
Role of Pancreatic Stellate Cell-Derived Exosomes in Pancreatic Cancer-Related Diabetes: A Novel Hypothesis


 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
- (i)
- (ii)
- Paradoxically experience marked weight loss, which starts even before the development of diabetes [14]; and
- (iii)
2. What Is Known about Pancreatic Cancer-Related Diabetes?
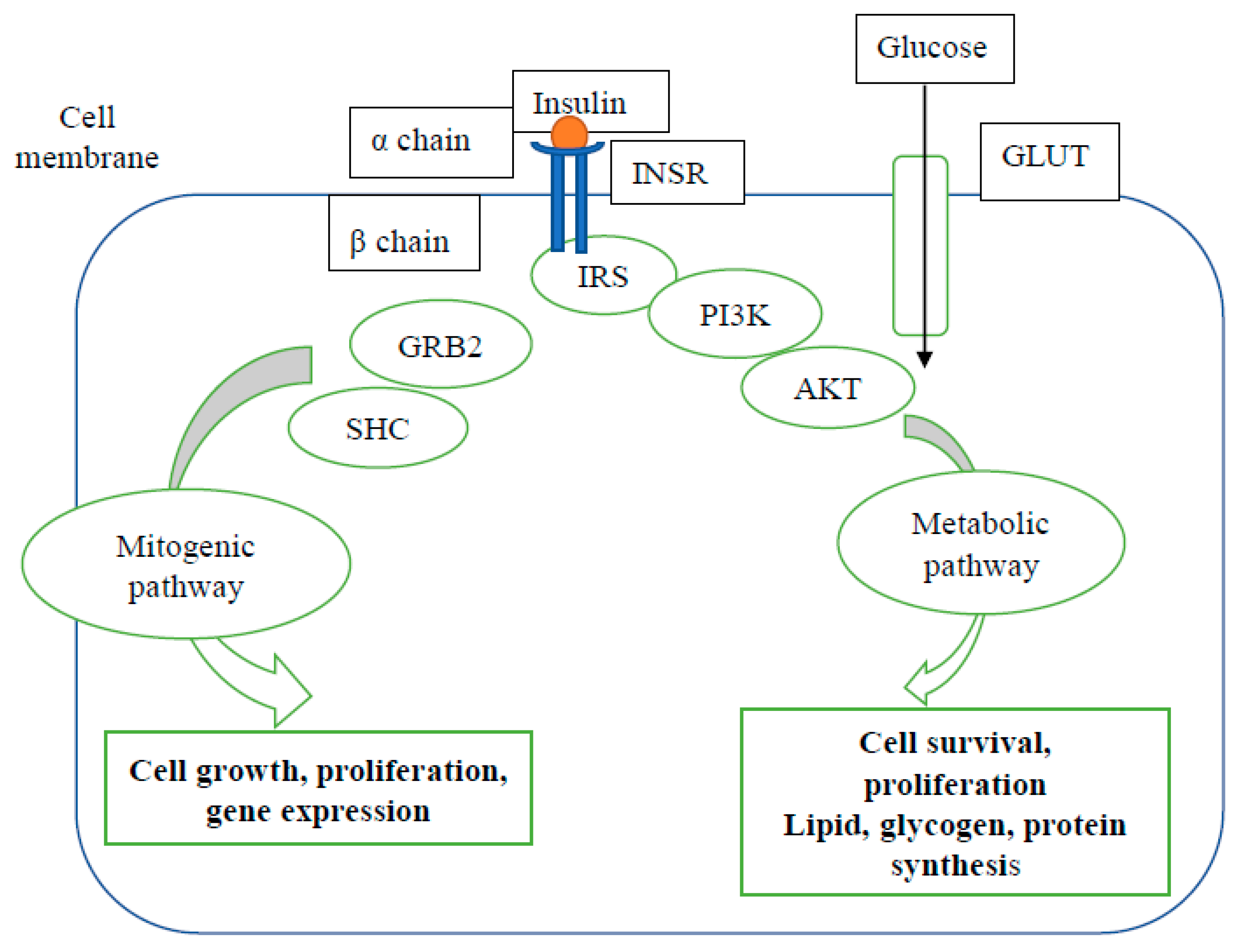
3. Mechanisms of Type 2 Diabetes Mellitus
4. Possible Mechanisms of Pancreatic Cancer-Related Diabetes (PCRD)
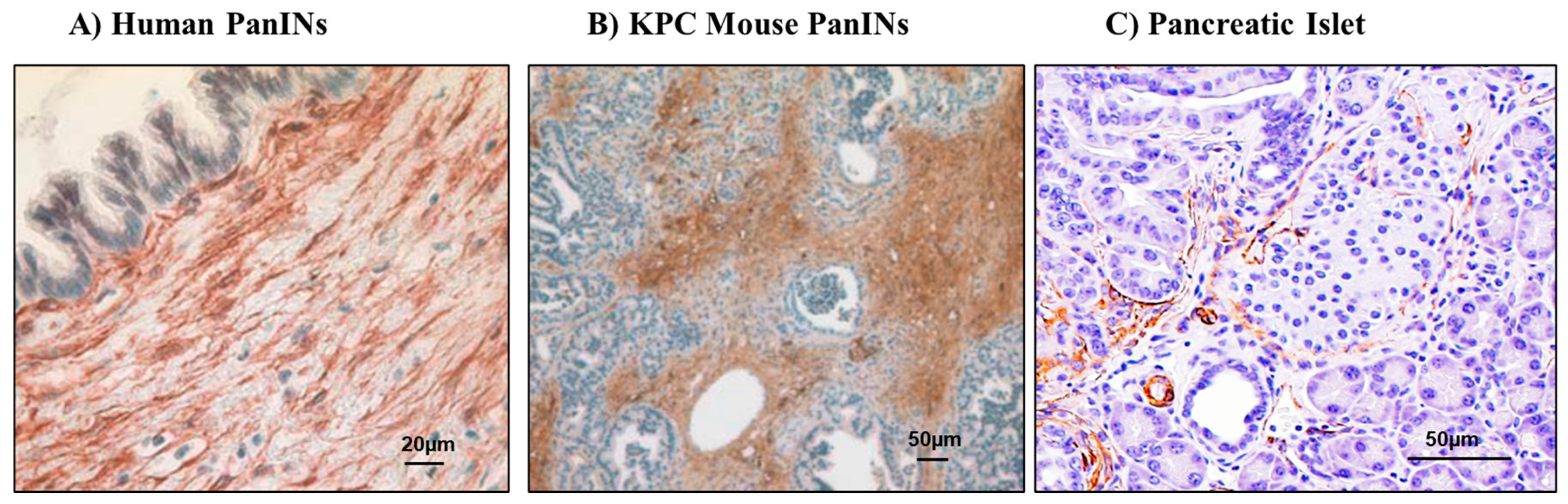
5. Pancreatic Stellate Cells and Their Role in PDAC and Diabetes
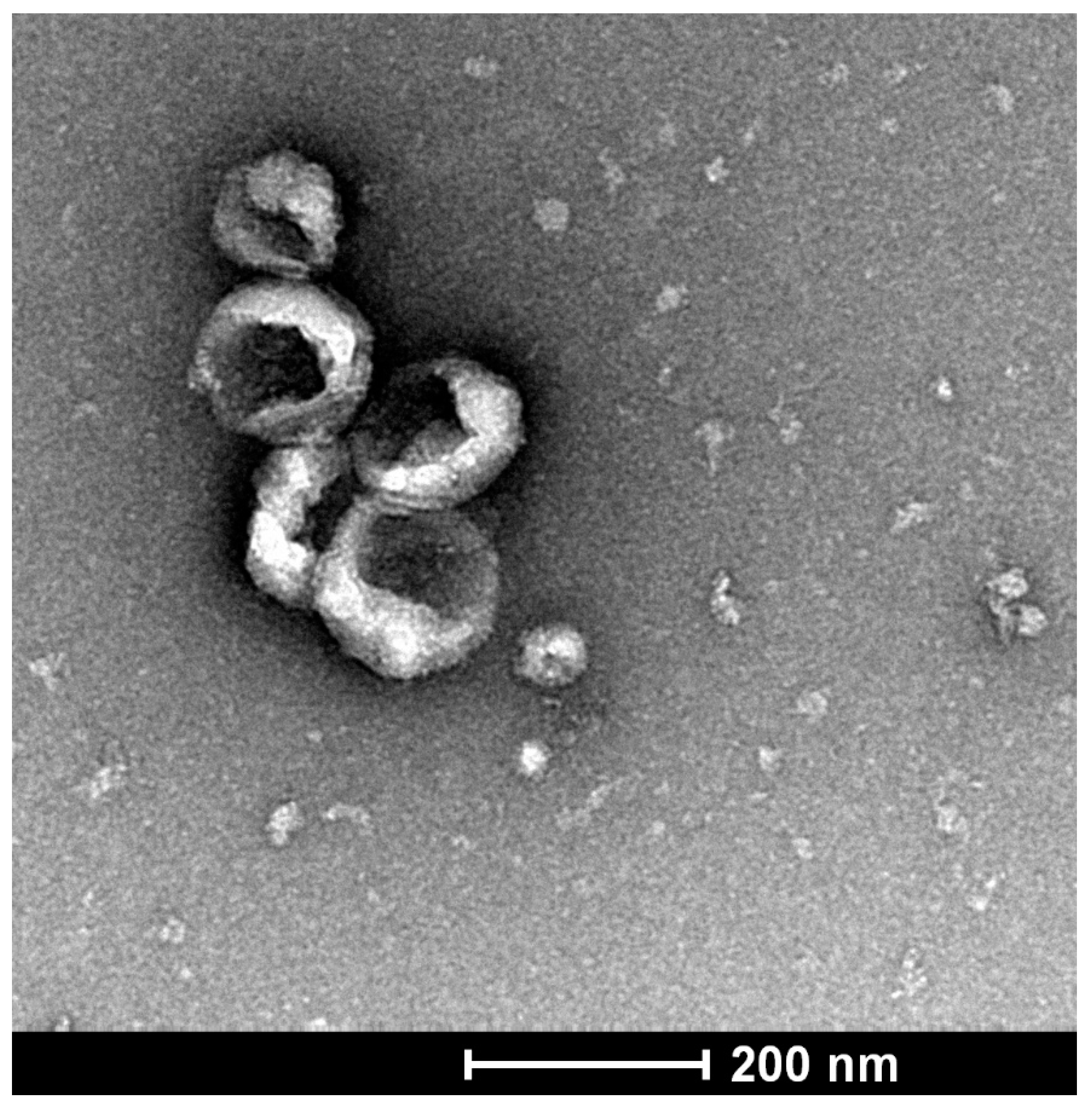
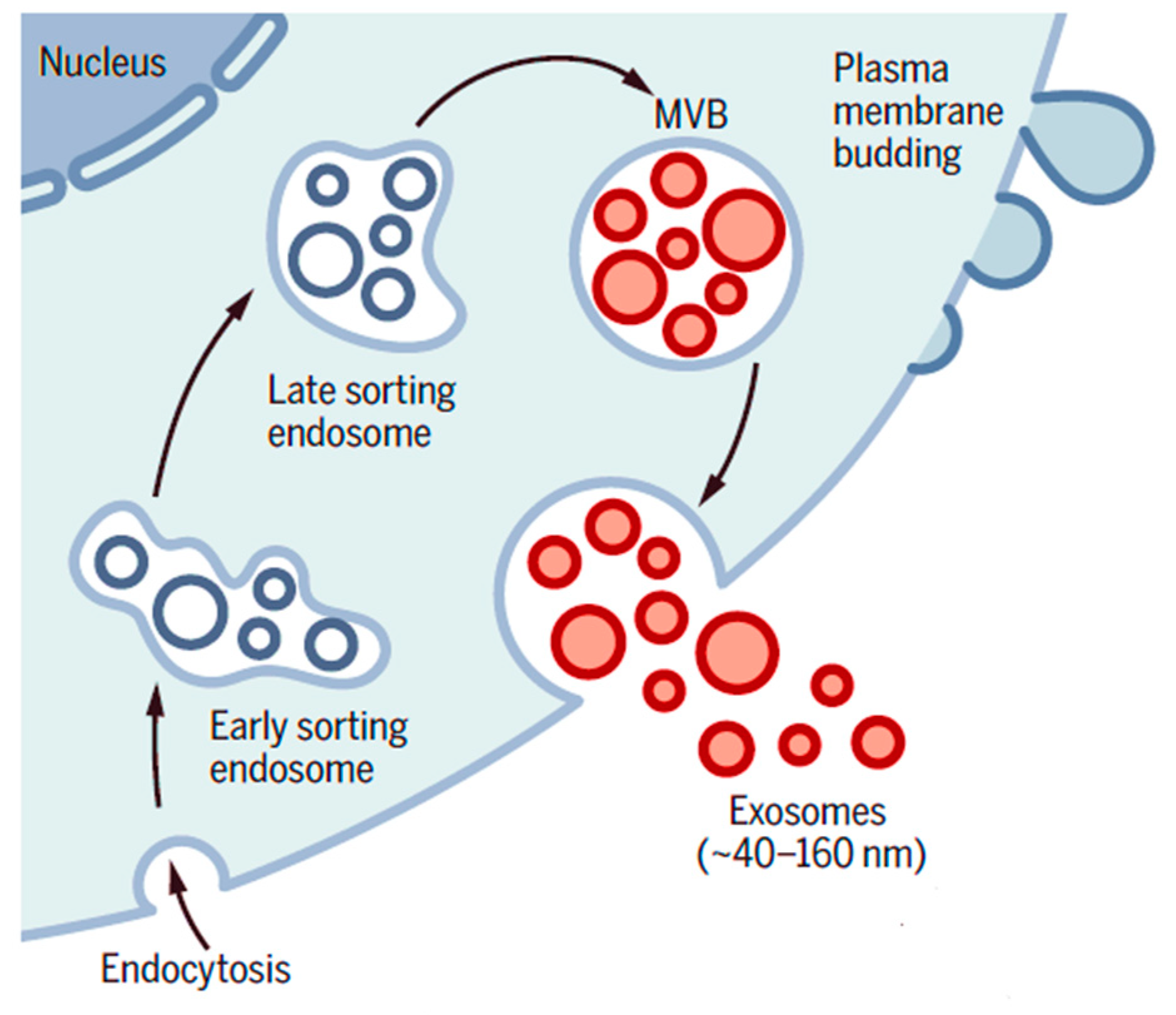
6. Exosomes and Their Role in Pancreatic Ductal Adenocarcinoma
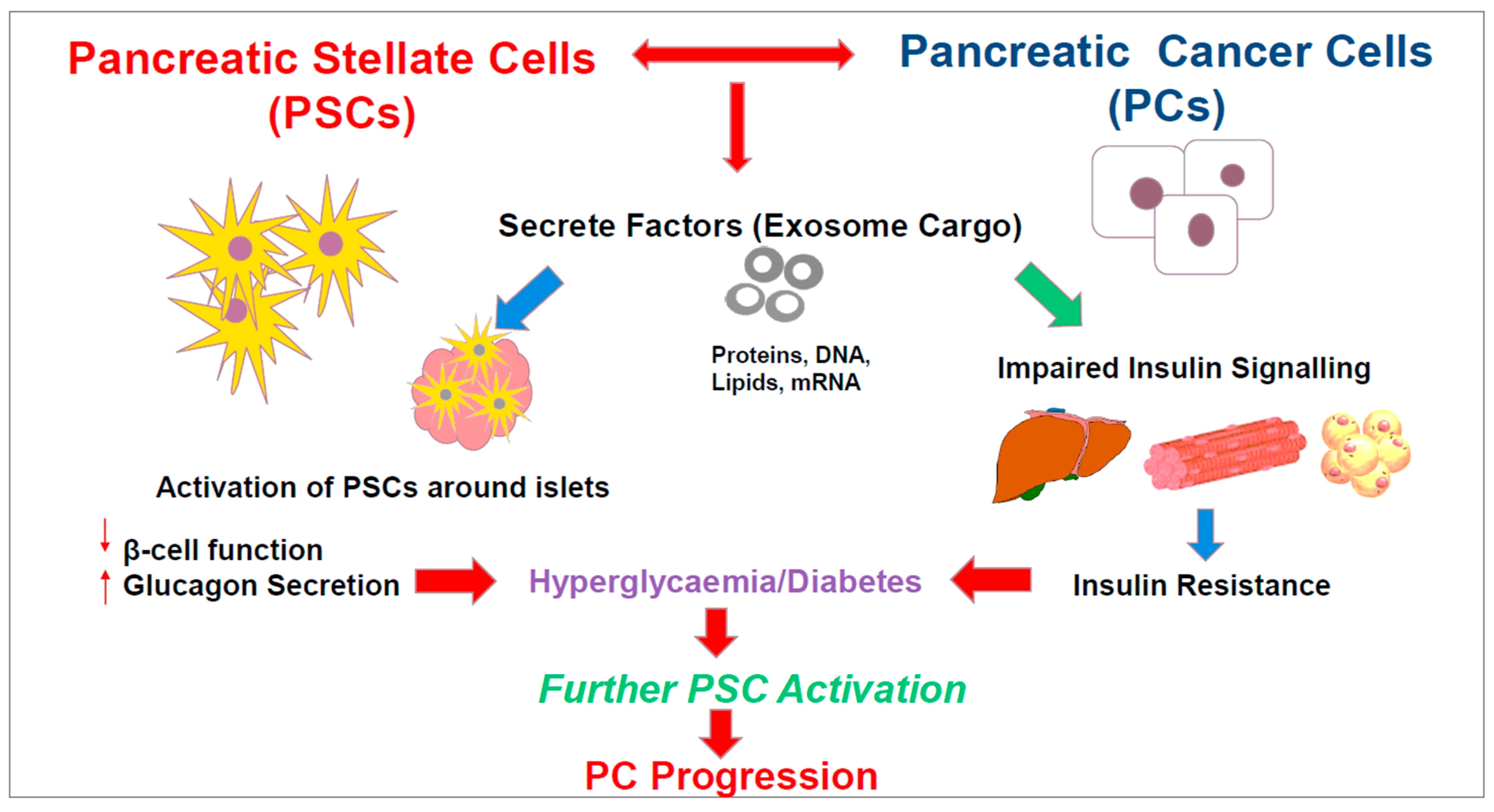
7. Role of Exosomes in Pancreatic Cancer-Related Diabetes
8. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ben, Q.; Xu, M.; Ning, X.; Liu, J.; Hong, S.; Huang, W.; Zhang, H.; Li, Z. Diabetes mellitus and risk of pancreatic cancer: A meta-analysis of cohort studies. Eur. J. Cancer 2011, 47, 1928–1937. [Google Scholar] [CrossRef]
- Huxley, R.; Ansary-Moghaddam, A.; Berrington de Gonzalez, A.; Barzi, F.; Woodward, M. Type-II diabetes and pancreatic cancer: A meta-analysis of 36 studies. Br. J. Cancer 2005, 92, 2076–2083. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; You, Y.; Guo, F.; Xu, J.; Dai, H.; Bie, P. Association of elevated risk of pancreatic cancer in diabetic patients: A systematic review and meta-analysis. Oncol. Lett. 2017, 13, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Paternoster, S.; Falasca, M. The intricate relationship between diabetes, obesity and pancreatic cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1873, 188326. [Google Scholar] [CrossRef]
- Association, A.D. 2. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes—2021. Diabetes Care 2021, 44, S15–S33. [Google Scholar] [CrossRef] [PubMed]
- Chari, S.T.; Leibson, C.L.; Rabe, K.G.; Timmons, L.J.; Ransom, J.; de Andrade, M.; Petersen, G.M. Pancreatic cancer-associated diabetes mellitus: Prevalence and temporal association with diagnosis of cancer. Gastroenterology 2008, 134, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pannala, R.; Basu, A.; Petersen, G.M.; Chari, S.T. New-onset diabetes: A potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol. 2009, 10, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Sah, R.P.; Nagpal, S.J.S.; Mukhopadhyay, D.; Chari, S.T. New insights into pancreatic cancer-induced paraneoplastic diabetes. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Kandlakunta, H.; Nagpal, S.J.S.; Feng, Z.; Hoos, W.; Petersen, G.M.; Chari, S.T. Model to determine risk of pancreatic cancer in patients with new-onset diabetes. Gastroenterology 2018, 155, 730–739.e3. [Google Scholar] [CrossRef]
- Boursi, B.; Finkelman, B.; Giantonio, B.J.; Haynes, K.; Rustgi, A.K.; Rhim, A.D.; Mamtani, R.; Yang, Y.-X. A clinical prediction model to assess risk for pancreatic cancer among patients with new-onset diabetes. Gastroenterology 2017, 152, 840–850.e3. [Google Scholar] [CrossRef] [Green Version]
- Chari, S.T.; Leibson, C.L.; Rabe, K.G.; Ransom, J.; De Andrade, M.; Petersen, G.M. Probability of pancreatic cancer following diabetes: A population-based study. Gastroenterology 2005, 129, 504–511. [Google Scholar] [CrossRef]
- Chari, S.T.; Zapiach, M.; Yadav, D.; Rizza, R.A. Beta-cell function and insulin resistance evaluated by HOMA in pancreatic cancer subjects with varying degrees of glucose intolerance. Pancreatology 2005, 5, 229–233. [Google Scholar] [CrossRef]
- Permert, J.; Larsson, J.; Fruin, A.B.; Tatemoto, K.; Herrington, M.K.; Adrian, T.E. Islet hormone secretion in pancreatic cancer patients with diabetes. Pancreas 1997, 15, 60–68. [Google Scholar] [CrossRef]
- Hart, P.A.; Kamada, P.; Rabe, K.G.; Srinivasan, S.; Basu, A.; Aggarwal, G.; Chari, S.T. Weight Loss Precedes Cancer Specific Symptoms in Pancreatic Cancer Associated Diabetes Mellitus. Pancreas 2011, 40, 768. [Google Scholar] [CrossRef] [Green Version]
- Pannala, R.; Leirness, J.B.; Bamlet, W.R.; Basu, A.; Petersen, G.M.; Chari, S.T. Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology 2008, 134, 981–987. [Google Scholar] [CrossRef] [Green Version]
- Permert, J.; Ihse, I.; Jorfeldt, L.; von Schenck, H.; Arnquist, H.J.; Larsson, J. Improved glucose metabolism after subtotal pancreatectomy for pancreatic cancer. Br. J. Surg. 1993, 80, 1047–1050. [Google Scholar] [CrossRef]
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early detection of pancreatic cancer: Opportunities and challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Yoon, Y.S.; Han, H.S.; Cho, J.Y.; Choi, Y.; Jang, J.Y.; Choi, H. Prognostic relevance of preoperative diabetes mellitus and the degree of hyperglycemia on the outcomes of resected pancreatic ductal adenocarcinoma. J. Surg. Oncol. 2016, 113, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression Model for Pancreatic Cancer. Clin. Cancer Res. 2000, 6, 2969. [Google Scholar] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.V.; Wilson, J.S. A multipronged approach to pancreatic cancer treatment. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 385–387. [Google Scholar] [CrossRef]
- Xu, R.; Greening, D.W.; Zhu, H.-J.; Takahashi, N.; Simpson, R.J. Extracellular vesicle isolation and characterization: Toward clinical application. J. Clin. Investig. 2016, 126, 1152–1162. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef]
- Aggarwal, G.; Ramachandran, V.; Javeed, N.; Arumugam, T.; Dutta, S.; Klee, G.G.; Klee, E.W.; Smyrk, T.C.; Bamlet, W.; Han, J.J. Adrenomedullin is up-regulated in patients with pancreatic cancer and causes insulin resistance in β cells and mice. Gastroenterology 2012, 143, 1510–1517. [Google Scholar] [CrossRef] [Green Version]
- Javeed, N.; Sagar, G.; Dutta, S.K.; Smyrk, T.C.; Lau, J.S.; Bhattacharya, S.; Truty, M.; Petersen, G.M.; Kaufman, R.J.; Chari, S.T. Pancreatic cancer–derived exosomes cause paraneoplastic β-cell dysfunction. Clin. Cancer Res. 2015, 21, 1722–1733. [Google Scholar] [CrossRef] [Green Version]
- Sagar, G.; Sah, R.P.; Javeed, N.; Dutta, S.K.; Smyrk, T.C.; Lau, J.S.; Giorgadze, N.; Tchkonia, T.; Kirkland, J.L.; Chari, S.T. Pathogenesis of pancreatic cancer exosome-induced lipolysis in adipose tissue. Gut 2016, 65, 1165–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, G.; Kamada, P.; Chari, S.T. Prevalence of diabetes mellitus in pancreatic cancer compared to common cancers. Pancreas 2013, 42, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Permert, J.; Larsson, J.; Westermark, G.T.; Herrington, M.K.; Christmanson, L.; Pour, P.M.; Westermark, P.; Adrian, T.E. Islet amyloid polypeptide in patients with pancreatic cancer and diabetes. N. Engl. J. Med. 1994, 330, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, V.W.; Stram, D.O.; Porcel, J.; Chari, S.T.; Maskarinec, G.; Le Marchand, L.; Wilkens, L.R.; Haiman, C.A.; Pandol, S.J.; Monroe, K.R. Pancreatic cancer following incident diabetes in African Americans and Latinos: The multiethnic cohort. J. Natl. Cancer Inst. 2019, 111, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.P.; Sharma, A.; Nagpal, S.; Patlolla, S.H.; Sharma, A.; Kandlakunta, H.; Anani, V.; Angom, R.S.; Kamboj, A.K.; Ahmed, N. Phases of metabolic and soft tissue changes in months preceding a diagnosis of pancreatic ductal adenocarcinoma. Gastroenterology 2019, 156, 1742–1752. [Google Scholar] [CrossRef]
- Permert, J.; Adrian, T.E.; Jacobsson, P.; Jorfelt, L.; Fruin, A.B.; Larsson, J. Is profound peripheral insulin resistance in patients with pancreatic cancer caused by a tumor-associated factor? Am. J. Surg. 1993, 165, 61–67. [Google Scholar] [CrossRef]
- Nagpal, S.J.S.; Kandlakunta, H.; Sharma, A.; Sannapaneni, S.; Velamala, P.; Majumder, S.; Matveyenko, A.; Chari, S.T. Endocrinopathy in Pancreatic Cancer Is Characterized by Reduced Islet Size and Density with Preserved Endocrine Composition as Compared to Type 2 Diabetes: Presidential Poster Award: 45. Am. J. Gastroenterol. 2018, 113, S26–S28. [Google Scholar] [CrossRef]
- Westermark, P. Quantitative studies of amyloid in the islets of Langerhans. Upsala J. Med Sci. 1972, 77, 91–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, F.; Koczan, D.; Adam, U.; Benz, S.; von Dobschuetz, E.; Prall, F.; Nizze, H.; Thiesen, H.-J.; Hopt, U.T.; Löbler, M. Expression of connexin26 in islets of Langerhans is associated with impaired glucose tolerance in patients with pancreatic adenocarcinoma. Pancreas 2004, 29, 284–290. [Google Scholar] [CrossRef]
- Huang, H.; Dong, X.; Kang, M.X.; Xu, B.; Chen, Y.; Zhang, B.; Chen, J.; Xie, Q.P.; Wu, Y.L. Novel blood biomarkers of pancreatic cancer–associated diabetes mellitus identified by peripheral blood–based gene expression profiles. Am. J. Gastroenterol. 2010, 105, 1661–1669. [Google Scholar] [CrossRef]
- Lee, M.; Wong, H.-L.; Tsang, E.S.; Addison, S.M.F.; Topham, J.T.; Karasinska, J.; Kalloger, S.; Loree, J.M.; Schaeffer, D.F.; Renouf, D.J. Clinicopathological features of pancreatic cancer-related diabetes. J. Clin. Oncol. 2020, 38, 675. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Bao, Y.; Qian, Z.R.; Wu, C.; Kraft, P.; Ogino, S.; Stampfer, M.J.; Sato, K.; Ma, J.; Buring, J.E. Hyperglycemia, insulin resistance, impaired pancreatic β-cell function, and risk of pancreatic cancer. J. Natl. Cancer Inst. 2013, 105, 1027–1035. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Investig. 2000, 106, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Kahn, C.R. Insulin resistance, insulin insensitivity, and insulin unresponsiveness: A necessary distinction. Metabolism 1978, 27, 1893–1902. [Google Scholar] [CrossRef]
- Kolterman, O.; Gray, R.S.; Griffin, J.; Burstein, P.; Insel, J.; Scarlett, J.A.; Olefsky, J.M. Receptor and postreceptor defects contribute to the insulin resistance in noninsulin-dependent diabetes mellitus. J. Clin. Investig. 1981, 68, 957–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olefsky, J.M.; Kolterman, O.G.; Scarlett, J.A. Insulin action and resistance in obesity and noninsulin-dependent type II diabetes mellitus. Am. J. Physiol.-Endocrinol. Metab. 1982, 243, E15–E30. [Google Scholar] [CrossRef] [PubMed]
- Höppener, J.W.; Ahrén, B.; Lips, C.J. Islet amyloid and type 2 diabetes mellitus. N. Engl. J. Med. 2000, 343, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Knezetic, J.A.; Strömmer, L.; Permert, J.; Larsson, J.; Adrian, T.E. The intracellular mechanism of insulin resistance in pancreatic cancer patients. J. Clin. Endocrinol. Metab. 2000, 85, 1232–1238. [Google Scholar] [CrossRef]
- Wang, F.; Larsson, J.; Abdiu, A.; Gasslander, T.; Westermark, P.; Adrian, T.E.; Permert, J. Dissociated secretion of islet amyloid polypeptide and insulin in serum-free culture media conditioned by human pancreatic adenocarcinoma cell lines. Int. J. Pancreatol. 1997, 21, 157–164. [Google Scholar] [CrossRef]
- Wang, F.; Adrian, T.E.; Westermark, G.; Gasslander, T.; Permert, J. Dissociated insulin and islet amyloid polypeptide secretion from isolated rat pancreatic islets cocultured with human pancreatic adenocarcinoma cells. Pancreas 1999, 18, 403–409. [Google Scholar] [CrossRef]
- Ahrén, B.; Andrén-Sandberg, Å. Glucose tolerance and insulin secretion in experimental pancreatic cancer in the Syrian hamster. Res. Exp. Med. 1993, 193, 21–26. [Google Scholar] [CrossRef]
- Cersosimo, E.; Pisters, P.W.; Pesola, G.; McDermott, K.; Bajorunas, D.; Brennan, M.F. Insulin secretion and action in patients with pancreatic cancer. Cancer 1991, 67, 486–493. [Google Scholar] [CrossRef]
- Basso, D.; Plebani, M.; Fogar, P.; Del Favero, G.; Briani, G.; Meggiato, T.; Panozzo, M.; Ferrara, C.; D’Angeli, F.; Burlina, A. Beta-cell function in pancreatic adenocarcinoma. Pancreas 1994, 9, 332–335. [Google Scholar] [CrossRef]
- Apte, M.; Haber, P.; Applegate, T.; Norton, I.; McCaughan, G.; Korsten, M.; Pirola, R.; Wilson, J. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133. [Google Scholar] [CrossRef]
- Lardon, J.; Rooman, I.; Bouwens, L. Nestin expression in pancreatic stellate cells and angiogenic endothelial cells. Histochem. Cell Biol. 2002, 117, 535–540. [Google Scholar] [CrossRef]
- Sparmann, G.; Kruse, M.-L.; Hofmeister-Mielke, N.; Koczan, D.; Jaster, R.; Liebe, S.; Wolff, D.; Emmrich, J. Bone marrow-derived pancreatic stellate cells in rats. Cell Res. 2010, 20, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.; McCarroll, J.; Park, S.; Wu, M.; Pirola, R.; Korsten, M.; Wilson, J.; Apte, M. Rat pancreatic stellate cells secrete matrix metalloproteinases: Implications for extracellular matrix turnover. Gut 2003, 52, 275–282. [Google Scholar] [CrossRef]
- Berna, M.J.; Seiz, O.; Nast, J.F.; Benten, D.; Bläker, M.; Koch, J.; Lohse, A.W.; Pace, A. CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J. Biol. Chem. 2010, 285, 38905–38914. [Google Scholar] [CrossRef] [Green Version]
- Phillips, P.A.; Yang, L.; Shulkes, A.; Vonlaufen, A.; Poljak, A.; Bustamante, S.; Warren, A.; Xu, Z.; Guilhaus, M.; Pirola, R. Pancreatic stellate cells produce acetylcholine and may play a role in pancreatic exocrine secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 17397–17402. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol. Cancer 2018, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.; Wilson, J. Alcohol-induced pancreatic injury. Best Pract. Res. Clin. Gastroenterol. 2003, 17, 593–612. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schünemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef]
- Pothula, S.P.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic stellate cells: Aiding and abetting pancreatic cancer progression. Pancreatology 2020, 20, 409–418. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Biankin, A.V.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Hepatocyte growth factor inhibition: A novel therapeutic approach in pancreatic cancer. Br. J. Cancer 2016, 114, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Merrett, N.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Targeting the HGF/c-MET pathway: Stromal remodelling in pancreatic cancer. Oncotarget 2017, 8, 76722. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596. [Google Scholar] [CrossRef]
- Apte, M.; Park, S.; Phillips, P.; Santucci, N.; Goldstein, D.; Kumar, R.; Ramm, G.; Buchler, M.; Friess, H.; McCarroll, J. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 2009, 11, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.L.; Cao, S.G.; Li, Y.; Sun, B.; Chen, D.; Wang, D.S.; Zhou, Y.B. Pancreatic stellate cells facilitate pancreatic cancer cell viability and invasion. Oncol. Lett. 2019, 17, 2057–2062. [Google Scholar] [CrossRef] [Green Version]
- Ikenaga, N.; Ohuchida, K.; Mizumoto, K.; Cui, L.; Kayashima, T.; Morimatsu, K.; Moriyama, T.; Nakata, K.; Fujita, H.; Tanaka, M. CD10+ pancreatic stellate cells enhance the progression of pancreatic cancer. Gastroenterology 2010, 139, 1041–1051.e8. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M. Inter-and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Noy, A.; Bilezikian, J.P. Clinical review 63: Diabetes and pancreatic cancer: Clues to the early diagnosis of pancreatic malignancy. J. Clin. Endocrinol. Metab. 1994, 79, 1223–1231. [Google Scholar]
- Jones, S.; Chen, W.-d.; Parmigiani, G.; Diehl, F.; Beerenwinkel, N.; Antal, T.; Traulsen, A.; Nowak, M.A.; Siegel, C.; Velculescu, V.E. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 4283–4288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, A.M.; Wolfgang, C.L.; Canto, M.I.; Klein, A.P.; Herman, J.M.; Goggins, M.; Fishman, E.K.; Kamel, I.; Weiss, M.J.; Diaz, L.A. The early detection of pancreatic cancer: What will it take to diagnose and treat curable pancreatic neoplasia? Cancer Res. 2014, 74, 3381–3389. [Google Scholar] [CrossRef] [Green Version]
- Brat, D.J.; Lillemoe, K.D.; Yeo, C.J.; Warfield, P.B.; Hruban, R.H. Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am. J. Surg. Pathol. 1998, 22, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.-C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 2016, 381, 194–200. [Google Scholar] [CrossRef]
- Mekapogu, A.; Pothula, S.; Pirola, R.; Wilson, J.; Apte, M. Multifunctional role of pancreatic stellate cells in pancreatic cancer. Ann. Pancreat. Cancer 2019, 2. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.S.; Pirola, R.C.; Apte, M. Stars and stripes in pancreatic cancer: Role of stellate cells and stroma in cancer progression. Front. Physiol. 2014, 5, 52. [Google Scholar] [CrossRef] [Green Version]
- Datar, S.; Bhonde, R. Islet-derived stellate-like cells as a novel source for islet neogenesis in chicks. Poult. Sci. 2009, 88, 654–660. [Google Scholar] [CrossRef]
- Zha, M.; Li, F.; Xu, W.; Chen, B.; Sun, Z. Isolation and characterization of islet stellate cells in rat. Islets 2014, 6, e28701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, M.; Xu, W.; Jones, P.M.; Sun, Z. Isolation and characterization of human islet stellate cells. Exp. Cell Res. 2016, 341, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Ko, S.H.; Hong, O.K.; Kim, J.W.; Ahn, Y.B.; Song, K.H.; Cha, B.Y.; Son, H.Y.; Kim, M.J.; Jeong, I.K.; Yoon, K.H. High glucose increases extracellular matrix production in pancreatic stellate cells by activating the renin–angiotensin system. J. Cell. Biochem. 2006, 98, 343–355. [Google Scholar] [CrossRef]
- Nomiyama, Y.; Tashiro, M.; Yamaguchi, T.; Watanabe, S.; Taguchi, M.; Asaumi, H.; Nakamura, H.; Otsuki, M. High glucose activates rat pancreatic stellate cells through protein kinase C and p38 mitogen-activated protein kinase pathway. Pancreas 2007, 34, 364–372. [Google Scholar] [CrossRef]
- Hong, O.K.; Lee, S.H.; Rhee, M.; Ko, S.H.; Cho, J.H.; Choi, Y.H.; Song, K.H.; Son, H.Y.; Yoon, K.H. Hyperglycemia and hyperinsulinemia have additive effects on activation and proliferation of pancreatic stellate cells: Possible explanation of islet-specific fibrosis in type 2 diabetes mellitus. J. Cell. Biochem. 2007, 101, 665–675. [Google Scholar] [CrossRef]
- Kikuta, K.; Masamune, A.; Hamada, S.; Takikawa, T.; Nakano, E.; Shimosegawa, T. Pancreatic stellate cells reduce insulin expression and induce apoptosis in pancreatic β-cells. Biochem. Biophys. Res. Commun. 2013, 433, 292–297. [Google Scholar] [CrossRef]
- Lee, E.; Ryu, G.R.; Ko, S.-H.; Ahn, Y.-B.; Yoon, K.-H.; Ha, H.; Song, K.-H. Antioxidant treatment may protect pancreatic beta cells through the attenuation of islet fibrosis in an animal model of type 2 diabetes. Biochem. Biophys. Res. Commun. 2011, 414, 397–402. [Google Scholar] [CrossRef]
- Saito, R.; Yamada, S.; Yamamoto, Y.; Kodera, T.; Hara, A.; Tanaka, Y.; Kimura, F.; Takei, I.; Umezawa, K.; Kojima, I. Conophylline suppresses pancreatic stellate cells and improves islet fibrosis in Goto-Kakizaki rats. Endocrinology 2012, 153, 621–630. [Google Scholar] [CrossRef]
- Lee, E.; Ryu, G.R.; Ko, S.-H.; Ahn, Y.-B.; Song, K.-H. A role of pancreatic stellate cells in islet fibrosis and β-cell dysfunction in type 2 diabetes mellitus. Biochem. Biophys. Res. Commun. 2017, 485, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.-L.; Lai, F.M.; Tong, P.C.; Zhong, D.-R.; Yang, D.; Tomlinson, B.; Chan, J.C. Prevalence and clinicopathological characteristics of islet amyloid in Chinese patients with type 2 diabetes. Diabetes 2003, 52, 2759–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Waldron, R.T.; Su, H.-Y.; Moro, A.; Chang, H.-H.; Eibl, G.; Ferreri, K.; Kandeel, F.R.; Lugea, A.; Li, L. Insulin promotes proliferation and fibrosing responses in activated pancreatic stellate cells. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 311, G675–G687. [Google Scholar] [CrossRef] [Green Version]
- Ko, S.-H.; Kwon, H.-S.; Kim, S.-R.; Moon, S.-D.; Ahn, Y.-B.; Song, K.-H.; Son, H.-S.; Cha, B.-Y.; Lee, K.-W.; Son, H.-Y. Ramipril treatment suppresses islet fibrosis in Otsuka Long–Evans Tokushima fatty rats. Biochem. Biophys. Res. Commun. 2004, 316, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Hama, K.; Ohnishi, H.; Yasuda, H.; Ueda, N.; Mashima, H.; Satoh, Y.; Hanatsuka, K.; Kita, H.; Ohashi, A.; Tamada, K. Angiotensin II stimulates DNA synthesis of rat pancreatic stellate cells by activating ERK through EGF receptor transactivation. Biochem. Biophys. Res. Commun. 2004, 315, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Ryu, G.R.; Lee, E.; Chun, H.-J.; Yoon, K.-H.; Ko, S.-H.; Ahn, Y.-B.; Song, K.-H. Oxidative stress plays a role in high glucose-induced activation of pancreatic stellate cells. Biochem. Biophys. Res. Commun. 2013, 439, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Horibe, S.; Tanahashi, T.; Kawauchi, S.; Murakami, Y.; Rikitake, Y. Mechanism of recipient cell-dependent differences in exosome uptake. BMC Cancer 2018, 18, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Stefanius, K.; Servage, K.; de Souza Santos, M.; Gray, H.F.; Toombs, J.E.; Chimalapati, S.; Kim, M.S.; Malladi, V.S.; Brekken, R.; Orth, K. Human pancreatic cancer cell exosomes, but not human normal cell exosomes, act as an initiator in cell transformation. eLife 2019, 8, e40226. [Google Scholar] [CrossRef]
- Abd Elmageed, Z.Y.; Yang, Y.; Thomas, R.; Ranjan, M.; Mondal, D.; Moroz, K.; Fang, Z.; Rezk, B.M.; Moparty, K.; Sikka, S.C. Neoplastic reprogramming of patient-derived adipose stem cells by prostate cancer cell-associated exosomes. Stem Cells 2014, 32, 983–997. [Google Scholar] [CrossRef] [Green Version]
- Nabet, B.Y.; Qiu, Y.; Shabason, J.E.; Wu, T.J.; Yoon, T.; Kim, B.C.; Benci, J.L.; DeMichele, A.M.; Tchou, J.; Marcotrigiano, J. Exosome RNA unshielding couples stromal activation to pattern recognition receptor signaling in cancer. Cell 2017, 170, 352–366.e13. [Google Scholar] [CrossRef] [Green Version]
- Tsilioni, I.; Theoharides, T.C. Extracellular vesicles are increased in the serum of children with autism spectrum disorder, contain mitochondrial DNA, and stimulate human microglia to secrete IL-1β. J. Neuroinflammation 2018, 15, 1–8. [Google Scholar] [CrossRef]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.F.; Li, S.; Chin, A.R. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Kahlert, C.; Kalluri, R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J. Mol. Med. 2013, 91, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Deng, C.-X. Current progresses of exosomes as cancer diagnostic and prognostic biomarkers. Int. J. Biol. Sci. 2019, 15, 1. [Google Scholar] [CrossRef]
- Casari, I.; Howard, J.A.; Robless, E.E.; Falasca, M. Exosomal integrins and their influence on pancreatic cancer progression and metastasis. Cancer Lett. 2021, 507, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidi, A.; Paladin, D.; Greening, D.W.; Falasca, M. Oncogenic and non-malignant pancreatic exosome cargo reveal distinct expression of oncogenic and prognostic factors involved in tumor invasion and metastasis. Proteomics 2019, 19, 1800158. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tao, Y.; Wang, X.; Jiang, P.; Li, J.; Peng, M.; Zhang, X.; Chen, K.; Liu, H.; Zhen, P. Tumor-secreted exosomal miR-222 promotes tumor progression via regulating P27 expression and re-localization in pancreatic cancer. Cell. Physiol. Biochem. 2018, 51, 610–629. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, X.; Chen, Q.; Liu, T.; Lu, C.; Yu, J.; Miao, Y.; Wei, J. Downregulated miR-98-5p promotes PDAC proliferation and metastasis by reversely regulating MAP4K4. J. Exp. Clin. Cancer Res. 2018, 37, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Milane, L.; Singh, A.; Mattheolabakis, G.; Suresh, M.; Amiji, M.M. Exosome mediated communication within the tumor microenvironment. J. Control. Release 2015, 219, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.-P.; Goh, B.-C. Exosome-mediated metastasis: From epithelial–mesenchymal transition to escape from immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Steinbichler, T.B.; Dudás, J.; Riechelmann, H.; Skvortsova, I.-I. The role of exosomes in cancer metastasis. Semin. Cancer Biol. 2017, 44, 170–181. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Nagathihalli, N.S.; Castellanos, J.A.; Shi, C.; Beesetty, Y.; Reyzer, M.L.; Caprioli, R.; Chen, X.; Walsh, A.J.; Skala, M.C.; Moses, H.L. Signal transducer and activator of transcription 3, mediated remodeling of the tumor microenvironment results in enhanced tumor drug delivery in a mouse model of pancreatic cancer. Gastroenterology 2015, 149, 1932–1943.e9. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Wu, H.; Xiao, Y.; Liang, Z.; Liu, T. Upregulation of exosomal microRNA-21 in pancreatic stellate cells promotes pancreatic cancer cell migration and enhances Ras/ERK pathway activity. Int. J. Oncol. 2020, 56, 1025–1033. [Google Scholar] [CrossRef]
- Charrier, A.; Chen, R.; Chen, L.; Kemper, S.; Hattori, T.; Takigawa, M.; Brigstock, D.R. Connective tissue growth factor (CCN2) and microRNA-21 are components of a positive feedback loop in pancreatic stellate cells (PSC) during chronic pancreatitis and are exported in PSC-derived exosomes. J. Cell Commun. Signal. 2014, 8, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Takikawa, T.; Masamune, A.; Yoshida, N.; Hamada, S.; Kogure, T.; Shimosegawa, T. Exosomes derived from pancreatic stellate cells: MicroRNA signature and effects on pancreatic cancer cells. Pancreas 2017, 46, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, H.; Wang, Q.; Chen, K.; Marko, K.; Tian, X.; Yang, Y. Pancreatic stellate cells derived exosomal miR-5703 promotes pancreatic cancer by downregulating CMTM4 and activating PI3K/Akt pathway. Cancer Lett. 2020, 490, 20–30. [Google Scholar] [CrossRef]
- Cao, W.; Zeng, Z.; He, Z.; Lei, S. Hypoxic pancreatic stellate cell-derived exosomal mirnas promote proliferation and invasion of pancreatic cancer through the PTEN/AKT pathway. Aging 2021, 13, 7120. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.; Bernard, V.; San Lucas, F.; Allenson, K.; Capello, M.; Kim, D.; Gascoyne, P.; Mulu, F.; Stephens, B.; Huang, J. Surfaceome profiling enables isolation of cancer-specific exosomal cargo in liquid biopsies from pancreatic cancer patients. Ann. Oncol. 2018, 29, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Que, R.; Ding, G.; Chen, J.; Cao, L. Analysis of serum exosomal microRNAs and clinicopathologic features of patients with pancreatic adenocarcinoma. World J. Surg. Oncol. 2013, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Joshi, G.K.; Deitz-McElyea, S.; Liyanage, T.; Lawrence, K.; Mali, S.; Sardar, R.; Korc, M. Label-free nanoplasmonic-based short noncoding RNA sensing at attomolar concentrations allows for quantitative and highly specific assay of microRNA-10b in biological fluids and circulating exosomes. ACS Nano 2015, 9, 11075–11089. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.-F.; Hannafon, B.N.; Zhao, Y.D.; Postier, R.G.; Ding, W.-Q. Plasma exosome miR-196a and miR-1246 are potential indicators of localized pancreatic cancer. Oncotarget 2017, 8, 77028. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, P.; Li, J.; Peng, M.; Zhao, X.; Zhang, X.; Chen, K.; Zhang, Y.; Liu, H.; Gan, L. Tumor-derived exosomal lnc-Sox2ot promotes EMT and stemness by acting as a ceRNA in pancreatic ductal adenocarcinoma. Oncogene 2018, 37, 3822–3838. [Google Scholar] [CrossRef]
- Yu, S.; Li, Y.; Liao, Z.; Wang, Z.; Wang, Z.; Li, Y.; Qian, L.; Zhao, J.; Zong, H.; Kang, B. Plasma extracellular vesicle long RNA profiling identifies a diagnostic signature for the detection of pancreatic ductal adenocarcinoma. Gut 2020, 69, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Taucher, V.; Mangge, H.; Haybaeck, J. Non-coding RNAs in pancreatic cancer: Challenges and opportunities for clinical application. Cell. Oncol. 2016, 39, 295–318. [Google Scholar] [CrossRef] [PubMed]
- Javeed, N.; Gustafson, M.P.; Dutta, S.K.; Lin, Y.; Bamlet, W.R.; Oberg, A.L.; Petersen, G.M.; Chari, S.T.; Dietz, A.B.; Mukhopadhyay, D. Immunosuppressive CD14+ HLA-DRlo/neg monocytes are elevated in pancreatic cancer and “primed” by tumor-derived exosomes. Oncoimmunology 2017, 6, e1252013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, B.; Zheng, W.; Kang, M.; Chen, Q.; Qin, W.; Li, C.; Zhang, Y.; Shao, Y.; Wu, Y. Exosomes derived from pancreatic cancer cells induce insulin resistance in C2C12 myotube cells through the PI3K/Akt/FoxO1 pathway. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Korc, M. Pancreatic cancer–associated diabetes is an “exosomopathy”. Clin. Cancer Res. 2015, 21, 1508–1510. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.K.; Tang, F.; Cheung, T.T.; Cheung, B.M.Y. Adrenomedullin and diabetes. World J. Diabetes 2014, 5, 364. [Google Scholar] [CrossRef]
- Kong, F.; Li, L.; Du, Y.; Zhu, H.; Li, Z.; Kong, X. Exosomal adrenomedullin derived from cancer-associated fibroblasts promotes lipolysis in adipose tissue. Gut 2018, 67, 2226–2227. [Google Scholar] [CrossRef]
- Perera, C.; Xu, Z.; Mekapogu, A.R.; Hosen, S.Z.; Pothula, S.; Greenfield, J.; Chari, S.; Goldstein, D.; Pirola, R.; Wilson, J. 1133 Pancreatic Stellate Cell and Cancer Cell Derived Exosomes Impair Beta Cell Function: Implications for Pancreatic Cancer Related Diabetes. Gastroenterology 2020, 158, S-221. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type 2 Diabetes Mellitus | Pancreatic Cancer Related Diabetes (PCRD) |
|---|---|
| Not associated with a tumour | Resolution of Diabetes upon surgical resection of the pancreatic tumour |
| Commonly associated with weight gain | Associated weight loss before the onset of diabetes |
| Diabetes often improves alongside weight loss | Glycaemic control worsens alongside weight loss |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, C.J.; Falasca, M.; Chari, S.T.; Greenfield, J.R.; Xu, Z.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Role of Pancreatic Stellate Cell-Derived Exosomes in Pancreatic Cancer-Related Diabetes: A Novel Hypothesis. Cancers 2021, 13, 5224. https://doi.org/10.3390/cancers13205224
Perera CJ, Falasca M, Chari ST, Greenfield JR, Xu Z, Pirola RC, Wilson JS, Apte MV. Role of Pancreatic Stellate Cell-Derived Exosomes in Pancreatic Cancer-Related Diabetes: A Novel Hypothesis. Cancers. 2021; 13(20):5224. https://doi.org/10.3390/cancers13205224
Chicago/Turabian StylePerera, Chamini J., Marco Falasca, Suresh T. Chari, Jerry R. Greenfield, Zhihong Xu, Romano C. Pirola, Jeremy S. Wilson, and Minoti V. Apte. 2021. "Role of Pancreatic Stellate Cell-Derived Exosomes in Pancreatic Cancer-Related Diabetes: A Novel Hypothesis" Cancers 13, no. 20: 5224. https://doi.org/10.3390/cancers13205224
APA StylePerera, C. J., Falasca, M., Chari, S. T., Greenfield, J. R., Xu, Z., Pirola, R. C., Wilson, J. S., & Apte, M. V. (2021). Role of Pancreatic Stellate Cell-Derived Exosomes in Pancreatic Cancer-Related Diabetes: A Novel Hypothesis. Cancers, 13(20), 5224. https://doi.org/10.3390/cancers13205224