. Immunology 2008, 123, 145–155. [Google Scholar] [CrossRef]
- Webb, T.J.; Li, X.; Giuntoli, R.L.; Lopez, P.H.H.; Heuser, C.; Schnaar, R.L.; Tsuji, M.; Kurts, C.; Oelke, M.; Schneck, J.P. Molecular identification of GD3 as a suppressor of the innate immune response in ovarian cancer. Cancer Res. 2012, 72, 3744–3752. [Google Scholar] [CrossRef] [Green Version]
- Makhlouf, A.M.; Fathalla, M.M.; Zakhary, M.A.; Makarem, M.H. Sulfatides in ovarian tumors: Clinicopathological correlates. Int. J. Gynecol. Cancer 2004, 14, 89–93. [Google Scholar] [CrossRef]
- Takahashi, T.; Suzuki, T. Role of sulfatide in normal and pathological cells and tissues. J. Lipid Res. 2012, 53, 1437–1450. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.A.; Ngamcherdtrakul, W.; Luoh, S.W.; Yantasee, W. Prognostic and therapeutic role of tumor-infiltrating lymphocyte subtypes in breast cancer. Cancer Metastasis Rev. 2021, 40, 519–536. [Google Scholar] [CrossRef]
- Hermans, I.F.; Silk, J.D.; Gileadi, U.; Mathew, B.; Ritter, G.; Schmidt, R.; Adrian, L.; Old, L.; Cerundolo, V. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J. Immunol. 2003, 171, 5140–5147. [Google Scholar] [CrossRef] [Green Version]
- Macho-Fernandez, E.; Cruz, L.J.; Ghinnagow, R.; Fontaine, J.; Bialecki, E.; Frisch, B.; Trottein, F.; Faveeuw, C. Targeted delivery of α-galactosylceramide to CD8+ dendritic cells optimizes type I NKT cell–based antitumor responses. J. Immunol. 2014, 193, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Reilly, E.C.; Thompson, E.A.; Aspeslagh, S.; Wands, J.R.; Elewaut, D.; Brossay, L. Activated iNKT cells promote memory CD8+ T cell differentiation during viral infection. PLoS ONE 2012, 7, e37991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H.; Johnston, B.; Butcher, E.C. Trafficking machinery of NKT cells: Shared and differential chemokine receptor expression among Vα24+Vβ11+ NKT cell subsets with distinct cytokine-producing capacity. Blood 2002, 100, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, B.; Kim, C.H.; Soler, D.; Emoto, M.; Butcher, E.C. Differential chemokine responses and homing patterns of murine TCRαβ NKT vell subsets. J. Immunol. 2003, 171, 2960–2969. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Courtney, A.N.; Jena, B.; Heczey, A.; Liu, D.; Marinova, E.; Guo, L.; Xu, X.; Torikai, H.; Mo, Q.; et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J. Clin. Invest. 2016, 126, 2341–2355. [Google Scholar] [CrossRef]
- Fuji, N.; Ueda, Y.; Fujiwara, H.; Itoh, T.; Yoshimura, T.; Yamagishi, H. Antitumor effect of α-galactosylceramide (KRN7000) on spontaneous hepatic metastases requires endogenous interleukin 12 in the liver. Clin. Cancer Res. 2000, 6, 3380–3387. [Google Scholar]
- Hayakawa, Y.; Takeda, K.; Yagita, H.; Smyth, M.J.; Van Kaer, L.; Okumura, K.; Saiki, I. IFN-γ–mediated inhibition of tumor angiogenesis by natural killer T-cell ligand, α-galactosylceramide. Blood 2002, 100, 1728–1733. [Google Scholar]
- Ito, H.; Ando, T.; Seishima, M. Inhibition of iNOS activity enhances the anti-tumor effects of α-galactosylceramide in established murine cancer model. Oncotarget 2015, 6, 41863–42874. [Google Scholar] [CrossRef] [Green Version]
- Kawano, T.; Cui, J.; Koezuka, Y.; Toura, I.; Kaneko, Y.; Sato, H.; Kondo, E.; Harada, M.; Koseki, H.; Nakayama, T.; et al. Natural killer-like nonspecific tumor cell lysis mediated by specific ligand-activated Vα14 NKT cells. Proc. Natl. Acad. Sci. USA 1998, 95, 5690–5693. [Google Scholar] [CrossRef] [Green Version]
- Mattarollo, S.R.; West, A.C.; Steegh, K.; Duret, H.; Paget, C.; Martin, B.; Matthews, G.M.; Shortt, J.; Chesi, M.; Bergsagel, P.L.; et al. NKT cell adjuvant-based tumor vaccine for treatment of myc oncogene-driven mouse B-cell lymphoma. Blood 2012, 120, 3019–3029. [Google Scholar] [CrossRef] [Green Version]
- Toura, I.; Kawano, T.; Akutsu, Y.; Nakayama, T.; Ochiai, T.; Taniguchi, M. Inhibition of experimental tumor metastasis by dendritic cells pulsed with α-galactosylceramide. J. Immunol. 1999, 163, 2387–2391. [Google Scholar]
- Uchida, T.; Horiguchi, S.; Tanaka, Y.; Yamamoto, H.; Kunii, N.; Motohashi, S.; Taniguchi, M.; Nakayama, T.; Okamoto, Y. Phase I study of α-galactosylceramide-pulsed antigen presenting cells administration to the nasal submucosa in unresectable or recurrent head and neck cancer. Cancer Immunol. Immunother. 2008, 57, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, A.; Motohashi, S.; Ishikawa, E.; Fuchida, H.; Higashino, K.; Otsuji, M.; Iizasa, T.; Nakayama, T.; Taniguchi, M.; Fujisawa, T. A phase I study of α-galactosylceramide (KRN7000)–pulsed dendritic cells in patients with advanced and recurrent non–small cell lung cancer. Clin. Cancer Res. 2005, 11, 1910–1917. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, T.; Kamata, T.; Tanaka, K.; Ihara, F.; Takami, M.; Suzuki, H.; Nakajima, T.; Ikeuchi, T.; Kawasaki, Y.; Hanaoka, H.; et al. Phase II study of α-galactosylceramide-pulsed antigen-presenting cells in patients with advanced or recurrent non-small cell lung cancer. J. Immunother. Cancer 2020, 8, e000316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, T.; Nakayama, T.; Akutsu, Y.; Motohashi, S.; Shibata, Y.; Harada, M.; Kamada, N.; Shimizu, C.; Shimizu, E.; Saito, T.; et al. Inhibition of tumor metastasis by adoptive transfer of IL-12-activated Vα14 NKT cells. Int. J. Cancer 2001, 91, 523–528. [Google Scholar] [CrossRef]
- Bagnara, D.; Ibatici, A.; Corselli, M.; Sessarego, N.; Tenca, C.; De Santanna, A.; Mazzarello, A.; Daga, A.; Corvò, R.; De Rossi, G.; et al. Adoptive immunotherapy mediated by ex vivo expanded natural killer T cells against CD1d-expressing lymphoid neoplasms. Haematologica 2009, 94, 967–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heczey, A.; Liu, D.; Tian, G.; Courtney, A.N.; Wei, J.; Marinova, E.; Gao, X.; Guo, L.; Yvon, E.; Hicks, J.; et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014, 124, 2824–2833. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Huang, W.; Heczey, A.; Liu, D.; Guo, L.; Wood, M.; Jin, J.; Courtney, A.N.; Liu, B.; Di Pierro, E.J.; et al. NKT cells coexpressing a GD2-specific chimeric antigen receptor and IL15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin. Cancer Res. 2019, 25, 7126–7138. [Google Scholar] [CrossRef] [Green Version]
- Heczey, A.; Courtney, A.N.; Montalbano, A.; Robinson, S.; Liu, K.; Li, M.; Ghatwai, N.; Dakhova, O.; Liu, B.; Raveh-Sadka, T.; et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: An interim analysis. Nat. Med. 2020, 26, 1686–1690. [Google Scholar] [CrossRef]
- Rotolo, A.; Caputo, V.S.; Holubova, M.; Baxan, N.; Dubois, O.; Chaudhry, M.S.; Xiao, X.; Goudevenou, K.; Pitcher, D.S.; Petevi, K.; et al. Enhanced anti-lymphoma activity of CAR19-iNKT cells underpinned by dual CD19 and CD1d targeting. Cancer Cell 2018, 34, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Simon, B.; Wiesinger, M.; März, J.; Wistuba-Hamprecht, K.; Weide, B.; Schuler-Thurner, B.; Schuler, G.; Dörrie, J.; Uslu, U. The generation of CAR-transfected natural Killer T nells for the immunotherapy of melanoma. Int. J. Mol. Sci. 2018, 19, 2365. [Google Scholar] [CrossRef] [Green Version]
- Corgnac, S.; Perret, R.; Derré, L.; Zhang, L.; Stirnemann, K.; Zauderer, M.; Speiser, D.E.; Mach, J.-P.; Romero, P.; Donda, A. CD1d-antibody fusion proteins target iNKT cells to the tumor and trigger long-term therapeutic responses. Cancer Immunol. Immunother. 2013, 62, 747–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stirnemann, K.; Romero, J.F.; Baldi, L.; Robert, B.; Cesson, V.; Besra, G.S.; Zauderer, M.; Wurm, F.; Corradin, G.; Mach, J.-P.; et al. Sustained activation and tumor targeting of NKT cells using a CD1d-anti-HER2-scFv fusion protein induce antitumor effects in mice. J. Clin. Invest. 2008, 118, 994–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Guan, P.; Wiener, S.J.; Patel, N.P.; Gohl, T.G.; Evans, E.; Zauderer, M.; Nichols, K.E. Enhancing the antitumor functions of invariant natural killer T cells using a soluble CD1d-CD19 fusion protein. Blood Adv. 2019, 3, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Kunii, N.; Horiguchi, S.; Motohashi, S.; Yamamoto, H.; Ueno, N.; Yamamoto, S.; Sakurai, D.; Taniguchi, M.; Nakayama, T.; Okamoto, Y. Combination therapy of in vitro-expanded natural killer T cells and α-galactosylceramide-pulsed antigen-presenting cells in patients with recurrent head and neck carcinoma. Cancer Sci. 2009, 100, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Parekh, V.V.; Lalani, S.; Kim, S.; Halder, R.; Azuma, M.; Yagita, H.; Kumar, V.; Wu, L.; Van Kaer, L. PD-1/PD-L1 blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J. Immunol. 2009, 182, 2816–2826. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Shimizu, M.; Kogo, H.; Negishi, Y.; Tamura, H.; Morita, R.; Takahashi, H. A combination of check-point blockade and α-galactosylceramide elicits long-lasting suppressive effects on murine hepatoma cell growth in vivo. Immunobiology 2020, 225, 151860. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bhave, M.S.; Yagita, H.; Cardell, S.L. Natural killer T-cell agonist α-galactosylceramide and PD-1 blockade synergize to reduce tumor development in a preclinical model of colon cancer. Front. Immunol. 2020, 11, 581301. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Sharkey, J.; McLaughlin, N.M.; Exley, M.A.; Smyth, M.J. CD1d-based combination therapy eradicates established tumors in mice. J. Immunol. 2009, 183, 1911–1920. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.W.L.; Westwood, J.A.; Darcy, P.K.; Sharkey, J.; Tsuji, M.; Franck, R.W.; Porcelli, S.A.; Besra, G.S.; Takeda, K.; Yagita, H.; et al. Combined natural killer T-cell–based immunotherapy eradicates established tumors in mice. Cancer Res. 2007, 67, 7495–7504. [Google Scholar] [CrossRef] [Green Version]
- Nakui, M.; Ohta, A.; Sekimoto, M.; Sato, M.; Iwakabe, K.; Yahata, T.; Kitamura, H.; Koda, T.; Kawano, T.; Makuuchi, H.; et al. Potentiation of antitumor effect of NKT cell ligand, α-galactosylceramide by combination with IL-12 on lung metastasis of malignant melanoma cells. Clin. Exp. Metastasis 2000, 18, 147–153. [Google Scholar] [CrossRef]
- Smyth, M.J.; Wallace, M.E.; Nutt, S.L.; Yagita, H.; Godfrey, D.I.; Hayakawa, Y. Sequential activation of NKT cells and NK cells provides effective innate immunotherapy of cancer. J. Exp. Med. 2005, 201, 1973–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara, M.L.; Jilesen, Z.; Stojdl, D.; Persano, S. Codelivery of mRNA with α-galactosylceramide using a new lipopolyplex formulation induces a strong antitumor response upon intravenous administration. ACS Omega 2019, 4, 13015–13026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzatti, M.B.; Sousa, M.E.P.; Tunissi, A.S.; Mortara, R.A.; de Oliveira, A.M.; Pereira Cerize, N.N.; de Keller, A.C. Nano spray dryer for vectorizing α-galactosylceramide in polymeric nanoparticles: A single step process to enhance invariant Natural Killer T lymphocyte responses. Int. J. Pharm. 2019, 565, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, U.; Hiltbrunner, S.; Georgoudaki, A.-M.; Karlsson, M.C.; Näslund, T.I.; Gabrielsson, S. Synergistic induction of adaptive antitumor immunity by codelivery of antigen with α-galactosylceramide on exosomes. Cancer Res. 2013, 73, 3865–3876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sainz, V.; Moura, L.I.F.; Peres, C.; Matos, A.I.; Viana, A.S.; Wagner, A.M.; Vela Ramirez, J.E.; Barata, T.; Gaspar, M.; Brocchini, S.; et al. α-Galactosylceramide and peptide-based nano-vaccine synergistically induced a strong tumor suppressive effect in melanoma. Acta Biomater. 2018, 76, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, J.; Stolk, D.A.; Nijen Twilhaar, M.K.; Ambrosini, M.; Storm, G.; van der Vliet, H.J.; de Gruijl, T.D.; van Kooyk, Y.; den Haan, J.M.M. Liposomal nanovaccine containing α-Galactosylceramide and ganglioside GM3 stimulates robust CD8+ T cell responses via CD169+ macrophages and cDC1. Vaccines 2021, 9, 56. [Google Scholar] [CrossRef]
- East, J.E.; Sun, W.; Webb, T.J. Artificial antigen presenting cell (aAPC) mediated activation and expansion of natural killer T cells. J. Vis. Exp. 2012, 70, 4333. [Google Scholar] [CrossRef]
- Thapa, P.; Zhang, G.; Xia, C.; Gelbard, A.; Overwijk, W.W.; Liu, C.; Hwu, P.; Chang, D.Z.; Courtney, A.; Sastry, J.K.; et al. Nanoparticle formulated α-galactosylceramide activates NKT cells without inducing anergy. Vaccine 2009, 27, 3484–3488. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Yun, Z.; Tagawa, T.; De la Maza, L.; Wu, M.O.; Yu, J.; Zhao, Y.; de Perrot, M. Activation of CD1d-restricted natural killer T cells can inhibit cancer cell proliferation during chemotherapy by promoting the immune responses in murine mesothelioma. Cancer Immunol. Immunother. 2014, 63, 1285–1296. [Google Scholar] [CrossRef]
- Aketa, H.; Tatsumi, T.; Kohga, K.; Tsunematsu, H.; Aono, S.; Shimizu, S.; Kodama, T.; Nawa, T.; Shigekawa, M.; Hikita, H.; et al. The combination therapy of α-galactosylceramide and 5-fluorouracil showed antitumor effect synergistically against liver tumor in mice. Int. J. Cancer 2013, 133, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.; Neparidze, N.; Zhang, L.; Nair, S.; Monesmith, T.; Sundaram, R.; Miesowicz, F.; Dhodapkar, K.M.; Dhodapkar, M.V. Clinical regressions and broad immune activation following combination therapy targeting human NKT cells in myeloma. Blood 2013, 121, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Sampath, P.; Rojas, J.J.; Thorne, S.H. Oncolytic virus-mediated targeting of PGE2 in the tumor alters the immune status and sensitizes established and resistant tumors to immunotherapy. Cancer Cell 2016, 30, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Gebremeskel, S.; Nelson, A.; Walker, B.; Oliphant, T.; Lobert, L.; Mahoney, D.; Johnston, B. Natural killer T cell immunotherapy combined with oncolytic vesicular stomatitis virus or reovirus treatments differentially increases survival in mouse models of ovarian and breast cancer metastasis. J. Immunother. Cancer 2021, 9, e002096. [Google Scholar] [CrossRef]
- Yamada, D.; Iyoda, T.; Vizcardo, R.; Shimizu, K.; Sato, Y.; Endo, T.A.; Kitahara, G.; Okoshi, M.; Kobayashi, M.; Sakurai, M.; et al. Efficient regeneration of human Vα24+ invariant natural killer T cells and their anti-tumor activity in vivo. Stem Cells 2016, 34, 2852–2860. [Google Scholar] [CrossRef] [PubMed]
- Watarai, H.; Fujii, S.; Yamada, D.; Rybouchkin, A.; Sakata, S.; Nagata, Y.; Iida-Kobayashi, M.; Sekine-Kondo, E.; Shimizu, K.; Shozaki, Y.; et al. Murine induced pluripotent stem cells can be derived from and differentiate into natural killer T cells. J. Clin. Invest. 2010, 120, 2610–2618. [Google Scholar] [CrossRef]
- Uchida, T.; Nakashima, H.; Yamagata, A.; Ito, S.; Ishikiriyama, T.; Nakashima, M.; Seki, S.; Kumagai, H.; Oshima, N. Repeated administration of α-galactosylceramide ameliorates experimental lupus nephritis in mice. Sci. Rep. 2018, 8, 8225. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-R.; Tsai, Y.-C.; Chang, Y.-J.; Wu, J.-C.; Hung, J.-T.; Lin, K.-H.; Wong, C.-H.; Yu, A.L. α-Galactosylceramide but not phenyl-glycolipids induced NKT cell anergy and IL-33–mediated myeloid-derived suppressor cell accumulation via upregulation of egr2/3. J. Immunol. 2014, 192, 1972–1981. [Google Scholar] [CrossRef] [Green Version]
- Giaccone, G.; Punt, C.J.A.; Ando, Y.; Ruijter, R.; Nishi, N.; Peters, M.; von Blomberg, B.M.E.; Scheper, R.J.; van der Vliet, H.J.J.; van den Eertwegh, A.J.M.; et al. A phase I study of the natural killer T-cell ligand α-galactosylceramide (KRN7000) in patients with solid tumors. Clin. Cancer Res. 2002, 8, 3702–3709. [Google Scholar] [PubMed]
- Nicol, A.J.; Tazbirkova, A.; Nieda, M. Comparison of clinical and immunological effects of intravenous and intradermal sdministration of α-GalactosylCeramide (KRN7000)-pulsed dendritic cells. Clin. Cancer Res. 2011, 17, 5140–5151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motohashi, S.; Nagato, K.; Kunii, N.; Yamamoto, H.; Yamasaki, K.; Okita, K.; Hanaoka, H.; Shimizu, N.; Suzuki, M.; Yoshino, I.; et al. A hhase I-II study of α-Galactosylceramide-pulsed IL-2/GM-CSF-cultured peripheral blood mononuclear cells in patients with advanced and recurrent non-small cell lung cancer. J. Immunol. 2009, 182, 2492–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, C.; Field, C.S.; Tang, C.-W.; Ferguson, P.M.; Compton, B.; Anderson, R.J.; Painter, G.F.; Weinkove, R.; Hermans, I.; Berridge, M.V. Vaccines adjuvanted with an NKT cell agonist induce effective T-cell responses in models of CNS lymphoma. Immunotherapy 2020, 12, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corgnac, S.; Perret, R.; Zhang, L.; Mach, J.-P.; Romero, P.; Donda, A. iNKT/CD1d-antitumor immunotherapy significantly increases the efficacy of therapeutic CpG/peptide-based cancer vaccine. J. Immunother. Cancer 2014, 2, 39. [Google Scholar] [CrossRef] [PubMed]
- Hunn, M.K.; Hermans, I.F. Exploiting invariant NKT cells to promote T-cell responses to cancer vaccines. Oncoimmunology 2013, 2, e23789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, S.-Y.; Ko, H.-J.; Chang, W.-S.; Park, S.-H.; Kweon, M.-N.; Kang, C.-Y. α-Galactosylceramide can act as a nasal vaccine adjuvant inducing protective immune responses against viral infection and tumor. J. Immunol. 2005, 175, 3309–3317. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Kurosawa, Y.; Taniguchi, M.; Steinman, R.M.; Fujii, S. Cross-presentation of glycolipid from tumor cells loaded with α-galactosylceramide leads to potent and long-lived T cell–mediated immunity via dendritic cells. J. Exp. Med. 2007, 204, 2641–2653. [Google Scholar] [CrossRef]
- Zhang, L.; Donda, A. α-Galactosylceramide/CD1d-antibody fusion proteins redirect invariant natural killer T cell immunity to solid tumors and promote prolonged therapeutic responses. Front. Immunol. 2017, 8, 1417. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, U.; Mahadeo, K.M.; Kebriaei, P.; Shpall, E.J.; Saini, N.Y. Chimeric Antigen receptor T-cells in B-acute lymphoblastic leukemia: State of the art and future directions. Front. Oncol. 2020, 10, 1594. [Google Scholar] [CrossRef]
- Cortés-Selva, D.; Dasgupta, B.; Singh, S.; Grewal, I.S. Innate and innate-like cells:the future of chimeric antigen receptor (CAR) cell therapy. Trends Pharmacol. Sci. 2021, 42, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Du, S.-H.; Li, Z.; Chen, C.; Tan, W.-K.; Chi, Z.; Kwang, T.W.; Xu, X.-H.; Wang, S. Co-expansion of cytokine-induced killer clls and Vγ9Vδ2 T cells for CAR T-cell therapy. PLoS ONE 2016, 11, e0161820. [Google Scholar] [CrossRef]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 expression in solid tumors and role as a Target for Cancer Therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Vantaku, V.; Donepudi, S.R.; Ambati, C.R.; Jin, F.; Putluri, V.; Nguyen, K.; Rajapakshe, K.; Coarfa, C.; Battula, V.L.; Lotan, Y.; et al. Expression of ganglioside GD2, reprogram the lipid metabolism and EMT phenotype in bladder cancer. Oncotarget 2017, 8, 95620–95631. [Google Scholar] [CrossRef] [Green Version]
- Orsi, G.; Barbolini, M.; Ficarra, G.; Tazzioli, G.; Manni, P.; Petrachi, T.; Mastrolia, I.; Orvieto, E.; Spano, C.; Prapa, M.; et al. GD2 expression in breast cancer. Oncotarget 2017, 8, 31592–31600. [Google Scholar] [CrossRef]
- Battula, V.L.; Shi, Y.; Evans, K.W.; Wang, R.-Y.; Spaeth, E.L.; Jacamo, R.O.; Guerra, R.; Sahin, A.A.; Marini, F.C.; Hortobagyi, G.; et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J. Clin. Invest. 2012, 122, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Song, L.; Wei, J.; Courtney, A.N.; Gao, X.; Marinova, E.; Guo, L.; Heczey, A.; Asgharzadeh, S.; Kim, E.; et al. IL-15 protects NKT cells from inhibition by tumor-associated macrophages and enhances antimetastatic activity. J. Clin. Invest. 2012, 122, 2221–2233. [Google Scholar] [CrossRef] [Green Version]
- Ranson, T.; Vosshenrich, C.A.J.; Corcuff, E.; Richard, O.; Laloux, V.; Lehuen, A.; Di Santo, J.P. IL-15 availability conditions homeostasis of peripheral natural killer T cells. Proc. Natl. Acad. Sci. USA 2003, 100, 2663–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Wu, X.; Yan, J.; Yu, H.; Xu, L.; Chi, Z.; Sheng, X.; Si, L.; Cui, C.; Dai, J.; et al. Anti-GD2/4-1BB chimeric antigen receptor T cell therapy for the treatment of Chinese melanoma patients. J. Hematol. Oncol. 2018, 11, 1. [Google Scholar] [CrossRef]
- Kuur Therapeutics. Kuur Therapeutics Announces Interm Clinical Data Suporting NKT Cell Therapy Platform. Available online: https://kuurtx.com/2021/01/kuur-therapeutics-announces-interim-clinical-data-supporting-car-nkt-cell-therapy-platform/ (accessed on 6 June 2021).
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019, 30, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sun, D.; Wang, J.; Han, C.; Qian, Y.; Chen, G.; Li, X.; Zhang, J.; Cui, P.; Du, W.; et al. Immune checkpoint inhibitors combined with chemotherapy for the treatment of advanced pancreatic cancer patients. Cancer Immunol. Immunother. 2020, 69, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-H.S.; Wang, C.-C.; Huang, Y.-C.; Pavlidis, S.; Liu, C.-Y.; Ko, H.-W.; Chung, F.-T.; Lin, T.-Y.; Wang, C.-L.; Guo, Y.-K.; et al. Comparison of a combination of chemotherapy and immune checkpoint inhibitors and immune checkpoint inhibitors alone for the treatment of advanced and metastatic non-small cell lung cancer. Thorac. Cancer 2019, 10, 1158–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addeo, A.; Banna, G.L.; Metro, G.; Di Maio, M. Chemotherapy in combination with immune checkpoint inhibitors for the first-line treatment of patients with advanced non-small cell lung cancer: A systematic review and literature-based meta-analysis. Front. Oncol. 2019, 9, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Su, W.; Lu, T.; Wang, Y.; Dong, Y.; Qin, Y.; Liu, D.; Sun, L.; Jiao, W. First-line immune-checkpoint inhibitors in non-small cell lung cancer: Current landscape and future progress. Front. Pharmacol. 2020, 11, 1591. [Google Scholar] [CrossRef]
- Chan, O.T.M.; Yang, L.X. The immunological effects of taxanes. Cancer Immunol. Immunother. 2000, 49, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Ahlmann, M.; Hempel, G. The effect of cyclophosphamide on the immune system: Implications for clinical cancer therapy. Cancer Chemother. Pharmacol. 2016, 78, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Tani, K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 2014, 21, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Opzoomer, J.W.; Sosnowska, D.; Anstee, J.E.; Spicer, J.F.; Arnold, J.N. Cytotoxic chemotherapy as an immune stimulus: A molecular perspective on turning up the immunological heat on cancer. Front. Immunol. 2019, 10, 1654. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef]
- Mattarollo, S.R.; Kenna, T.; Nieda, M.; Nicol, A.J. Chemotherapy pretreatment sensitizes solid tumor-derived cell lines to Vα24+ NKT cell-mediated cytotoxicity. Int. J. Cancer 2006, 119, 1630–1637. [Google Scholar] [CrossRef]
- Fallarini, S.; Paoletti, T.; Orsi Battaglini, N.; Lombardi, G. Invariant NKT cells increase drug-induced osteosarcoma cell death. Br. J. Pharmacol. 2012, 167, 1533–1549. [Google Scholar] [CrossRef] [Green Version]
- Gebremeskel, S.; Lobert, L.; Tanner, K.; Walker, B.; Oliphant, T.; Clarke, L.E.; Dellaire, G.; Johnston, B. Natural killer T-cell immunotherapy in combination with chemotherapy-induced immunogenic cell death targets metastatic breast cancer. Cancer Immunol. Res. 2017, 5, 1086–1097. [Google Scholar] [CrossRef] [Green Version]
- Noubade, R.; Majri-Morrison, S.; Tarbell, K.V. Beyond cDC1: Emerging roles of DC crosstalk in cancer immunity. Front. Immunol. 2019, 10, 1014. [Google Scholar] [CrossRef] [Green Version]
- Verhelle, D.; Corral, L.G.; Wong, K.; Mueller, J.H.; Moutouh-de Parseval, L.; Jensen-Pergakes, K.; Schafer, P.H.; Chen, R.; Glezer, E.; Ferguson, G.D.; et al. Lenalidomide and CC-4047 inhibit the proliferation of malignant B cells while expanding normal CD34+ progenitor cells. Cancer Res. 2007, 67, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Teo, S.K. Properties of thalidomide and its analogues: Implications for anticancer therapy. AAPS J. 2005, 7, E14–E19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotla, V.; Goel, S.; Nischal, S.; Heuck, C.; Vivek, K.; Das, B.; Verma, A. Mechanism of action of lenalidomide in hematological malignancies. J. Hematol. Oncol. 2009, 2, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.C.; Neeson, P.; Leeansyah, E.; Tainton, K.; Quach, H.; Prince, H.M.; Godfrey, D.I.; Ritchie, D.; Berzins, S.P. Testing the NKT cell hypothesis in lenalidomide-treated myelodysplastic syndrome patients. Leukemia 2010, 24, 592–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammayappan, A.; Peng, K.-W.; Russell, S.J. Characteristics of oncolytic vesicular stomatitis virus displaying tumor-targeting ligands. J. Virol. 2013, 87, 13543–13555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojdl, D.F.; Lichty, B.D.; TenOever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef]
- Wong, H.H.; Lemoine, R.N.; Wang, Y. Oncolytic viruses for cancer therapy: Overcoming the obstacles. Viruses 2010, 2, 78–106. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and Iimproves anti-PD-1 immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melaiu, O.; Lucarini, V.; Giovannoni, R.; Fruci, D.; Gemignani, F. News on immune checkpoint inhibitors as immunotherapy strategies in adult and pediatric solid tumors. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Lounici, Y.; Wajda, P.; Barrin, S.; Caux, C.; Dubois, B.; Ray-Coquard, I. Neoadjuvant immune checkpoint inhibitors in cancer, current state of the art. Crit. Rev. Oncol. Hematol. 2021, 157, 103172. [Google Scholar] [CrossRef]
- Bally, A.P.R.; Lu, P.; Tang, Y.; Austin, J.W.; Scharer, C.D.; Ahmed, R.; Boss, J.M. NF-κB regulates PD-1 expression in macrophages. J. Immunol. 2015, 194, 4545–4554. [Google Scholar] [CrossRef] [Green Version]
- Hegel, J.K.; Knieke, K.; Kolar, P.; Reiner, S.L.; Brunner-Weinzierl, M.C. CD152 (CTLA-4) regulates effector functions of CD8+ T lymphocytes by repressing Eomesodermin. Eur. J. Immunol. 2009, 39, 883–893. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Bruno, T.C.; Meeker, A.K.; De Marzo, A.M.; Isaacs, W.B.; Drake, C.G. Human prostate-infiltrating CD8+ T lymphocytes are oligoclonal and PD-1+. Prostate 2009, 69, 1694–1703. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Harrison, D.L.; Song, Y.; Ji, J.; Huang, J.; Hui, E. Antigen-presenting cell-intrinsic PD-1 neutralizes PD-L1 in cis to sttenuate PD-1 dignaling in T cells. Cell Rep. 2018, 24, 379–390.e6. [Google Scholar] [CrossRef] [Green Version]
- Kamata, T.; Suzuki, A.; Mise, N.; Ihara, F.; Takami, M.; Makita, Y.; Horinaka, A.; Harada, K.; Kunii, N.; Yoshida, S.; et al. Blockade of programmed death-1/programmed death ligand pathway enhances the antitumor immunity of human invariant natural killer T cells. Cancer Immunol. Immunother. 2016, 65, 1477–1489. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.-A.; Seo, H.; Kim, B.-S.; Choi, J.; Jeon, I.; Shin, K.-S.; Koh, C.-H.; Song, B.; Kim, I.-K.; Min, B.S.; et al. Activation of NKT cells in an anti-PD-1–resistant tumor model enhances antitumor immunity by reinvigorating exhausted CD8 T cells. Cancer Res. 2018, 78, 5315–5326. [Google Scholar] [CrossRef] [Green Version]
- Hakanen, H.H.E.; Hernberg, M.; Mäkelä, S.; Yadav, B.; Brück, O.; Juteau, S.; Kohtamäki, L.; Ilander, M.; Mustjoki, S.; Anna, K. Metastatic melanoma patients responding to PD1 therapy have higher proportion of peripheral blood NKT-cells. Cancer Immunol. Res. 2019, 7, A130. [Google Scholar] [CrossRef]
- Pilones, K.A.; Kawashima, N.; Yang, A.M.; Babb, J.S.; Formenti, S.C.; Demaria, S. Invariant natural killer T cells regulate breast cancer response to radiation and CTLA-4 blockade. Clin. Cancer Res. 2009, 15, 597–606. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; Dutcher, J.P.; Anderson, J.E.; Eckhardt, S.G.; Stephans, K.F.; Razvillas, B.; Garl, S.; Butine, M.D.; Perry, V.P.; Armitage, R.J.; et al. Phase I study of recombinant human CD40 ligand in cancer patients. J. Clin. Oncol. 2001, 19, 3280–3287. [Google Scholar] [CrossRef]
- Arsene, D.; Galais, M.-P.; Bouhier-Leporrier, K.; Reimund, J.-M. Recent developments in colorectal cancer treatment by monoclonal antibodies. Expert Opin. Biol. Ther. 2006, 6, 1175–1192. [Google Scholar] [CrossRef]
- Melero, I.; Shuford, W.W.; Newby, S.A.; Aruffo, A.; Ledbetter, J.A.; Hellström, K.E.; Mittler, R.S.; Chen, L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med. 1997, 3, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-K.; Wang, W. Characteristics and clinical trial results of agonistic anti-CD40 antibodies in the treatment of malignancies. Oncol. Lett. 2020, 20, 176. [Google Scholar] [CrossRef] [PubMed]
- Attarwala, H. TGN1412: From discovery to disaster. J. Young Pharm. 2010, 2, 332–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parekh, V.V.; Wilson, M.T.; Olivares-Villagómez, D.; Singh, A.K.; Wu, L.; Wang, C.-R.; Joyce, S.; Van Kaer, L. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J. Clin. Invest. 2005, 115, 2572–2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macho Fernandez, E.; Chang, J.; Fontaine, J.; Bialecki, E.; Rodriguez, F.; Werkmeister, E.; Krieger, V.; Ehret, C.; Heurtault, B.; Fournel, S.; et al. Activation of invariant natural killer T lymphocytes in response to the α-galactosylceramide analogue KRN7000 encapsulated in PLGA-based nanoparticles and microparticles. Int. J. Pharm. 2012, 423, 45–54. [Google Scholar] [CrossRef]
- Fujii, S.; Shimizu, K.; Hemmi, H.; Fukui, M.; Bonito, A.J.; Chen, G.; Franck, R.W.; Tsuji, M.; Steinman, R.M. Glycolipid α-C-galactosylceramide is a distinct inducer of dendritic cell function during innate and adaptive immune responses of mice. Proc. Natl. Acad. Sci. USA 2006, 103, 11252–11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Song, L.; Metelitsa, L.S.; Bittman, R. Synthesis and evaluation of an α-C-Galactosylceramide analogue that induces Th1-biased responses in human natural killer T cells. ChemBioChem 2006, 7, 1750–1756. [Google Scholar] [CrossRef] [PubMed]
- Patel, O.; Cameron, G.; Pellicci, D.G.; Liu, Z.; Byun, H.-S.; Beddoe, T.; McCluskey, J.; Franck, R.W.; Castaño, A.R.; Harrak, Y.; et al. NKT TCR recognition of CD1d-α-C-galactosylceramide. J. Immunol. 2011, 187, 4705–4713. [Google Scholar] [CrossRef] [Green Version]
- Schmieg, J.; Yang, G.; Franck, R.W.; Tsuji, M. Superior protection against malaria and melanoma metastases by a C-glycoside analogue of the natural killer T cell ligand α-Galactosylceramide. J. Exp. Med. 2003, 198, 1631–1641. [Google Scholar] [CrossRef]
- Guillaume, J.; Seki, T.; Decruy, T.; Venken, K.; Elewaut, D.; Tsuji, M.; Van Calenbergh, S. Synthesis of C6′′-modified α-C-GalCer analogues as mouse and human iNKT cell agonists. Org. Biomol. Chem. 2017, 15, 2217–2225. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chen, G.; Garcia-Navarro, R.; Franck, R.W.; Tsuji, M. Identification of C-glycoside analogues that display a potent biological activity against murine and human invariant natural killer T cells. Immunology 2009, 127, 216–225. [Google Scholar] [CrossRef]
- Li, X.; Fujio, M.; Imamura, M.; Wu, D.; Vasan, S.; Wong, C.-H.; Ho, D.D.; Tsuji, M. Design of a potent CD1d-binding NKT cell ligand as a vaccine adjuvant. Proc. Natl. Acad. Sci. USA 2010, 107, 13010–13015. [Google Scholar] [CrossRef] [Green Version]
- Padte, N.N.; Boente-Carrera, M.; Andrews, C.D.; McManus, J.; Grasperge, B.F.; Gettie, A.; Coelho-dos-Reis, J.G.; Li, X.; Wu, D.; Bruder, J.T.; et al. A glycolipid adjuvant, 7DW8-5, enhances CD8+ T cell responses Induced by an adenovirus-vectored malaria vaccine in non-human primates. PLoS ONE 2013, 8, e78407. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Nakajima, N.; Wu, L.; Yamashita, M.; Lopes, T.J.S.; Tsuji, M.; Hasegawa, H.; Watanabe, T.; Kawaoka, Y. A gycolipid adjuvant, 7DW8-5, enhances the protective immune response to the current split influenza vaccine in mice. Front. Microbiol. 2019, 10, 2157. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.S.; Chan, A.C.; Kyparissoudis, K.; De Rose, R.; Godfrey, D.I.; Kent, S.J. Peripheral NKT cells in simian immunodeficiency virus-infected macaques. J. Virol. 2009, 83, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Gansuvd, B.; Goodwin, J.; Asiedu, C.K.; Jiang, X.L.; Jargal, U.; Andrades, P.; Exley, M.A.; Thomas, J.M. Invariant natural killer T cells from rhesus macaque spleen and peripheral blood are phenotypically and functionally distinct populations. J. Med. Primatol. 2008, 37, 1–11. [Google Scholar] [CrossRef]
- Tefit, J.N.; Crabé, S.; Orlandini, B.; Nell, H.; Bendelac, A.; Deng, S.; Savage, P.B.; Teyton, L.; Serra, V. Efficacy of ABX196, a new NKT agonist, in prophylactic human vaccination. Vaccine 2014, 32, 6138–6145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabe, S.; Scherrer, D.; Ehrlich, H.; Pouletty, P. Combinations Including ABX196 for the Treatment of Cancer. U.S. Patent 20190328747 2019. [Google Scholar]
- Dashtsoodol, N.; Shigeura, T.; Tashiro, T.; Aihara, M.; Chikanishi, T.; Okada, H.; Hanada, K.; Sano, H.; Kurogi, A.; Taniguchi, M. Natural killer T cell-targeted immunotherapy mediating long-term memory responses and strong antitumor activity. Front. Immunol. 2017, 8, 1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Abbar, A.; Ngai, S.C.; Nograles, N.; Alhaji, S.Y.; Abdullah, S. Induced pluripotent stem cells: Reprogramming platforms and applications in cell replacement therapy. Biores. Open Access 2020, 9, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Pasquet, L.; Camara, K.; Bloom, A.C.; Richardson, S.K.; Howell, A.R.; Terabe, M.; Berzofsky, J.A. The ceramide structure of sulfatide-analogues influences the functional activity of type II NKT cells. J. Immunol. 2018, 200, 20–57. [Google Scholar]
- Zhao, J.; Weng, X.; Bagchi, S.; Wang, C.-R. Polyclonal type II natural killer T cells require PLZF and SAP for their development and contribute to CpG-mediated antitumor response. Proc. Natl. Acad. Sci. USA 2014, 111, 2674–2679. [Google Scholar] [CrossRef] [Green Version]
- Marschner, A.; Rothenfusser, S.; Hornung, V.; Prell, D.; Krug, A.; Kerkmann, M.; Wellisch, D.; Poeck, H.; Greinacher, A.; Giese, T.; et al. CpG ODN enhance antigen-specific NKT cell activation via plasmacytoid dendritic cells. Eur. J. Immunol. 2005, 35, 2347–2357. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.G.; Vasilakos, J.P.; Tross, D.; Smirnov, D.; Klinman, D.M. Combination therapy targeting toll like receptors 7, 8 and 9 eliminates large established tumors. J. Immunother. Cancer 2014, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Paget, C.; Bialecki, E.; Fontaine, J.; Vendeville, C.; Mallevaey, T.; Faveeuw, C.; Trottein, F. Role of invariant NK T lymphocytes in immune responses to CpG oligodeoxynucleotides. J. Immunol. 2009, 182, 1846–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, R.R.; Villanueva, A.I.; Read, L.R.; Brisbin, J.T.; Bhaumik, S.K.; LaMarre, J.; Murali-Krishna, K.; Sharif, S. CpG oligonucleotide-mediated co-stimulation of mouse invariant natural killer T cells negatively regulates their activation status. Cell Tissue Res. 2017, 369, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Sfondrini, L.; Besusso, D.; Zoia, M.T.; Rodolfo, M.; Invernizzi, A.M.; Taniguchi, M.; Nakayama, T.; Colombo, M.P.; Ménard, S.; Balsari, A. Absence of the CD1 molecule up-regulates antitumor activity induced by CpG oligodeoxynucleotides in mice. J. Immunol. 2002, 169, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Li, X.; He, Y.; Zhu, W.; Gao, L.; Liu, Y.; Gao, L.; Wen, Q.; Zhong, J.F.; Zhang, C.; et al. Recent advances in CAR-T cell engineering. J. Hematol. Oncol. 2020, 13, 86. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Schultz, L.M.; Davis, K.L.; Baggott, C.; Chaudry, C.; Marcy, A.C.; Mavroukakis, S.; Sahaf, B.; Kong, K.A.; Muffly, L.S.; Kim, S.; et al. Phase 1 study of CD19/CD22 bispecific chimeric antigen receptor (CAR) therapy in children and young adults with B cell acute lymphoblastic leukemia (ALL). Blood 2018, 132, 898. [Google Scholar] [CrossRef]
- Shah, N.N.; Johnson, B.D.; Schneider, D.; Zhu, F.; Szabo, A.; Keever-Taylor, C.A.; Krueger, W.; Worden, A.A.; Kadan, M.J.; Yim, S.; et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: A phase 1 dose escalation and expansion trial. Nat. Med. 2020, 26, 1569–1575. [Google Scholar] [CrossRef]
- Feldmann, A.; Arndt, C.; Bergmann, R.; Loff, S.; Cartellieri, M.; Bachmann, D.; Aliperta, R.; Hetzenecker, M.; Ludwig, F.; Albert, S.; et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget 2017, 8, 31368–31385. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells, and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Caruso, H.G.; Tanaka, R.; Liang, J.; Ling, X.; Sabbagh, A.; Henry, V.K.; Collier, T.L.; Heimberger, A.B. Shortened ex vivo manufacturing time of EGFRvIII-specific chimeric antigen receptor (CAR) T cells reduces immune exhaustion and enhances antiglioma therapeutic function. J. Neurooncol. 2019, 145, 429–439. [Google Scholar] [CrossRef]
- Kebriaei, P.; Huls, H.; Neel, S.L.; Olivares, S.; Orozco, A.F.; Su, S.; Maiti, S.N.; Smith, A.; De Groot, E.; Kantarjian, H.M.; et al. Shortening the time to manufacture CAR+ T cells with sleeping beauty system supports T-cell engraftment and anti-tumor effects in patients with refractory CD19+ tumors. Blood 2017, 130, 2060. [Google Scholar] [CrossRef]
- Morita, D.; Nishio, N.; Saito, S.; Tanaka, M.; Kawashima, N.; Okuno, Y.; Suzuki, S.; Matsuda, K.; Maeda, Y.; Wilson, M.H.; et al. Enhanced expression of anti-CD19 chimeric antigen receptor in piggyBac transposon-rngineered T cells. Mol. Ther.—Methods Clin. Dev. 2018, 8, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Strategy | Therapy Regimens | Preclinical Models or Clinical Trials | Outcome | References |
|---|---|---|---|---|
| Free glycolipids | ||||
| α-GalCer (i.v.) | Multiple cancer models | Activation of iNKT, NK, T cells, increased IFN-γ and IL-12 | [97,98] | |
| α-GalCer (i.p.) | Multiple cancer models | NK and iNKT mediated tumor cell elimination, elevated IFN-γ | [99,100] | |
| Adoptive transfers | ||||
| Tumor cells irradiated and loaded with α-GalCer | A20 lymphoma, Meth A sarcoma, VK*Myc model, Eµ-myc model | iNKT cells and effector T cells promote anti-tumor immunity, elevated IFN-γ and IL-12 | [101] | |
| α-GalCer loaded dendritic cells | Multiple cancer models | Activation of iNKT cells | [51,52,102] | |
| α-GalCer loaded -APCs | Myeloma, NSCLC, and Head and neck cancer | Increased iNKT expansion and IFNγ production leading to stable disease | [103,104,105] | |
| Ex-vivo expanded iNKT cells | Multiple cancer models | Increased iNKT cytotoxicity, tumor regression and overall survival. Was model dependent | [17,52,106,107] | |
| CAR-NKT cells | ||||
| GD2 CAR NKT | Melanoma and neuroblastoma models | Cytotoxicity against GD2 positive tumors, increased Th1 cytokines and localization to tumor site | [108,109,110] | |
| CD62L+ CD19 CAR NKT cells | B cell lymphoma model | Prolonged persistence in vivo | [96,111] | |
| CSPG4 CAR NKT cells | Melanoma | Increased iNKT pro-inflammatory cytokine production | [112] | |
| CD1d-antibody fusion proteins | ||||
| Anti-HER2 | Melanoma and Colon carcinoma | Increased iNKT pro-inflammatory cytokine production and cytotoxicity. Increase DC, NK, and CD8 T cells recruitment Increased tumor regression. Limited off-target effects. | [113,114] | |
| Anti-CEA | Colon carcinoma | Increased iNKT pro-inflammatory cytokine production and cytotoxicity. Increased tumor regression. | [113] | |
| Anti-CD19 | Melanoma and Colon carcinoma | Increased iNKT pro-inflammatory cytokine production and cytotoxicity. Increased tumor regression. | [115] | |
| Combination therapies | ||||
| α-GalCer-loaded DCs + expanded iNKT cells | Head and neck cancer | Increased circulating iNKT cell number and IFNγ production. | [17,116] | |
| α-GalCer -loaded DCs + anti-PD-1 or anti-PD-L1 mAbs | Melanoma metastasis, Colon cancer, Hepatoma model | Prevented iNKT cell anergy and enhanced anti-tumor function overcomes CD8 T cell exhaustion in PD-1 resistant tumors | [117,118,119] | |
| α-GalCer + anti-4-1BB, anti-CD40, or anti-DR5 | Renal and breast cancer models | Stimulate robust anti-tumor immunity | [120,121] | |
| α-GalCer + IL-12 or IL-21 | Melanoma and breast cancer models | Increased tumor regression and overall survival, increased iNKT cell cytotoxicity | [122,123] | |
| Vector bound α-GalCer | B16 melanoma models | Increased iNKT cell expansion and cytokine release, prevented NKT cell anergy | [124,125,126,127,128,129,130] | |
| α-GalCer + Cisplatin | Mesothelioma Model | Increased pro-inflammatory cytokine gene expression and tumor regression | [131] | |
| α-GalCer + 5-FU | Colon cancer model | Increased NK cell cytotoxicity and coreceptor expression | [132] | |
| α-GalCer loaded APCs and lenalidomide | Multiple myeloma patients | Decreased cancer cell proliferation, angiogenesis. Increased T cell, NK cell and iNKT cell activation and expansion. Elevated iNKT cell cytokine production. Well tolerated in patients. | [133,134] | |
| α-GalCer loaded APCs and anti-DEC205 nanoparticles | B16-F10 melanoma model | iNKT cell-mediated activation of NK cells, DCs, and γδ T cells | [92] | |
| α-GalCer-loaded APCs and oncolytic VSV | 4T1 breast cancer and ID8 ovarian cancer models | Induced immunogenic cell death and lead to increased overall survival | [134,135] | |
| α-GalCer + iPSC-iNKT cells | EL4 T cell lymphoma | Enhanced anti-tumor activity and tumor regression | [136,137] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelson, A.; Lukacs, J.D.; Johnston, B. The Current Landscape of NKT Cell Immunotherapy and the Hills Ahead. Cancers 2021, 13, 5174. https://doi.org/10.3390/cancers13205174
Nelson A, Lukacs JD, Johnston B. The Current Landscape of NKT Cell Immunotherapy and the Hills Ahead. Cancers. 2021; 13(20):5174. https://doi.org/10.3390/cancers13205174
Chicago/Turabian StyleNelson, Adam, Jordan D. Lukacs, and Brent Johnston. 2021. "The Current Landscape of NKT Cell Immunotherapy and the Hills Ahead" Cancers 13, no. 20: 5174. https://doi.org/10.3390/cancers13205174
APA StyleNelson, A., Lukacs, J. D., & Johnston, B. (2021). The Current Landscape of NKT Cell Immunotherapy and the Hills Ahead. Cancers, 13(20), 5174. https://doi.org/10.3390/cancers13205174