
Antigen Presenting Cells from Tumor and Colon of Colorectal Cancer Patients Are Distinct in Activation and Functional Status, but Comparably Responsive to Activated T Cells

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. APCs in Both Tumor and Adjacent Colon Are Predominantly CD64+CD14+ MPs
2.2. The Level of MPs Relative to TILs in MSS Tumors Exceeds That of MSI-H Tumors
2.3. MPs in Tumor and Colon Inversely Express Co-Stimulatory CD80 and Co-Inhibitory PD-L1
2.4. Tissue-Resident Memory T Cells in Tumor and Colon Express Opposed PD-1 and CD69 Levels
2.5. Association of Activation Status between Particular APCs and T cells Are Tissue-Specific
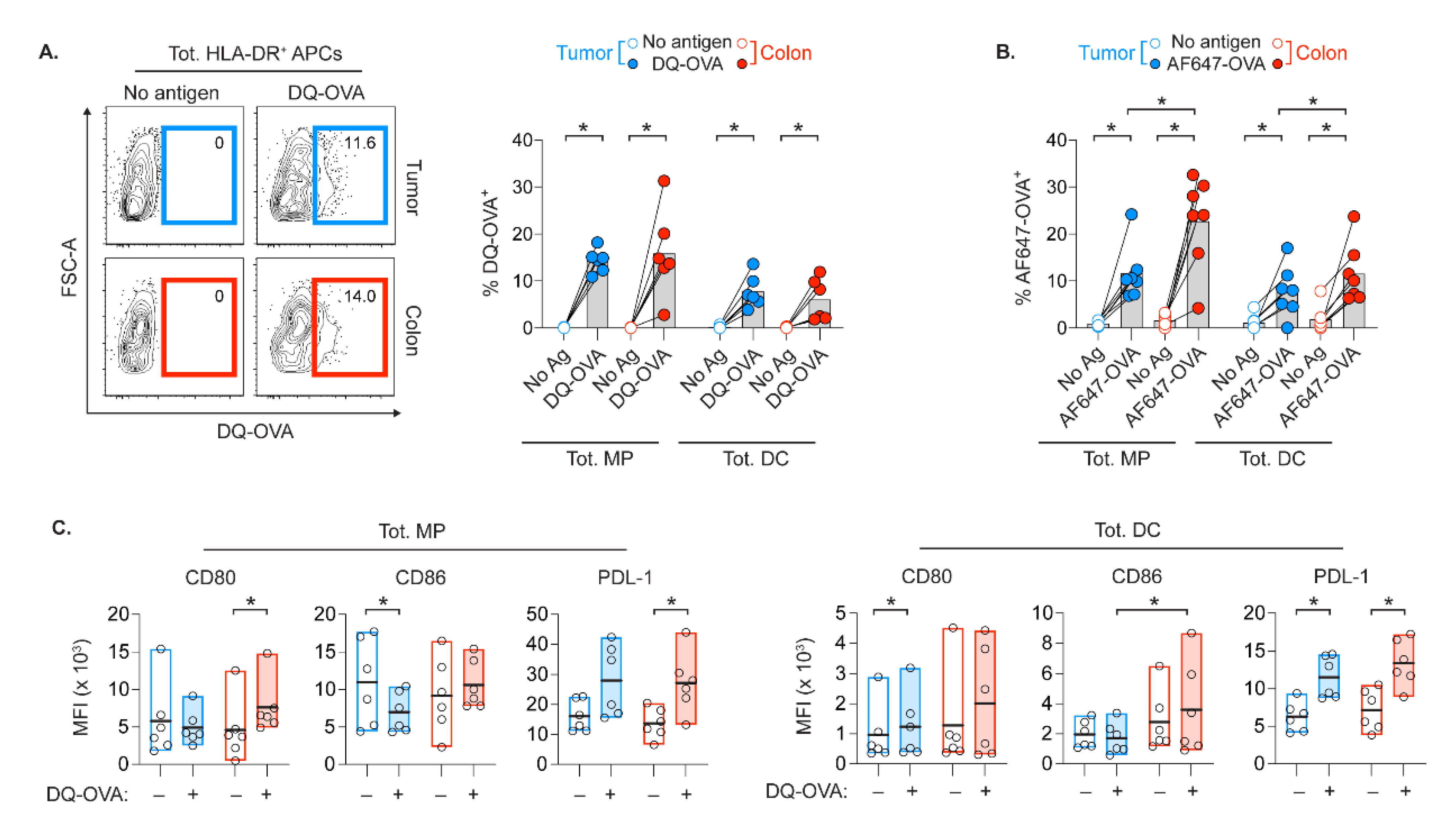
2.6. APCs in Tumor and Colon Display Opposed Co-Stimulatory Capacity upon Protein Digestion
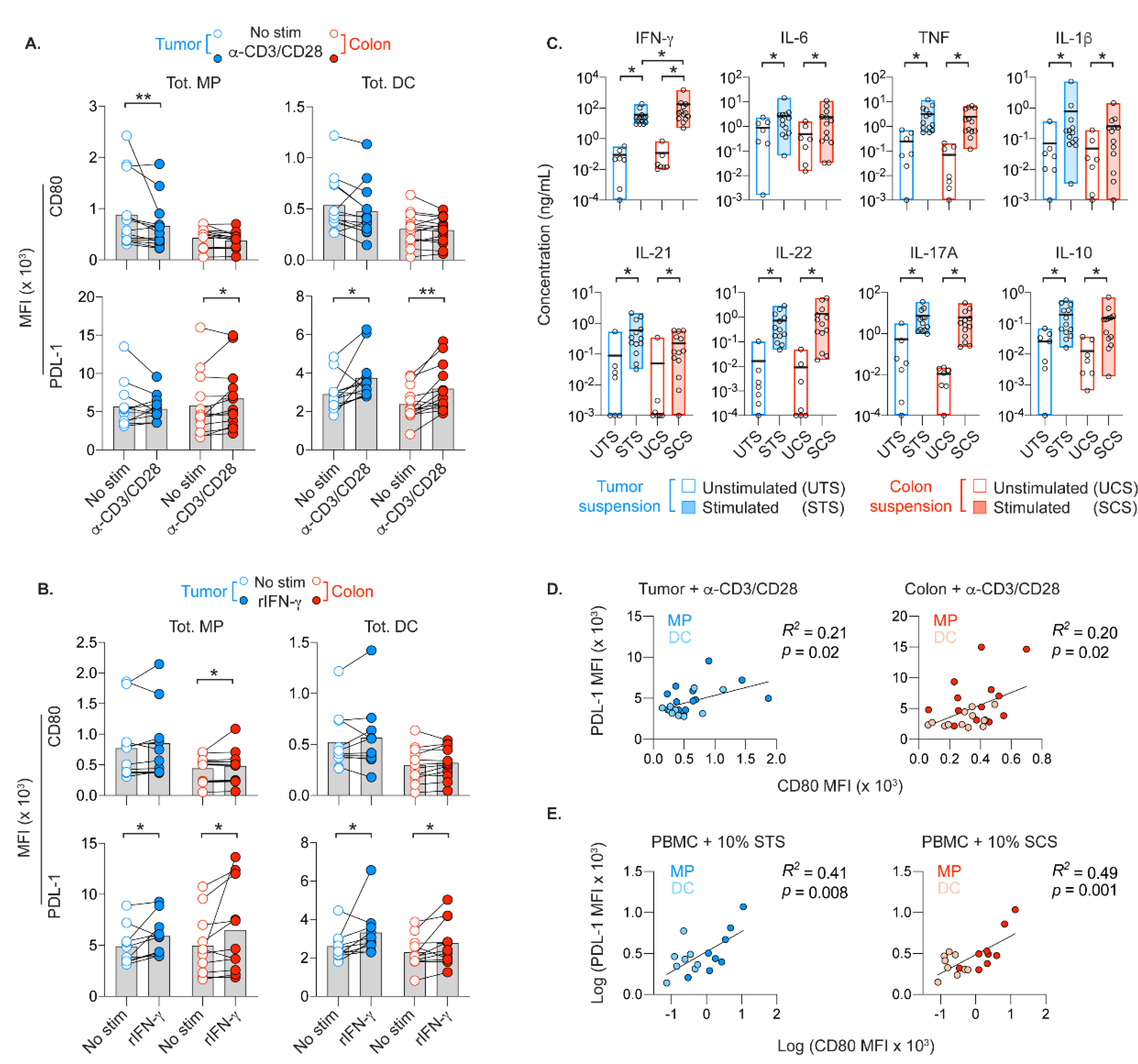
2.7. APCs Co-Express CD80 and PD-L1 in Presence of Activated T Cells In Vitro
3. Discussion
4. Materials and Methods
4.1. CRC Patients
4.2. Immunohistochemistry
4.3. Generation of Single Cell Suspensions
4.4. OVA Uptake and Degradation
4.5. Stimulation of APCs by Activated T Cells
4.6. Supernatant Cytokine Analyses
4.7. Flow Cytometry
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pages, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef]
- Li, X.; Ling, A.; Kellgren, T.G.; Lundholm, M.; Lofgren-Burstrom, A.; Zingmark, C.; Rutegard, M.; Ljuslinder, I.; Palmqvist, R.; Edin, S. A Detailed Flow Cytometric Analysis of Immune Activity Profiles in Molecular Subtypes of Colorectal Cancer. Cancers 2020, 12, 3440. [Google Scholar] [CrossRef] [PubMed]
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; de Miranda, N.F.; Goodall, C.P.; Blair, T.C.; Fox, B.A.; McDermott, J.E.; et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018, 9, 2724. [Google Scholar] [CrossRef] [Green Version]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019, 9, 1512. [Google Scholar] [CrossRef] [Green Version]
- Norton, S.E.; Dunn, E.T.; McCall, J.L.; Munro, F.; Kemp, R.A. Gut macrophage phenotype is dependent on the tumor microenvironment in colorectal cancer. Clin. Transl. Immunol. 2016, 5, e76. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Cueto, F.J.; Martinez-Lopez, M.; Labiano, S.; Morales-Kastresana, A.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Forssell, J.; Oberg, A.; Henriksson, M.L.; Stenling, R.; Jung, A.; Palmqvist, R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin. Cancer Res. 2007, 13, 1472–1479. [Google Scholar] [CrossRef] [Green Version]
- Gulubova, M.V.; Ananiev, J.R.; Vlaykova, T.I.; Yovchev, Y.; Tsoneva, V.; Manolova, I.M. Role of dendritic cells in progression and clinical outcome of colon cancer. Int. J. Colorectal Dis. 2012, 27, 159–169. [Google Scholar] [CrossRef]
- Pinto, M.L.; Rios, E.; Duraes, C.; Ribeiro, R.; Machado, J.C.; Mantovani, A.; Barbosa, M.A.; Carneiro, F.; Oliveira, M.J. The Two Faces of Tumor-Associated Macrophages and Their Clinical Significance in Colorectal Cancer. Front. Immunol. 2019, 10, 1875. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Xu, F.; Zhang, J.; Wang, L.; Zheng, Y.; Wu, X.; Wang, J.; Huang, Q.; Lai, M. Tumor-associated macrophages remodeling EMT and predicting survival in colorectal carcinoma. Oncoimmunology 2018, 7, e1380765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.J.; Koh, J.; Kim, S.; Jeon, S.R.; Chie, E.K.; Kim, K.; Kang, G.H.; Han, S.W.; Kim, T.Y.; Jeong, S.Y.; et al. Chemoradiation-Induced Alteration of Programmed Death-Ligand 1 and CD8(+) Tumor-Infiltrating Lymphocytes Identified Patients With Poor Prognosis in Rectal Cancer: A Matched Comparison Analysis. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 1216–1224. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.; Platell, C.; Iacopetta, B. Tumor-infiltrating lymphocytes and perforation in colon cancer predict positive response to 5-fluorouracil chemotherapy. Clin. Cancer Res. 2008, 14, 1413–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibutani, M.; Maeda, K.; Nagahara, H.; Fukuoka, T.; Iseki, Y.; Matsutani, S.; Kashiwagi, S.; Tanaka, H.; Hirakawa, K.; Ohira, M. Tumor-infiltrating Lymphocytes Predict the Chemotherapeutic Outcomes in Patients with Stage IV Colorectal Cancer. Vivo 2018, 32, 151–158. [Google Scholar] [CrossRef]
- Teng, F.; Meng, X.; Kong, L.; Mu, D.; Zhu, H.; Liu, S.; Zhang, J.; Yu, J. Tumor-infiltrating lymphocytes, forkhead box P3, programmed death ligand-1, and cytotoxic T lymphocyte-associated antigen-4 expressions before and after neoadjuvant chemoradiation in rectal cancer. Transl. Res. 2015, 166, 721–732.e1. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Alloatti, A.; Rookhuizen, D.C.; Joannas, L.; Carpier, J.M.; Iborra, S.; Magalhaes, J.G.; Yatim, N.; Kozik, P.; Sancho, D.; Albert, M.L.; et al. Critical role for Sec22b-dependent antigen cross-presentation in antitumor immunity. J. Exp. Med. 2017, 214, 2231–2241. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Godfrey, V.; Liu, Z.; Han, Y.; Liu, L.; Peng, H.; Weichselbaum, R.R.; Zaki, H.; Fu, Y.X. The AIM2 and NLRP3 inflammasomes trigger IL-1-mediated antitumor effects during radiation. Sci. Immunol. 2021, 6, eabc6998. [Google Scholar] [CrossRef]
- Fang, H.; Ang, B.; Xu, X.; Huang, X.; Wu, Y.; Sun, Y.; Wang, W.; Li, N.; Cao, X.; Wan, T. TLR4 is essential for dendritic cell activation and anti-tumor T-cell response enhancement by DAMPs released from chemically stressed cancer cells. Cell Mol. Immunol. 2014, 11, 150–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalanxhi, E.; Meltzer, S.; Schou, J.V.; Larsen, F.O.; Dueland, S.; Flatmark, K.; Jensen, B.V.; Hole, K.H.; Seierstad, T.; Redalen, K.R.; et al. Systemic immune response induced by oxaliplatin-based neoadjuvant therapy favours survival without metastatic progression in high-risk rectal cancer. Br. J. Cancer 2018, 118, 1322–1328. [Google Scholar] [CrossRef] [Green Version]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Cantero-Cid, R.; Casas-Martin, J.; Hernandez-Jimenez, E.; Cubillos-Zapata, C.; Varela-Serrano, A.; Avendano-Ortiz, J.; Casarrubios, M.; Montalban-Hernandez, K.; Villacanas-Gil, I.; Guerra-Pastrian, L.; et al. PD-L1/PD-1 crosstalk in colorectal cancer: Are we targeting the right cells? BMC Cancer 2018, 18, 945. [Google Scholar] [CrossRef]
- Strasser, K.; Birnleitner, H.; Beer, A.; Pils, D.; Gerner, M.C.; Schmetterer, K.G.; Bachleitner-Hofmann, T.; Stift, A.; Bergmann, M.; Oehler, R. Immunological differences between colorectal cancer and normal mucosa uncover a prognostically relevant immune cell profile. Oncoimmunology 2019, 8, e1537693. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Ahlmanner, F.; Sundstrom, P.; Akeus, P.; Eklof, J.; Borjesson, L.; Gustavsson, B.; Lindskog, E.B.; Raghavan, S.; Quiding-Jarbrink, M. CD39(+) regulatory T cells accumulate in colon adenocarcinomas and display markers of increased suppressive function. Oncotarget 2018, 9, 36993–37007. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.H.; Lerner, H.; et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017, 20, 2921–2934. [Google Scholar] [CrossRef] [Green Version]
- Okla, K.; Farber, D.L.; Zou, W. Tissue-resident memory T cells in tumor immunity and immunotherapy. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Liang, F.; Rezapour, A.; Falk, P.; Angenete, E.; Yrlid, U. Cryopreservation of Whole Tumor Biopsies from Rectal Cancer Patients Enable Phenotypic and In Vitro Functional Evaluation of Tumor-Infiltrating T Cells. Cancers 2021, 13, 2428. [Google Scholar] [CrossRef]
- Peng, Q.; Qiu, X.; Zhang, Z.; Zhang, S.; Zhang, Y.; Liang, Y.; Guo, J.; Peng, H.; Chen, M.; Fu, Y.X.; et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 2020, 11, 4835. [Google Scholar] [CrossRef]
- Hu, W.; Sun, R.; Chen, L.; Zheng, X.; Jiang, J. Prognostic significance of resident CD103(+)CD8(+)T cells in human colorectal cancer tissues. Acta Histochem 2019, 121, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Hofving, T.; Liang, F.; Karlsson, J.; Yrlid, U.; Nilsson, J.A.; Nilsson, O.; Nilsson, L.M. The Microenvironment of Small Intestinal Neuroendocrine Tumours Contains Lymphocytes Capable of Recognition and Activation after Expansion. Cancers 2021, 13, 4305. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, F.; Rezapour, A.; Szeponik, L.; Alsén, S.; Wettergren, Y.; Bexe Lindskog, E.; Quiding-Järbrink, M.; Yrlid, U. Antigen Presenting Cells from Tumor and Colon of Colorectal Cancer Patients Are Distinct in Activation and Functional Status, but Comparably Responsive to Activated T Cells. Cancers 2021, 13, 5247. https://doi.org/10.3390/cancers13205247
Liang F, Rezapour A, Szeponik L, Alsén S, Wettergren Y, Bexe Lindskog E, Quiding-Järbrink M, Yrlid U. Antigen Presenting Cells from Tumor and Colon of Colorectal Cancer Patients Are Distinct in Activation and Functional Status, but Comparably Responsive to Activated T Cells. Cancers. 2021; 13(20):5247. https://doi.org/10.3390/cancers13205247
Chicago/Turabian StyleLiang, Frank, Azar Rezapour, Louis Szeponik, Samuel Alsén, Yvonne Wettergren, Elinor Bexe Lindskog, Marianne Quiding-Järbrink, and Ulf Yrlid. 2021. "Antigen Presenting Cells from Tumor and Colon of Colorectal Cancer Patients Are Distinct in Activation and Functional Status, but Comparably Responsive to Activated T Cells" Cancers 13, no. 20: 5247. https://doi.org/10.3390/cancers13205247
APA StyleLiang, F., Rezapour, A., Szeponik, L., Alsén, S., Wettergren, Y., Bexe Lindskog, E., Quiding-Järbrink, M., & Yrlid, U. (2021). Antigen Presenting Cells from Tumor and Colon of Colorectal Cancer Patients Are Distinct in Activation and Functional Status, but Comparably Responsive to Activated T Cells. Cancers, 13(20), 5247. https://doi.org/10.3390/cancers13205247