
Nicotinic Acetylcholine Receptor Subunit α7 Mediates Cigarette Smoke-Induced PD-L1 Expression in Human Bronchial Epithelial Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. In Silico Gene Expression Analysis
2.2. Cell Culture
2.3. RNA Extraction and Quantitation of mRNA Expression
2.4. Protein Extraction and Immunoblot Analysis
2.5. Immunofluorescence Microscopy
2.6. Transient Transfection of Small Interfering RNA (siRNA)
2.7. Cignal 45-Pathway Reporter Arrays
2.8. Statistical Analyses
3. Results
3.1. CHRNA7 Expression Is Associated with Lung Cancer
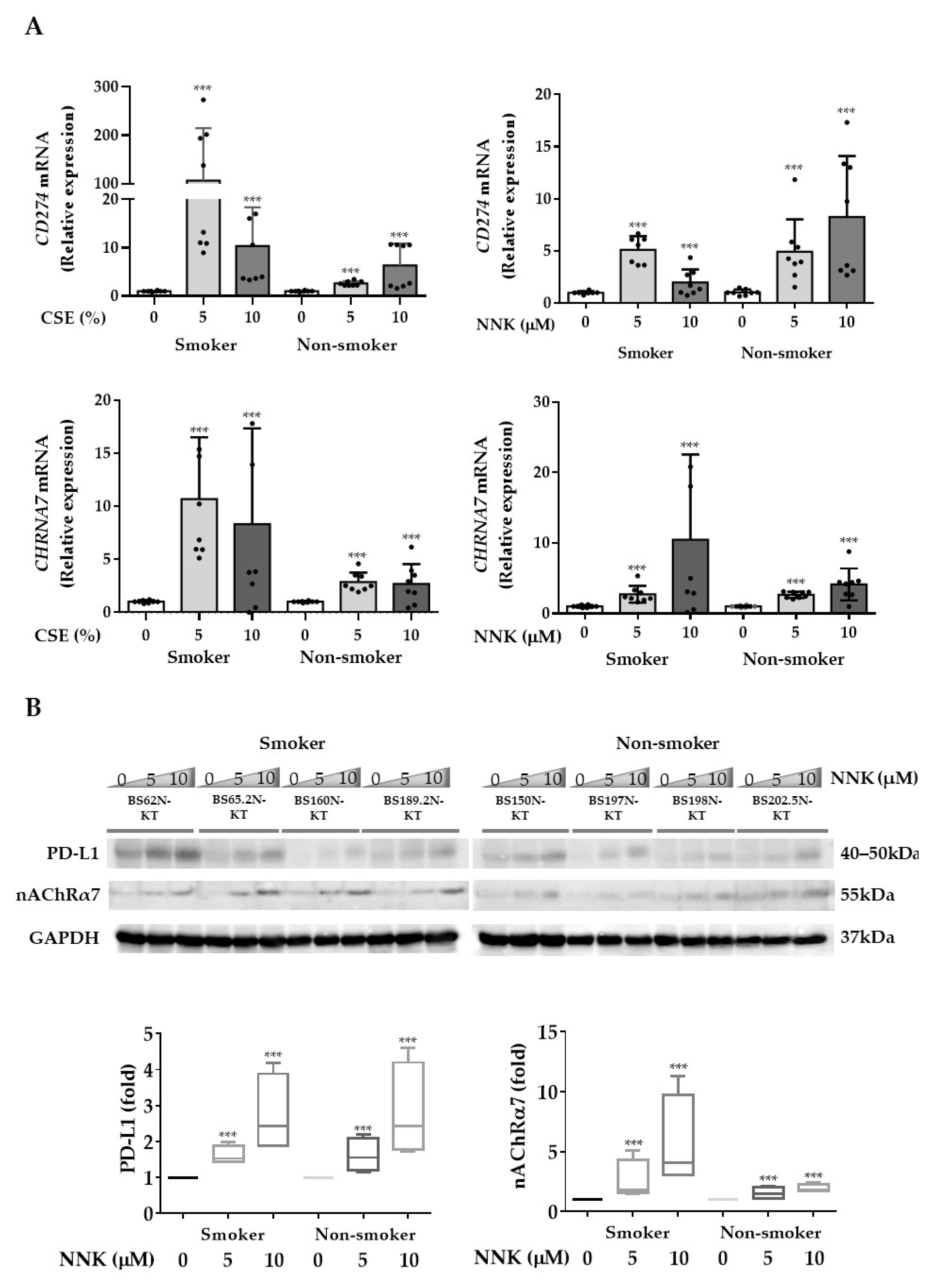
3.2. nAChRα7-Mediated Smoking-Induced PD-L1 Expression in HBECs
3.3. Antioxidant and STAT3 Pathways Were Involved in Mediating nAChRα7-Regulated PD-L1 Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hecht, S.S. Lung carcinogenesis by tobacco smoke. Int. J. Cancer 2012, 131, 2724–2732. [Google Scholar] [CrossRef] [Green Version]
- Khuder, S.A. Effect of cigarette smoking on major histological types of lung cancer: A meta-analysis. Lung Cancer 2001, 31, 139–148. [Google Scholar] [CrossRef]
- Hecht, S.S. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol. 1998, 11, 559–603. [Google Scholar] [CrossRef]
- Bosse, Y.; Amos, C.I. A Decade of GWAS Results in Lung Cancer. Cancer Epidemiol. Prev. Biomark. 2018, 27, 363–379. [Google Scholar] [CrossRef] [Green Version]
- Le Marchand, L.; Derby, K.S.; Murphy, S.E.; Hecht, S.S.; Hatsukami, D.; Carmella, S.G.; Tiirikainen, M.; Wang, H. Smokers with the CHRNA lung cancer-associated variants are exposed to higher levels of nicotine equivalents and a carcinogenic tobacco-specific nitrosamine. Cancer Res. 2008, 68, 9137–9140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Lu, X.; Qiu, F.; Fang, W.; Zhang, L.; Huang, D.; Xie, C.; Zhong, N.; Ran, P.; Zhou, Y.; et al. Duplicated copy of CHRNA7 increases risk and worsens prognosis of COPD and lung cancer. Eur. J. Hum. Genet. 2015, 23, 1019–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, D.C.; Luo, S.Y.; Fu, K.H.; Lui, M.M.; Chan, K.H.; Wistuba, I.I.; Gao, B.; Tsao, S.W.; Ip, M.S.; Minna, J.D. Nicotinic acetylcholine receptor expression in human airway correlates with lung function. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L232–L239. [Google Scholar] [CrossRef] [Green Version]
- Gahring, L.C.; Myers, E.J.; Dunn, D.M.; Weiss, R.B.; Rogers, S.W. Lung epithelial response to cigarette smoke and modulation by the nicotinic alpha 7 receptor. PLoS ONE 2017, 12, e0187773. [Google Scholar] [CrossRef]
- Mo, J.; Hu, X.; Gu, L.; Chen, B.; Khadaroo, P.A.; Shen, Z.; Dong, L.; Lv, Y.; Chitumba, M.N.; Liu, J. Smokers or non-smokers: Who benefits more from immune checkpoint inhibitors in treatment of malignancies? An up-to-date meta-analysis. World J. Surg. Oncol. 2020, 18, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.A.; Chae, Y.K.; Agte, S.; Pan, A.; Mohindra, N.A.; Villaflor, V.M.; Giles, F.J. Association of tumor mutational burden with smoking and mutation status in non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2017, 35, 24. [Google Scholar] [CrossRef]
- Norum, J.; Nieder, C. Tobacco smoking and cessation and PD-L1 inhibitors in non-small cell lung cancer (NSCLC): A review of the literature. ESMO Open 2018, 3, e000406. [Google Scholar] [CrossRef] [Green Version]
- Suresh, S.; Chen, B.; Zhu, J.; Golden, R.J.; Lu, C.; Evers, B.M.; Novaresi, N.; Smith, B.; Zhan, X.; Schmid, V.; et al. eIF5B drives integrated stress response-dependent translation of PD-L1 in lung cancer. Nat. Cancer 2020, 1, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Shay, J.W.; Minna, J.D. Immortalized normal human lung epithelial cell models for studying lung cancer biology. Respir. Investig. 2020, 58, 344–354. [Google Scholar] [CrossRef]
- Cai, L.; Lin, S.; Girard, L.; Zhou, Y.; Yang, L.; Ci, B.; Zhou, Q.; Luo, D.; Yao, B.; Tang, H.; et al. LCE: An open web portal to explore gene expression and clinical associations in lung cancer. Oncogene 2019, 38, 2551–2564. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Ramirez, R.D.; Sheridan, S.; Girard, L.; Sato, M.; Kim, Y.; Pollack, J.; Peyton, M.; Zou, Y.; Kurie, J.M.; Dimaio, J.M.; et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004, 64, 9027–9034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thai, P.; Statt, S.; Chen, C.H.; Liang, E.; Campbell, C.; Wu, R. Characterization of a Novel Long Noncoding RNA, SCAL1, Induced by Cigarette Smoke and Elevated in Lung Cancer Cell Lines. Am. J. Respir. Cell Mol. Biol. 2013, 49, 204–211. [Google Scholar] [CrossRef] [Green Version]
- Boo, H.-J.; Min, H.-Y.; Jang, H.-J.; Yun, H.J.; Smith, J.K.; Jin, Q.; Lee, H.-J.; Liu, D.; Kweon, H.-S.; Behrens, C.; et al. The tobacco-specific carcinogen-operated calcium channel promotes lung tumorigenesis via IGF2 exocytosis in lung epithelial cells. Nat. Commun. 2016, 7, 12961. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.C.; Kwok, H.H.; Yu, W.C.; Ko, F.W.; Tam, C.Y.; Lau, A.C.; Fong, D.Y.; Ip, M.S. CC16 levels correlate with cigarette smoke exposure in bronchial epithelial cells and with lung function decline in smokers. BMC Pulm. Med. 2018, 18, 47. [Google Scholar] [CrossRef] [Green Version]
- Solis, L.M.; Behrens, C.; Dong, W.; Suraokar, M.; Ozburn, N.C.; Moran, C.A.; Corvalan, A.H.; Biswal, S.; Swisher, S.G.; Bekele, B.N.; et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 2010, 16, 3743–3753. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef] [Green Version]
- Krysan, K.; Tran, L.M.; Grimes, B.S.; Fishbein, G.A.; Seki, A.; Gardner, B.K.; Walser, T.C.; Salehi-Rad, R.; Yanagawa, J.; Lee, J.M.; et al. The Immune Contexture Associates with the Genomic Landscape in Lung Adenomatous Premalignancy. Cancer Res. 2019, 79, 5022–5033. [Google Scholar] [CrossRef]
- Dave, K.; Ali, A.; Magalhaes, M. Increased expression of PD-1 and PD-L1 in oral lesions progressing to oral squamous cell carcinoma: A pilot study. Sci. Rep. 2020, 10, 9705. [Google Scholar] [CrossRef]
- Wang, J.; Xie, T.; Wang, B.; William, W.N.; Heymach, J.V.; El-Naggar, A.K.; Myers, J.N.; Caulin, C. PD-1 Blockade Prevents the Development and Progression of Carcinogen-Induced Oral Premalignant Lesions. Cancer Prev. Res. 2017, 10, 684–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinkus, M.L.; Graw, S.; Freedman, R.; Ross, R.G.; Lester, H.A.; Leonard, S. The human CHRNA7 and CHRFAM7A genes: A review of the genetics, regulation, and function. Neuropharmacology 2015, 96, 274–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, P.; Bonassi, S.; Giacconi, R.; Malavolta, M.; Tomino, C.; Maggi, F. COVID-19 and smoking: Is nicotine the hidden link? Eur. Respir. J. 2020, 55, 2001116. [Google Scholar] [CrossRef]
- Olds, J.L.; Kabbani, N. Is nicotine exposure linked to cardiopulmonary vulnerability to COVID-19 in the general population? FEBS J. 2020, 287, 3651–3655. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Hu, Y. α7 nicotinic acetylcholine receptors in lung cancer. Oncol. Lett. 2018, 16, 1375–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.D.; Liao, Y.C.; Ho, Y.S.; Chen, L.C.; Chang, H.W.; Cheng, T.C.; Liu, D.; Lee, W.R.; Shen, S.C.; Wu, C.H.; et al. The α9 Nicotinic Acetylcholine Receptor Mediates Nicotine-Induced PD-L1 Expression and Regulates Melanoma Cell Proliferation and Migration. Cancers 2019, 11, 1991. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Tang, L.; Chen, S.; Yin, C.; Peng, S.; Li, X.; Liu, T.; Liu, W.; Han, C.; Stawski, L.; et al. Targeting the upstream transcriptional activator of PD-L1 as an alternative strategy in melanoma therapy. Oncogene 2018, 37, 4941–4954. [Google Scholar] [CrossRef]
- Best, S.A.; De Souza, D.P.; Kersbergen, A.; Policheni, A.N.; Dayalan, S.; Tull, D.; Rathi, V.; Gray, D.H.; Ritchie, M.E.; McConville, M.J.; et al. Synergy between the KEAP1/NRF2 and PI3K Pathways Drives Non-Small-Cell Lung Cancer with an Altered Immune Microenvironment. Cell Metab. 2018, 27, 935–943.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.-Z.; Zhang, L.; Zhao, X.-C.; Gao, S.-H.; Qu, L.-W.; Yu, H.; Fang, W.-F.; Zhou, Y.-C.; Liang, F.; Zhang, C.; et al. The Aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat. Commun. 2019, 10, 1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desrichard, A.; Kuo, F.; Chowell, D.; Lee, K.-W.; Riaz, N.; Wong, R.J.; Chan, T.A.; Morris, L.G.T. Tobacco Smoking-Associated Alterations in the Immune Microenvironment of Squamous Cell Carcinomas. J. Natl. Cancer Inst. 2018, 110, 1386–1392. [Google Scholar] [CrossRef]
- Wu, C.-H.; Lee, C.-H.; Ho, Y.-S. Nicotinic Acetylcholine Receptor-Based Blockade: Applications of Molecular Targets for Cancer Therapy. Clin. Cancer Res. 2011, 17, 3533–3541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajiasgharzadeh, K.; Somi, M.H.; Sadigh-Eteghad, S.; Mokhtarzadeh, A.; Shanehbandi, D.; Mansoori, B.; Mohammadi, A.; Doustvandi, M.A.; Baradaran, B. The dual role of alpha7 nicotinic acetylcholine receptor in inflammation-associated gastrointestinal cancers. Heliyon 2020, 6, e03611. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwok, H.-H.; Gao, B.; Chan, K.-H.; Ip, M.S.-M.; Minna, J.D.; Lam, D.C.-L. Nicotinic Acetylcholine Receptor Subunit α7 Mediates Cigarette Smoke-Induced PD-L1 Expression in Human Bronchial Epithelial Cells. Cancers 2021, 13, 5345. https://doi.org/10.3390/cancers13215345
Kwok H-H, Gao B, Chan K-H, Ip MS-M, Minna JD, Lam DC-L. Nicotinic Acetylcholine Receptor Subunit α7 Mediates Cigarette Smoke-Induced PD-L1 Expression in Human Bronchial Epithelial Cells. Cancers. 2021; 13(21):5345. https://doi.org/10.3390/cancers13215345
Chicago/Turabian StyleKwok, Hoi-Hin, Boning Gao, Koon-Ho Chan, Mary Sau-Man Ip, John Dorrance Minna, and David Chi-Leung Lam. 2021. "Nicotinic Acetylcholine Receptor Subunit α7 Mediates Cigarette Smoke-Induced PD-L1 Expression in Human Bronchial Epithelial Cells" Cancers 13, no. 21: 5345. https://doi.org/10.3390/cancers13215345
APA StyleKwok, H. -H., Gao, B., Chan, K. -H., Ip, M. S. -M., Minna, J. D., & Lam, D. C. -L. (2021). Nicotinic Acetylcholine Receptor Subunit α7 Mediates Cigarette Smoke-Induced PD-L1 Expression in Human Bronchial Epithelial Cells. Cancers, 13(21), 5345. https://doi.org/10.3390/cancers13215345