
Identification of MicroRNA–mRNA Networks in Melanoma and Their Association with PD-1 Checkpoint Blockade Outcomes

 , , ,
, , ,  , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
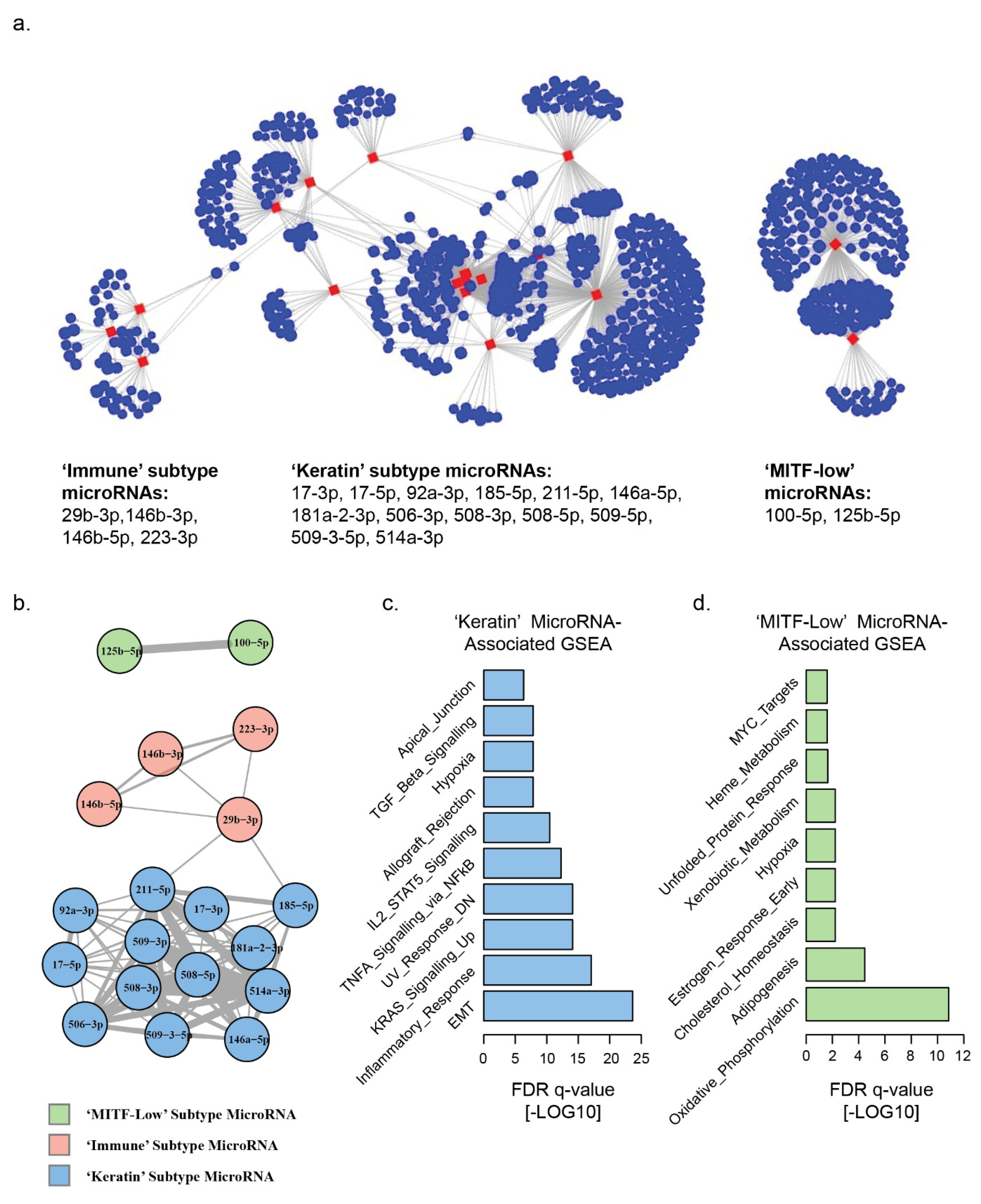
3.1. Landscape of MicroRNA–mRNA Associations in TCGA Melanomas
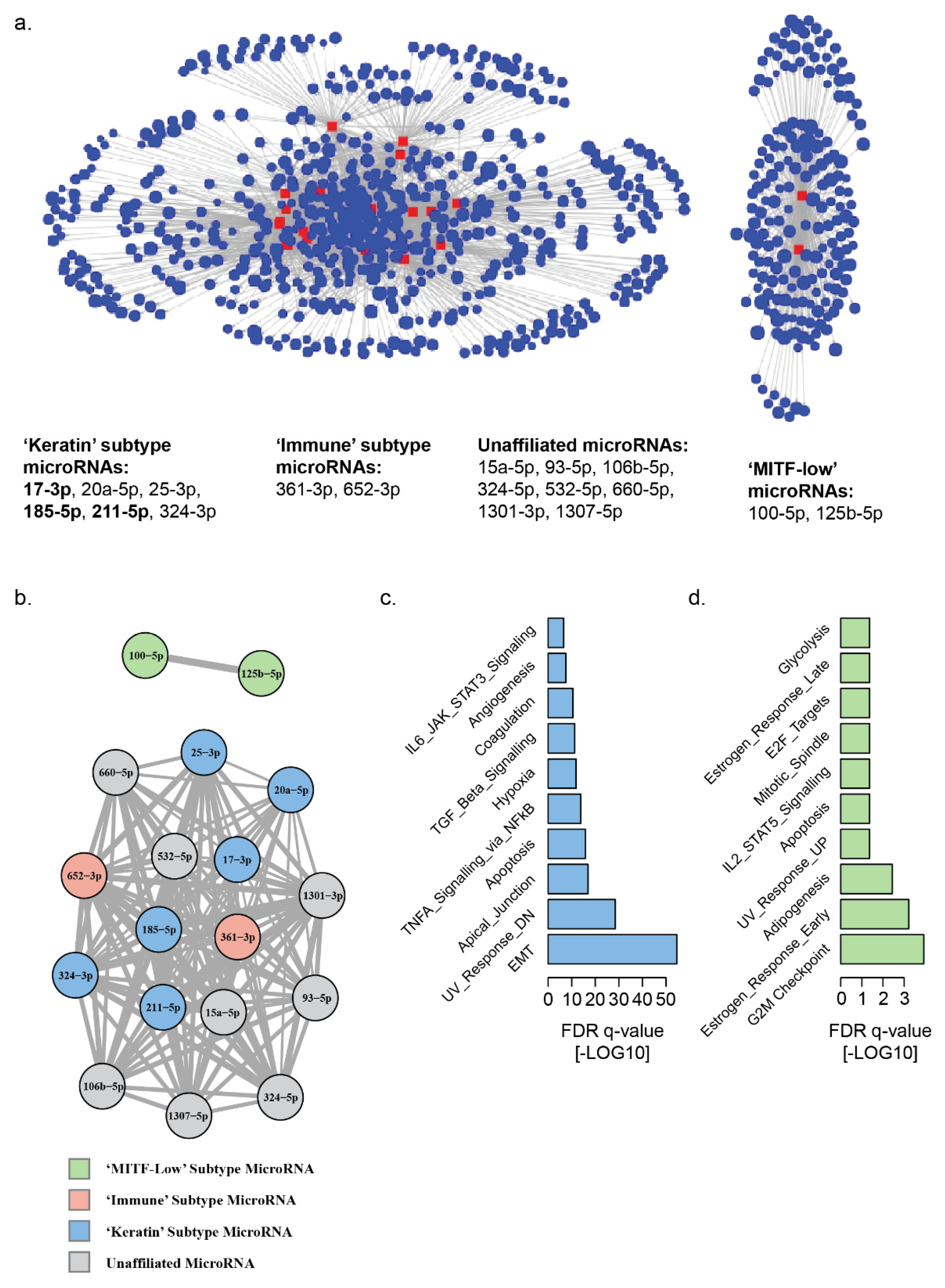
3.2. Landscape of MicroRNA–mRNA Associations in Patient Derived Melanoma Cell Lines
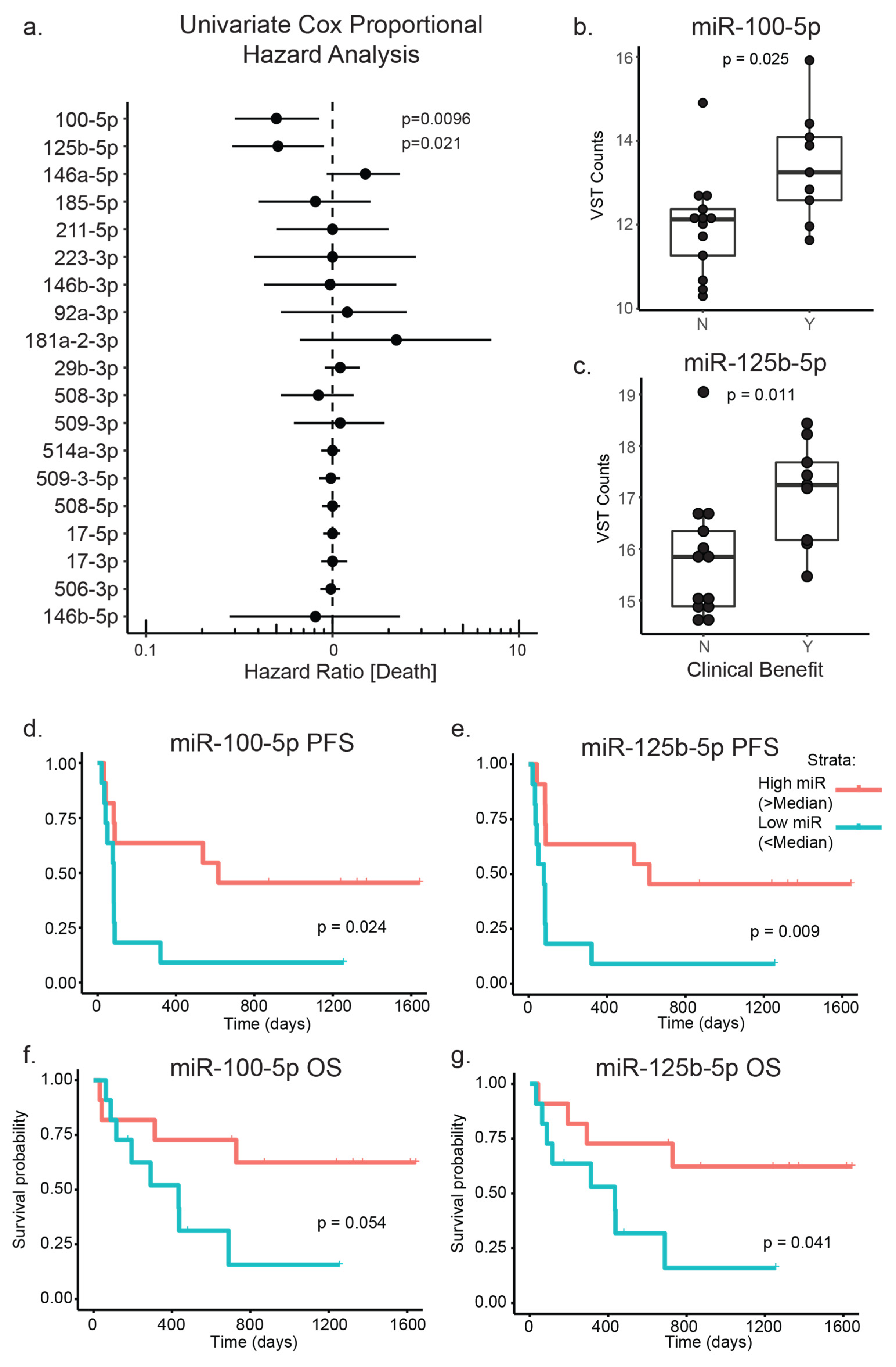
3.3. PD-1 Treated Patient Cohort
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data from Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [Green Version]
- Serrone, L.; Zeuli, M.; Sega, F.M.; Cognetti, F. Dacarbazine-based chemotherapy for metastatic melanoma: Thirty-year experience overview. J. Exp. Clin. Cancer Res. 2000, 19, 21–34. [Google Scholar] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearly, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilain, R.E.; Menzies, A.M.; Wilmott, J.S.; Kakavand, H.; Madore, J.; Guminski, A.; Liniker, E.; Kong, B.Y.; Cooper, A.J.; Howle, J.R.; et al. Dynamic Changes in PD-L1 Expression and Immune Infiltrates Early During Treatment Predict Response to PD-1 Blockade in Melanoma. Clin. Cancer Res. 2017, 23, 5024–5033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16. [Google Scholar] [CrossRef] [Green Version]
- Auslander, N.; Zhang, G.; Lee, J.S.; Frederick, D.T.; Miao, B.; Moll, T.; Tian, T.; Wei, Z.; Madan, S.; Sullivan, R.J.; et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med. 2018, 24, 1545–1549. [Google Scholar] [CrossRef]
- Prat, A.; Navarro, A.; Paré, L.; Reguart, N.; Galván, P.; Pascual, T.; Martínez, A.; Nuciforo, P.; Comerma, L.; Alos, L.; et al. Immune-Related Gene Expression Profiling After PD-1 Blockade in Non-Small Cell Lung Carcinoma, Head and Neck Squamous Cell Carcinoma, and Melanoma. Cancer Res. 2017, 77, 3540–3550. [Google Scholar] [CrossRef] [Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Tsoi, J.; Onyshchenko, M.; Abril-Rodriguez, G.; Ross-Macdonald, P.; Wind-Rotolo, M.; Champhekar, A.; Medina, E.; Torrejon, D.Y.; Shin, D.S.; et al. Conserved Interferon-γ Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 2020, 38, 500–515.e3. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.F.; Wei, W.; Gupta, S.; Smithy, J.W.; Zelterman, D.; Kluger, H.M.; Rimm, D.L. Multiplex quantitative analysis of cancer-associated fibroblasts and immunotherapy outcome in metastatic melanoma. J. Immunother. Cancer 2019, 7, 194. [Google Scholar] [CrossRef] [Green Version]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenetics 2019, 11, 25. [Google Scholar] [CrossRef]
- Stark, M.S.; Tyagi, S.; Nancarrow, D.J.; Boyle, G.M.; Cook, A.L.; Whiteman, D.C.; Parsons, P.G.; Schmidt, C.; Sturm, R.A.; Hayward, N.K. Characterization of the Melanoma miRNAome by Deep Sequencing. PLoS ONE 2010, 5, e9685. [Google Scholar] [CrossRef]
- Bonazzi, V.F.; Stark, M.S.; Hayward, N.K. MicroRNA regulation of melanoma progression. Melanoma Res. 2012, 22, 101–113. [Google Scholar] [CrossRef]
- Stark, M.S.; Klein, K.; Weide, B.; Haydu, L.E.; Pflugfelder, A.; Tang, Y.H.; Palmer, J.M.; Whiteman, D.C.; Scolyer, R.A.; Mann, G.J.; et al. The Prognostic and Predictive Value of Melanoma-related MicroRNAs Using Tissue and Serum: A MicroRNA Expression Analysis. EBioMedicine 2015, 2, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Caramuta, S.; Egyházi, S.; Rodolfo, M.; Witten, D.; Hansson, J.; Larsson, C.; Lui, W.-O. MicroRNA Expression Profiles Associated with Mutational Status and Survival in Malignant Melanoma. J. Investig. Dermatol. 2010, 130, 2062–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pencheva, N.; Tran, H.; Buss, C.; Huh, D.; Drobnjak, M.; Busam, K.; Tavazoie, S.F. Convergent Multi-miRNA Targeting of ApoE Drives LRP1/LRP8-Dependent Melanoma Metastasis and Angiogenesis. Cell 2012, 151, 1068–1082. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Yang, W. MiR-211 is epigenetically regulated by DNMT1 mediated methylation and inhibits EMT of melanoma cells by targeting RAB22A. Biochem. Biophys. Res. Commun. 2016, 476, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Messer, A.R.; Amitai-Lange, A.; Melamed, Z.; Ohana, R.; Bell, R.E.; Kapitansky, O.; Lerman, G.; Greenberger, S.; Khaled, M.; et al. Interactions of Melanoma Cells with Distal Keratinocytes Trigger Metastasis via Notch Signaling Inhibition of MITF. Mol. Cell 2015, 59, 664–676. [Google Scholar] [CrossRef] [Green Version]
- Vergani, E.; Di Guardo, L.; Dugo, M.; Rigoletto, S.; Tragni, G.; Ruggeri, R.; Perrone, F.; Tamborini, E.; Gloghini, A.; Arienti, F.; et al. Overcoming melanoma resistance to vemurafenib by targeting CCL2-induced miR-34a, miR-100 and miR-125b. Oncotarget 2016, 7, 4428–4441. [Google Scholar] [CrossRef] [Green Version]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Wang, F.; Kong, L.-Y.; Xu, S.; Doucette, T.; Ferguson, S.D.; Yang, Y.; McEnery, K.; Jethwa, K.; Gjyshi, O.; et al. miR-124 Inhibits STAT3 Signaling to Enhance T Cell–Mediated Immune Clearance of Glioma. Cancer Res. 2013, 73, 3913–3926. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Mastroianni, J.; Stickel, N.; Andrlova, H.; Hanke, K.; Melchinger, W.; Duquesne, S.; Schmidt, D.; Falk, M.; Andrieux, G.; Pfeifer, D.; et al. miR-146a Controls Immune Response in the Melanoma Microenvironment. Cancer Res. 2019, 79, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Nduom, E.K.; Kong, L.-Y.; Hashimoto, Y.; Xu, S.; Gabrusiewicz, K.; Ling, X.; Huang, N.; Qiao, W.; Zhou, S.; et al. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro-Oncology 2016, 18, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Wrzesinski, C.; Yu, Z.; Hu, J.; Gautam, S.; Hawk, N.V.; Telford, W.G.; Palmer, D.C.; Franco, Z.; Sukumar, M.; et al. miR-155 augments CD8+ T-cell antitumor activity in lymphoreplete hosts by enhancing responsiveness to homeostatic γc cytokines. Proc. Natl. Acad. Sci. USA 2015, 112, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Dudda, J.C.; Salaun, B.; Ji, Y.; Palmer, D.C.; Monnot, G.C.; Merck, E.; Boudousquie, C.; Utzschneider, D.T.; Escobar, T.M.; Perret, R.; et al. MicroRNA-155 Is Required for Effector CD8+ T Cell Responses to Virus Infection and Cancer. Immunity 2013, 38, 742–753. [Google Scholar] [CrossRef] [Green Version]
- Venza, I.; Visalli, M.; Beninati, C.; Benfatto, S.; Teti, D.; Venza, M. IL-10Rα expression is post-transcriptionally regulated by miR-15a, miR-185, and miR-211 in melanoma. BMC Med. Genom. 2015, 8, 81. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, L.; Fan, J.; Donye, D.; Dominguez, D.; Zhang, Y.; Curiel, T.J.; Fang, D.; Kuzel, T.M.; Zhang, B. Host miR155 Promotes Tumor Growth through a Myeloid-Derived Suppressor Cell–Dependent Mechanism. Cancer Res. 2015, 75, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Dragomir, M.; Mafra, A.C.P.; Dias, S.M.G.; Vasilescu, C.; Calin, G.A. Using microRNA Networks to Understand Cancer. Int. J. Mol. Sci. 2018, 19, 1871. [Google Scholar] [CrossRef] [Green Version]
- Oba, J.; Kim, S.-H.; Wang, W.-L.; Macedo, M.P.; Carapeto, F.; Mckean, M.A.; Van Arnam, J.; Eterovic, A.K.; Sen, S.; Kale, C.R.; et al. Targeting the HGF/MET Axis Counters Primary Resistance to KIT Inhibition in KIT-Mutant Melanoma. JCO Precis. Oncol. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Talukder, A.H.; Lim, S.A.; Kim, K.; Pan, K.; Melendez, B.; Bradley, S.D.; Jackson, K.R.; Khalili, J.S.; Wang, J.; et al. SLC45A2: A Melanoma Antigen with High Tumor Selectivity and Reduced Potential for Autoimmune Toxicity. Cancer Immunol. Res. 2017, 5, 618–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The igraph software package for complex network research. Available online: http://static1.squarespace.com/static/5b68a4e4a2772c2a206180a1/t/5cd1e3cbb208fc26c99de080/1557259212150/c1602a3c126ba822d0bc4293371c.pdf (accessed on 16 September 2021).
- Mazar, J.; Deyoung, K.; Khaitan, D.; Meister, E.; Almodovar, A.; Goydos, J.; Ray, A.; Perera, R.J. The Regulation of miRNA-211 Expression and Its Role in Melanoma Cell Invasiveness. PLoS ONE 2010, 5, e13779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duroux-Richard, I.; Roubert, C.; Ammari, M.; Présumey, J.; Grün, J.R.; Häupl, T.; Grützkau, A.; Lecellier, C.-H.; Boitez, V.; Codogno, P.; et al. miR-125b controls monocyte adaptation to inflammation through mitochondrial metabolism and dynamics. Blood 2016, 128, 3125–3136. [Google Scholar] [CrossRef] [Green Version]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF Regulates Oxidative Metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Dobbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Wei, W.; Robert, L.; Xue, M.; Tsoi, J.; Garcia-Diaz, A.; Moreno, B.H.; Kim, J.; Ng, R.; Lee, J.W.; et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc. Natl. Acad. Sci. USA 2017, 114, 13679–13684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, G.M.; Gopal, Y.N.V.; McQuade, J.L.; Peng, W.; DeBerardinis, R.J.; Davies, M.A. Metabolic strategies of melanoma cells: Mechanisms, interactions with the tumor microenvironment, and therapeutic implications. Pigment. Cell Melanoma Res. 2018, 31, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.-W.; Wentzel, E.A.; Mendell, J.T. Cell–cell contact globally activates microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 7016–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P.; et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5505–5516. [Google Scholar] [CrossRef] [Green Version]
- Fischer, G.M.; Jalali, A.; Kircher, D.A.; Lee, W.-C.; McQuade, J.L.; Haydu, L.E.; Joon, A.; Reuben, A.; De Macedo, M.P.; Carapeto, F.C.L.; et al. Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer Discov. 2019, 9, 628–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garutti, M.; Bonin, S.; Buriolla, S.; Bertoli, E.; Pizzichetta, M.A.; Zalaudek, I.; Puglisi, F. Find the Flame: Predictive Biomarkers for Immunotherapy in Melanoma. Cancers 2021, 13, 1819. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | PD-1 Inhibitor No Clinical Benefit (n = 13) | PD-1 Inhibitor Clinical Benefit (n = 9) |
|---|---|---|
| Sex | ||
| Male | 8 (62%) | 8 (89%) |
| Female | 5 (38%) | 1 (11%) |
| Melanoma Type | ||
| Cutaneous unspecified | 5 (38%) | 4 (44%) |
| Superficial spreading | 2 (15%) | - |
| Nodular | 4 (31%) | - |
| Acral lentiginous | 1 (8%) | 1 (11%) |
| Unknown primary | 1 (8%) | 4 (44%) |
| Disease State (AJCCv8) | ||
| IIIa/b | - | - |
| IIIc/d | 2 (15%) | 3 (33%) |
| IVa | 2 (15%) | - |
| IVb | 1 (8%) | - |
| IVc | 8 (62%) | 6 (67%) |
| IVd | - | - |
| Elevated Serum LDH (n (%) > ULN) | 6 (46%) | 5 (56%) |
| Prior Ipilimumab | ||
| Yes | 9 (69%) | 4 (44%) |
| No | 4 (31%) | 5 (56%) |
| Best Overall Response (BOR, RECIST 1.1) | ||
| CR | - | 3 (33%) |
| PR | - | 4 (44%) |
| SD | - | 2 (22%) |
| PD | 13 (100%) | - |
| PFS (median, range; days) | 78 (20–87) | 538 (321–NA) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sloane, R.A.S.; White, M.G.; Witt, R.G.; Banerjee, A.; Davies, M.A.; Han, G.; Burton, E.; Ajami, N.; Simon, J.M.; Bernatchez, C.; et al. Identification of MicroRNA–mRNA Networks in Melanoma and Their Association with PD-1 Checkpoint Blockade Outcomes. Cancers 2021, 13, 5301. https://doi.org/10.3390/cancers13215301
Sloane RAS, White MG, Witt RG, Banerjee A, Davies MA, Han G, Burton E, Ajami N, Simon JM, Bernatchez C, et al. Identification of MicroRNA–mRNA Networks in Melanoma and Their Association with PD-1 Checkpoint Blockade Outcomes. Cancers. 2021; 13(21):5301. https://doi.org/10.3390/cancers13215301
Chicago/Turabian StyleSloane, Robert A. Szczepaniak, Michael G. White, Russell G. Witt, Anik Banerjee, Michael A. Davies, Guangchun Han, Elizabeth Burton, Nadim Ajami, Julie M. Simon, Chantale Bernatchez, and et al. 2021. "Identification of MicroRNA–mRNA Networks in Melanoma and Their Association with PD-1 Checkpoint Blockade Outcomes" Cancers 13, no. 21: 5301. https://doi.org/10.3390/cancers13215301
APA StyleSloane, R. A. S., White, M. G., Witt, R. G., Banerjee, A., Davies, M. A., Han, G., Burton, E., Ajami, N., Simon, J. M., Bernatchez, C., Haydu, L. E., Tawbi, H. A., Gershenwald, J. E., Keung, E., Ross, M., McQuade, J., Amaria, R. N., Wani, K., Lazar, A. J., ... Wargo, J. A. (2021). Identification of MicroRNA–mRNA Networks in Melanoma and Their Association with PD-1 Checkpoint Blockade Outcomes. Cancers, 13(21), 5301. https://doi.org/10.3390/cancers13215301