
Molecular-Genetic Portrait of Breast Cancer with Triple Negative Phenotype


Abstract
:Simple Summary
Abstract
1. Introduction
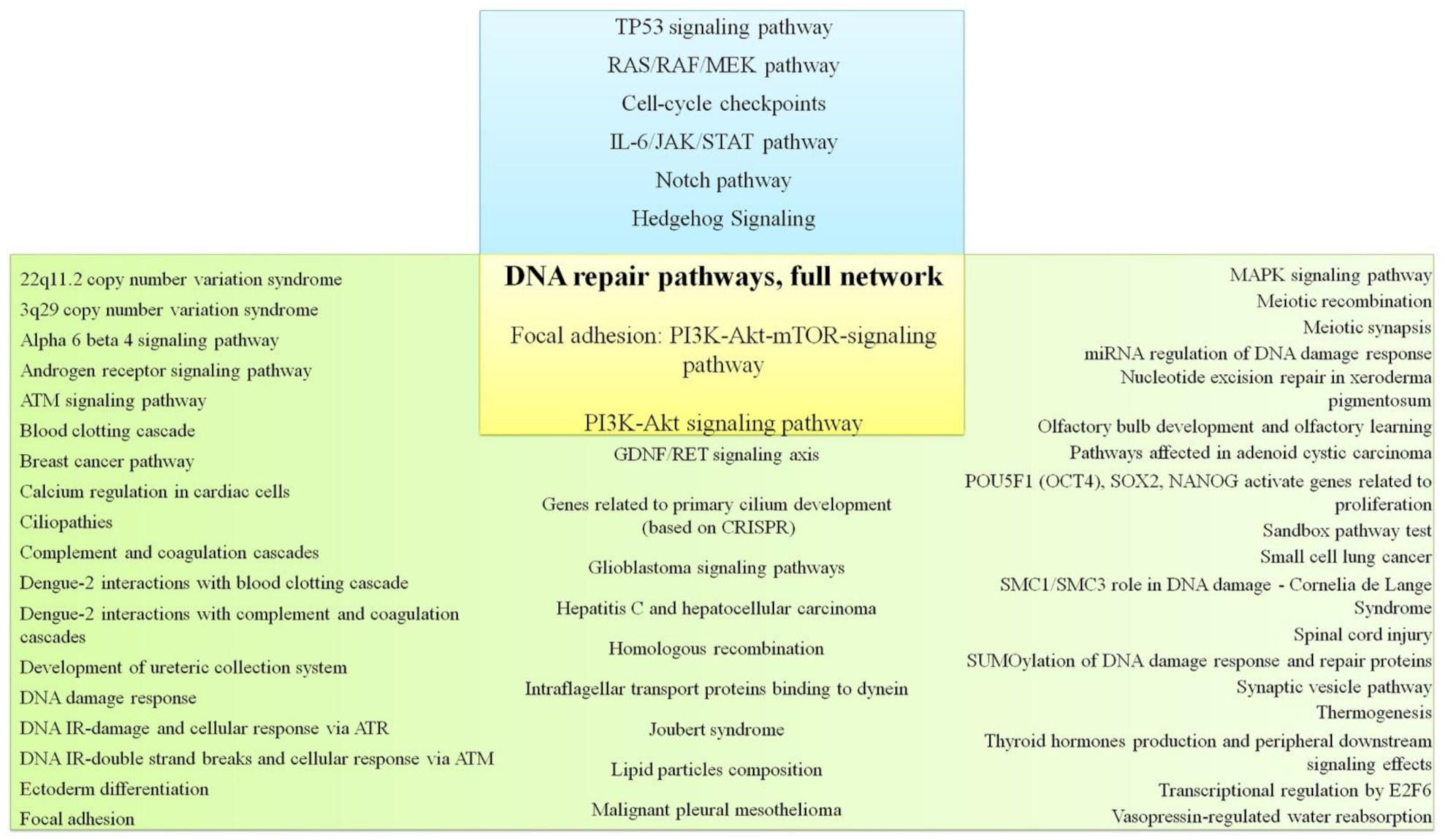
2. Molecular-Genetic Mechanisms Presenting TNBC
2.1. DNA Repair Pathway
2.2. TP53 Signaling Pathway
2.3. PI3K/AKT/mTOR Pathway
2.4. RAS/MAPK, RAS/RAF/MEK Pathways
2.5. Cell-Cycle Checkpoints
2.6. IL-6/JAK/STAT Pathway
2.7. NOTCH Pathway
2.8. Hedgehog Signaling
3. Molecular Markers for Immunotherapy of TNBC
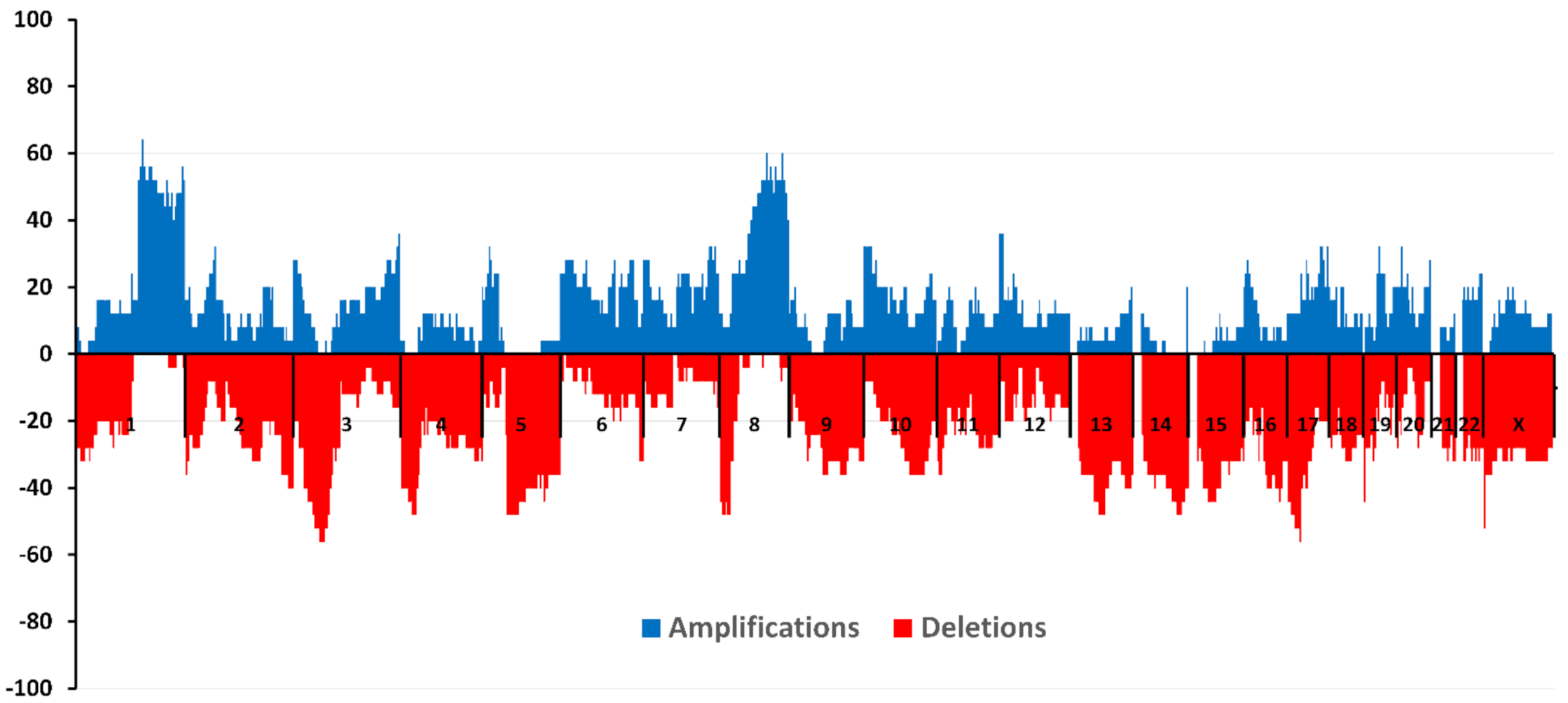
4. CNA and TN Breast Cancer
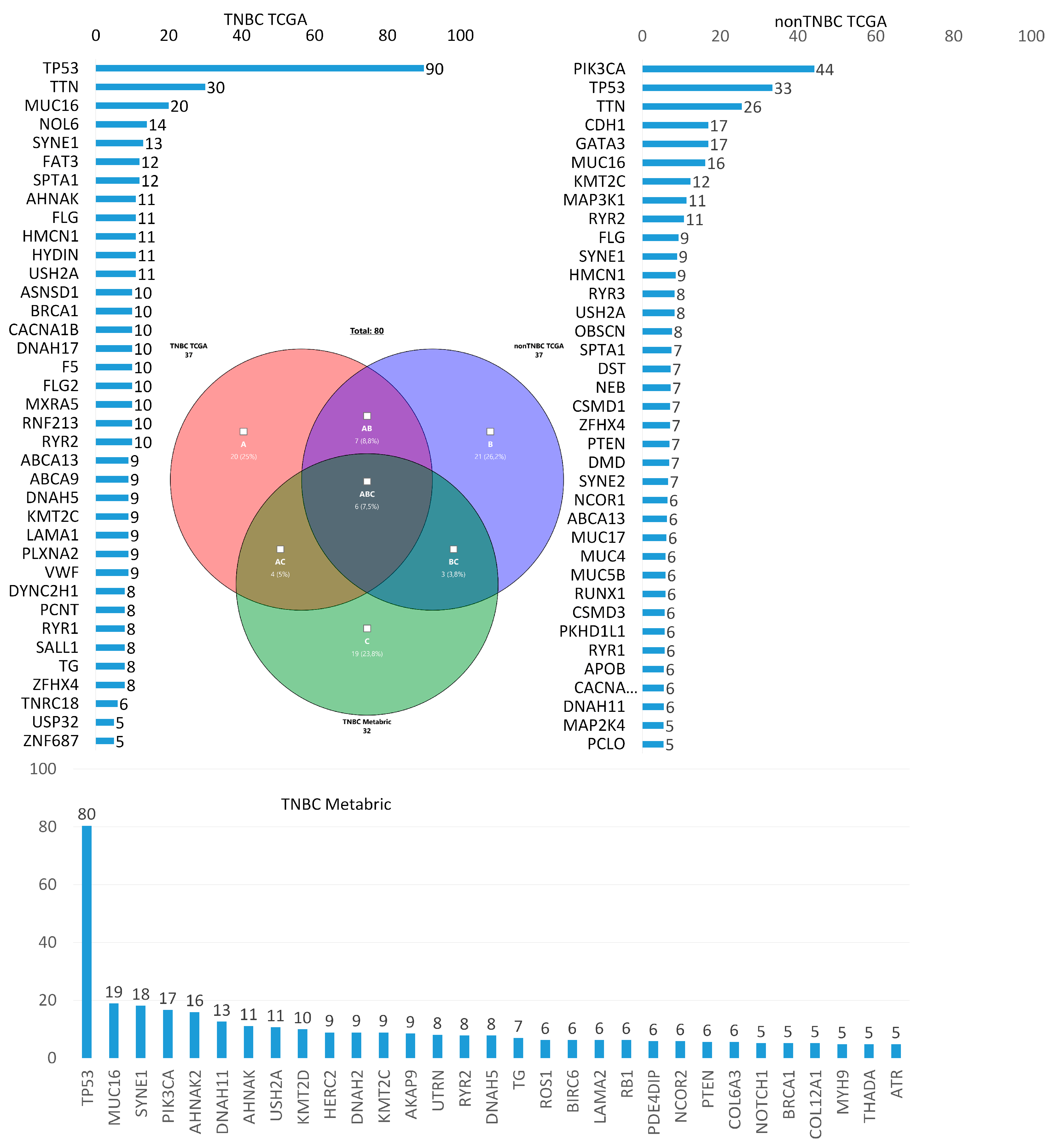
5. Own Research
Ethics Approval and Consent to Participate
6. CNA and TNBC: Relationship with Hematogenous Metastasis
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M. Molecular stratification of triple-negative breast cancers. Oncologist 2011, 16 (Suppl. Sl1), 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Xue, X.; Hu, C.; Xu, H.; Kou, D.; Li, R.; Li, M. Comparison of clinicopathological features and prognosis in triple-negative and non-triple negative breast cancer. J. Cancer 2016, 7, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Semiglazov, V.F. Breast cancer: Multidisciplinary approach to treatment. Pract. Oncol. 2015, 16, 49–54. [Google Scholar]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoajuvant therapy and long-term survival in patients with triple negative breast cancer. J. Clin. Oncol. 2008, 6, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [Green Version]
- Yuan, N.; Meng, M.; Liu, C.; Feng, L.; Hou, L.; Ning, Q.; Xin, G.; Pei, L.; Gu, S.; Li, X.; et al. Clinical characteristics and prognostic analysis of triple-negative breast cancer patients. Mol. Clin. Oncol. 2014, 2, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Engebraaten, O.; Vollan, H.K.; Børresen-Dale, A.-L. Triple-negative breast cancer and the need for new therapeutic targets. Am. J. Pathol. 2013, 183, 1064–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Fan, M.; Xie, J.; Wang, Z.; Wang, B.; Zhang, S.; Wang, L.; Cao, J.; Tao, Z.; Li, T.; et al. Chemotherapy of metastatic triple negative breast cancer: Experience of using platinum-based chemotherapy. Oncotarget 2015, 6, 43135–43143. [Google Scholar] [CrossRef]
- Audeh, M.W. Novel treatment strategies in triple-negative breast cancer: Specific role of poly(adenosine diphosphate-ribose) polymerase inhibition. Pharmgenomics Pers. Med. 2014, 7, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Crozier, J.A.; Advani, P.P.; LaPlant, B.; Hobday, T.; Jaslowski, A.J.; Moreno-Aspitia, A.; Perez, E.A. N0436 (Alliance): A Phase II Trial of Irinotecan with Cetuximab in Patients With Metastatic Breast Cancer Previously Exposed to Anthracycline and/or Taxane-Containing Therapy. Clin. Breast Cancer 2016, 16, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Nabholtz, J.M.; Chalabi, N.; Radosevic-Robin, N.; Dauplat, M.M.; Mouret-Reynier, M.A.; Van Praagh, I.; Servent, V.; Jacquin, J.P.; Benmammar, K.E.; Kullab, S.; et al. Multicentric neoadjuvant pilot Phase II study of cetuximab combined with docetaxel in operable triple negative breast cancer. Int. J. Cancer 2016, 138, 2274–2280. [Google Scholar] [CrossRef]
- Yadav, B.S.; Sharma, S.C.; Chanana, P.; Jhamb, S. Systemic treatment strategies for triple-negative breast cancer. World J. Clin. Oncol. 2014, 5, 125–133. [Google Scholar] [CrossRef]
- Di Desidero, T.; Xu, P.; Man, S.; Bocci, G.; Kerbel, R.S. Potent efficacy of metronomic topotecan and pazopanib combination therapy in preclinical models of primary or late stage metastatic triple-negative breast cancer. Oncotarget 2015, 6, 42396–42410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, U.D.; Castillo, A.F.; Dattilo, M.A.; Solano, A.R.; Maloberti, P.M.; Podesta, E.J. Acyl-CoA synthetase-4, a new regulator of mTOR and a potential therapeutic target for enhanced estrogen receptor function in receptor-positive and -negative breast cancer. Oncotarget 2015, 6, 42632–42650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.H.; Jung, H.H.; Ahn, J.S.; Im, Y.-H. Statin induces inhibition of triple negative breast cancer (TNBC) cells via PI3K pathway. Biochem. Biophys. Res. Commun. 2013, 439, 275–279. [Google Scholar] [CrossRef]
- Yang, T.; Yao, H.; He, G.; Song, L.; Liu, N.; Wang, Y.; Yang, Y.; Keller, E.T.; Deng, X. Effects of Lovastatin on MDA-MB-231 Breast Cancer Cells: An Antibody Microarray Analysis. J. Cancer 2016, 7, 192–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semiglazov, V.F.; Paltuev, R.M.; Semiglazov, V.V.; Dashyan, G.A.; Semiglazova, T.Y.; Krivorotko, P.V.; Nikolaev, K.S. General recommendations for the treatment of early breast cancer St. Gallen-2015, adapted by experts from the Russian Society of Mammological Oncology. Tumors Female Reprod. Syst. 2015, 11, 43–60. [Google Scholar] [CrossRef] [Green Version]
- Mersin, H.; Yildirim, E.; Berberoglu, U.; Gülben, K. The prognostic importance of triple negative breast carcinoma. Breast 2008, 17, 341–346. [Google Scholar] [CrossRef]
- Yang, C.-Q.; Liu, J.; Shi-Qi, Z.; Kun, Z.; Zi-Qian, G.; Ran, X.; Hui-Meng, L.; Ren-Bin, Z.; Gang, Z.; Da-Chuan, Y.; et al. Recent treatment progress of triple negative breast cancer. Prog. Biophys. Mol. Biol. 2020, 151, 40–53. [Google Scholar]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Frederick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Schafer, J.M.; Pendleton, C.S.; Tang, L.; Johnson, K.C.; Chen, X.; Balko, J.M.; Gómez, H.; Arteaga, C.L.; et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014, 16, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Manié, E.; Popova, T.; Battistella, A.; Tarabeux, J.; Caux-Moncoutier, V.; Golmard, L.; Smith, N.K.; Mueller, C.R.; Mariani, O.; Sigal-Zafrani, B.; et al. Genomic hallmarks of homologous recombination deficiency in invasive breast carcinomas. Cancer Genet. Epigenetics 2016, 138, 891–900. [Google Scholar] [CrossRef] [Green Version]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Wu, Y.H.; Hong, C.W.; Wang, Y.-C.; Huang, W.-J.; Yeh, Y.-L.; Wang, B., Jr.; Wang, Y.-J.; Chiu, H.-W. A novel histone deacetylase inhibitor TMU-35435 enhances etoposide cytotoxicity through the proteasomal degradation of DNA-PKcs in triple-negative breast cancer. Cancer Lett. 2017, 400, 79–88. [Google Scholar] [CrossRef]
- Lin, M.Z.; Martin, J.L.; Martin, J.L.; Baxter, R.C. The role of insulin-like growth factor binding protein-3 in the breast cancer cell response to DNA-damaging agents. Oncogene 2012, 33, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Silva, H.C.; Lin, M.Z.; Phillips, L.; Martin, J.L.; Baxter, R.C. IGFBP-3 interacts with NONO and SFPQ in PARP-dependent DNA damage repair in triple-negative breast cancer. Cell. Mol. Life Sci. 2019, 76, 2015–2030. [Google Scholar] [CrossRef]
- Ribeiro, E.; Ganzinelli, M.; Andreis, D.; Bertoni, R.; Giardini, R.; Fox, S.B.; Broggini, M.; Bottini, A.; Zanoni, V.; Bazzola, L.; et al. Triple Negative Breast Cancers Have a Reduced Expression of DNA Repair Genes. PLoS ONE 2013, 8, e66243. [Google Scholar] [CrossRef] [Green Version]
- Ollier, M.; Radosevic-Robin, N.; Kwiatkowski, F.; Ponelle, F.; Viala, S.; Privat, M.; Uhrhammer, N.; Bernard-Gallon, D.; Penault-Llorca, F.; Bignon, Y.-J.; et al. DNA repair genes implicated in triple negative familial non-BRCA1/2 breast cancer predisposition. Am. J. Cancer Res. 2015, 5, 2113–2126. [Google Scholar]
- Turner, N.; Moretti, E.; Siclari, O.; Migliaccio, I.; Santarpia, L.; D’Incalci, M.; Piccolo, S.; Veronesi, A.; Zambelli, A.; Del Sal, G.; et al. Targeting triple negative breast cancer: Is p53 the answer? Cancer Treat. Rev. 2013, 39, 541–550. [Google Scholar] [CrossRef]
- Shi, H.; Tan, S.J.; Zhong, H.; Hu, W.; Levine, A.; Xiao, C.J.; Peng, Y.; Qi, X.B.; Shou, W.H.; Run-lin, Z.M.; et al. Winter temperature and UV are tightly linked to genetic changes in the p53 tumor suppressor pathway in Eastern Asia. Am. J. Hum. Genet. 2009, 84, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Cui, J. Dynamics of P53 in response to DNA damage: Mathematical modeling and perspective. Prog. Biophys. Mol. Biol. 2015, 119, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Cossu-Rocca, P.; Orrù, S.; Muroni, M.R.; Sanges, F.; Sotgiu, G.; Ena, S.; Pira, G.; Murgia, L.; Manca, A.; Uras, M.G.; et al. Analysis of PIK3CA mutations and activation pathways in triple negative breast cancer. PLoS ONE 2015, 10, e0141763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooms, L.M.; Binge, L.C.; Davies, E.M.; Rahman, P.; Conway, J.R.W.; Gurung, R.; Ferguson, D.T.; Papa, A.; Fedele, C.G.; Vieusseux, J.L.; et al. The inositol polyphosphate 5-phosphatase PIPP regulates AKT1-dependent breast cancer growth and metastasis. Cancer Cell 2015, 28, 155–169. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Murphy, C.J.; Karreth, F.A.; Emdal, K.B.; White, F.M.; Elemento, O.; Toker, A.; Wulf, G.M.; Cantley, L.C. Identifying and targeting sporadic oncogenic genetic aberrations in mouse models of triple negative breast cancer. Cancer Discov. 2017, 8, 354–371. [Google Scholar] [CrossRef] [Green Version]
- Deng, M.; Wang, J.; Chen, Y.; Zhang, L.; Liu, D. Combination of SF1126 and gefitinib induces apoptosis of triple-negative breast cancer cells through the PI3K/AKTemTOR pathway. Anti-Cancer Drugs 2015, 26, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.R.; Yoshida, T.; Marusyk, A.; Beck, A.H.; Polyak, K.; Toker, A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014, 74, 964–973. [Google Scholar] [CrossRef] [Green Version]
- Ueng, S.H.; Chen, S.C.; Chang, Y.-S.; Hsueh, S.; Lin, Y.-C.; Chien, H.-P.; Lo, Y.-F.; Shen, S.-C.; Hsueh, C. Phosphorylated mTOR expression correlates with poor outcome in early-stage triple negative breast carcinomas. Int. J. Clin. Exp. Pathol. 2012, 5, 806–813. [Google Scholar]
- Ripple, M.O.; Kim, N.; Springett, R. Acute mitochondrial inhibition by mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK) 1/2 inhibitors regulates proliferation. J. Biol. Chem. 2013, 288, 2933–2940. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.W.; O’Shaughnessy, J.A.; Kiefer, J.A.; Aldrich, J.; Sinari, S.; Moses, T.M.; Wong, S.; Dinh, J.; Christoforides, A.; Blum, J.L.; et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol. Cancer Ther. 2013, 12, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Balko, J.M.; Cook, R.S.; Vaught, D.B.; Kuba, M.G.; Miller, T.W.; Bhola, N.E.; Sanders, M.E.; Granja-Ingram, N.M.; Smith, J.J.; Meszoely, I.M.; et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat. Med. 2012, 18, 1052–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Ding, H.; Liu, C.; Li, X.; Wei, L.; Yu, J.; Liu, M.; Ying, M.; Gao, W.; Jiang, H.; et al. A novel recurrent CHEK2 Y390C mutation identified in high-risk Chinese breast cancer patients impairs its activity and is associated with increased breast cancer risk. Oncogene 2015, 34, 5198–5205. [Google Scholar] [CrossRef]
- Heijink, A.M.; Everts, M.; Honeywell, M.E.; Richards, R.; Kok, Y.P.; de Vries, E.G.E.; Lee, M.J.; van Vugt, M.A.T.M. Modeling of Cisplatin-Induced Signaling Dynamics in Triple-Negative Breast Cancer Cells Reveals Mediators of Sensitivity. Cell Rep. 2019, 28, 2345–2357. [Google Scholar] [CrossRef] [Green Version]
- Pouliot, L.M.; Chen, Y.C.; Bai, J.; Guha, R.; Martin, S.E.; Gottesman, M.M.; Hall, M.D. Cisplatin sensitivity mediated by WEE1 and CHK1 is mediated by miR-155 and the miR-15 family. Cancer Res. 2012, 72, 5945–5955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Gao, W.; Wang, J.; Wang, D.; Peng, X.; Jia, Z.; Jiang, Y.; Li, G.; Tang, D.; Wang, Y. Study on the Mechanism of Cell Cycle Checkpoint Kinase 2 (CHEK2) Gene Dysfunction in Chemotherapeutic Drug Resistance of Triple Negative Breast Cancer Cells. Med. Sci. Monit. 2018, 24, 3176–3183. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Litchfield, L.M.; Webster, Y.; Chio, L.-C.; Wong, S.S.; Stewart, T.R.; Dowless, M.; Dempsey, J.; Zeng, Y.; Torres, R.; et al. Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell 2017, 32, 761–776. [Google Scholar] [CrossRef] [Green Version]
- Condorelli, R.; Spring, L.; O’Shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.J.; Lanman, R.B.; Iafrate, A.J.; et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol. 2018, 29, 640–645. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Hu, Y.; Gao, J.; Wang, M.; Li, M. Potential Prospect of CDK4/6 Inhibitors in Triple-Negative Breast Cancer. Cancer Manag. Res. 2021, 13, 5223–5237. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24− stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Utama, F.E.; Lin, J.; Yang, N.; Sjolund, A.B.; Ryder, A.; Johnson, K.J.; Neilson, L.M.; Liu, C.; Brill, K.L.; et al. Prolactin inhibits BCL6 expression in breast cancer through a Stat5a-dependent mechanism. Cancer Res. 2010, 70, 1711–1721. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Herrmann, A.; Reckamp, K.; Zhang, W.; Pal, S.; Hedvat, M.; Zhang, C.; Liang, W.; Scuto, A.; Weng, S.; et al. Anti-angiogenic and anti-metastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011, 71, 6601–6610. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.-M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.A.; Iyer, M.; Maher, C.A.; Grasso, C.S.; et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 2011, 17, 1646–1651. [Google Scholar] [CrossRef] [Green Version]
- Kontomanolis, E.; Panteliadou, M.; Giatromanolaki, A.; Pouliliou, S.; Efremidou, E.; Limberis, V.; Galazios, G.; Sivridis, E.; Koukourakis, M.I. Delta-like ligand 4 (DLL4) in the plasma and neoplastic tissues from breast cancer patients: Correlation with metastasis. Med. Oncol. 2014, 31, 945. [Google Scholar] [CrossRef]
- Speiser, J.; Foreman, K.; Drinka, E.; Godellas, C.; Perez, C.; Salhadar, A.; Erşahin, Ç.; Rajan, P. Notch-1 and Notch-4 biomarker expression in triple-negative breast cancer. Int. J. Surg. Pathol. 2012, 20, 139–145. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Davis, N.M.; Abrams, S.L.; Montalto, G.; Cervello, M.; Libra, M.; Nicoletti, F.; D’Assoro, A.B.; Cocco, L.; Martelli, A.M.; et al. Targeting breast cancer initiating cells: Advances in breast cancer research and therapy. Adv. Biol. Regul. 2014, 56, 81–107. [Google Scholar] [CrossRef]
- Suman, S.; Das, T.P.; Damodaran, C. Silencing NOTCH signaling causes growth arrest in both breast cancer stem cells and breast cancer cells. Br. J. Cancer 2013, 109, 2587–2596. [Google Scholar] [CrossRef]
- Nagamatsu, I.; Onishi, H.; Matsushita, S.; Kubo, M.; Kai, M.; Imaizumi, A.; Nakano, K.; Hattori, M.; Oda, Y.; Tanaka, M.; et al. NOTCH4 is a potential therapeutic target for triple-negative breast cancer. Anticancer Res. 2014, 34, 69–80. [Google Scholar]
- Chen, C.F.; Dou, X.W.; Liang, Y.-K.; Lin, H.-Y.; Bai, J.-W.; Zhang, X.-X.; Wei, X.-L.; Li, Y.-C.; Zhang, G.-J. Notch3 overexpression causes arrest of cell cycle progression by inducing Cdh1 expression in human breast cancer cells. Cell Cycle 2016, 15, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, D.; Wu, X.; Zeng, Y.; Li, L.; Hou, Y.; Li, W.; Liu, Z. Long non-coding RNA (LncRNA) RMST in triple-negative breast cancer (TNBC): Expression analysis and biological roles research. J. Cell. Physiol. 2018, 233, 6603–6612. [Google Scholar] [CrossRef]
- Xia, S.; Zhang, X.; Li, C.; Guan, H. Oridonin inhibits breast cancer growth and metastasis through blocking the Notch signaling. Saudi Pharm. J. 2017, 25, 638–643. [Google Scholar] [CrossRef]
- Sims-Mourtada, J.; Opdenaker, L.M.; Davis, J.; Arnold, K.M.; Flynn, D. Taxane-induced hedgehog signaling is linked to expansion of breast cancer stem-like populations after chemotherapy. Mol. Carcinog. 2014, 54, 1480–1493. [Google Scholar] [CrossRef]
- Colavito, S.A.; Zou, M.R.; Yan, Q.; Nguyen, D.X.; Stern, D.F. Significance of glioma-associated oncogene homolog 1 (GLI1) expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway. Breast Cancer Res. 2014, 16, 444. [Google Scholar] [CrossRef] [Green Version]
- Goel, H.L.; Pursell, B.; Chang, C.; Shaw, L.M.; Mao, J.; Simin, K.; Kumar, P.; Vander Kooi, C.W.; Shultz, L.D.; Greiner, D.L.; et al. GLI1 regulates a novel neuropilin-2/α6β1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO Mol. Med. 2013, 5, 488–508. [Google Scholar] [CrossRef]
- Han, B.; Qu, Y.; Jin, Y.; Yu, Y.; Deng, N.; Wawrowsky, K.; Zhang, X.; Li, N.; Bose, S.; Wang, Q.; et al. FOXC1 activates smoothened-independent Hedgehog signaling in basal-like breast cancer. Cell Rep. 2015, 13, 1046–1058. [Google Scholar] [CrossRef] [Green Version]
- Ke, Z.; Caiping, S.; Qing, Z.; Xiaojing, W. Sonic hedgehog-Gli1 signals promote epithelial-mesenchymal transition in ovarian cancer by mediating PI3K/AKT pathway. Med. Oncol. 2015, 32, 368. [Google Scholar] [CrossRef]
- Yamamichi, F.; Shigemura, K.; Behnsawy, H.M.; Meligy, F.Y.; Huang, W.C.; Li, X.; Yamanaka, K.; Hanioka, K.; Miyake, H.; Tanaka, K.; et al. Sonic hedgehog and androgen signaling in tumor and stromal compartments drives epithelial-mesenchymal transition in prostate cancer. Scand. J. Urol. 2014, 48, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Yue, D.; Li, H.; Che, J.; Zhang, Y.; Tseng, H.-H.K.; Jin, J.Q.; Luh, T.M.; Giroux-Leprieur, E.; Mo, M.; Zheng, Q.; et al. Hedgehog/Gli promotes epithelial-mesenchymal transition in lung squamous cell carcinomas. J. Exp. Clin. Cancer Res. 2014, 33, 34. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Fan, L.; Wei, G.; Chen, X.; Duan, W.; Xu, Q.; Sheng, W.; Wang, K.; Li, X. Gli-1 is crucial for hypoxia-induced epithelial-mesenchymal transition and invasion of breast cancer. Tumour Biol. 2015, 36, 3119–3126. [Google Scholar] [CrossRef]
- Das, S.; Samant, R.S. Nonclassical activation of Hedgehog signaling enhances multidrug resistance and makes cancer cells refractory to Smoothened-targeting Hedgehog inhibition. J. Biol. Chem. 2013, 288, 11824–11833. [Google Scholar] [CrossRef] [Green Version]
- García-Zaragoza, E.; Pérez-Tavarez, R.; Ballester, A.; Lafarga, V.; Jiménez-Reinoso, A.; Ramírez, A.; Murillas, R.; Gallego, M.I. Intraepithelial paracrine Hedgehog signaling induces the expansion of ciliated cells that express diverse progenitor cell markers in the basal epithelium of the mouse mammary gland. Dev. Biol. 2012, 372, 28–44. [Google Scholar] [CrossRef] [Green Version]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Loi, S.; Drubay, D.; Adams, S.; Pruneri, G.; Francis, P.A.; Lacroix-Triki, M.; Joensuu, H.; Dieci, M.V.; Badve, S.; Demaria, S.; et al. Tumor-infiltrating lymphocytes and prognosis: A pooled individual patient analysis of early-stage triple-negative breast cancers. J. Clin. Oncol. 2019, 37, 559. [Google Scholar] [CrossRef]
- Jiang, Y.-Z.; Liu, Y.; Xiao, Y.; Hu, X.; Jiang, L.; Zuo, W.-J.; Ma, D.; Ding, J.; Zhu, X.; Zou, J.; et al. Molecular subtyping and genomic profiling expand precision medicine in refractory metastatic triple-negative breast cancer: The FUTURE trial. Cell Res. 2021, 31, 178–186. [Google Scholar] [CrossRef]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Prev. Biomark. 2014, 23, 2965–2970. [Google Scholar] [CrossRef] [Green Version]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Liu, S.; Lachapelle, J.; Leung, S.; Gao, D.; Foulkes, W.D.; Nielsen, T.O. CD8+ lymphocyte infiltration is an independent favorable prognostic indicator in basal-like breast cancer. Breast Cancer Res. 2012, 14, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, M.; Jiang, Z.; Jiang, Z.; Wang, X. A comprehensive immunologic portrait of triple-negative breast cancer. Transl. Oncol. 2018, 11, 311–329. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.-T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef] [Green Version]
- Santa-Maria, C.A.; Kato, T.; Park, J.-H.; Kiyotani, K.; Rademaker, A.; Shah, A.N.; Gross, L.; Blanco, L.Z.; Jain, S.; Flaum, L.; et al. A pilot study of durvalumab and tremelimumab and immunogenomic dynamics in metastatic breast cancer. Oncotarget 2018, 9, 18985. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.-Y.; Song, Y.; Wang, J.; Chen, L.-Y.; Pang, J.-Y.; Zhou, L.-R.; Shen, S.-J.; Cao, X.; Wang, Y.-X.; Shao, M.-M.; et al. Mismatch repair deficiency and microsatellite instability in triple-negative breast cancer: A retrospective study of 440 patients. Front. Oncol. 2021, 11, 570623. [Google Scholar] [CrossRef]
- Kurata, K.; Kubo, M.; Kai, M.; Mori, H.; Kawaji, H.; Kaneshiro, K.; Yamada, M.; Nishimura, R.; Osako, T.; Arima, N.; et al. Microsatellite instability in Japanese female patients with triple-negative breast cancer. Breast Cancer 2020, 27, 490–498. [Google Scholar] [CrossRef] [Green Version]
- Karn, T.; Denkert, C.; Weber, K.E.; Holtrich, U.; Hanusch, C.; Sinn, B.V.; Higgs, B.W.; Jank, P.; Sinn, H.P.; Huober, J.; et al. Tumor mutational burden and immune infiltration as independent predictors of response to neoadjuvant immune checkpoint inhibition in early TNBC in GeparNuevo. Ann. Oncol. 2020, 31, 1216–1222. [Google Scholar] [CrossRef]
- Winer, E.P.; Lipatov, O.; Im, S.-A.; Goncalves, A.; Muñoz-Couselo, E.; Seok Lee, K.; Schmid, P.; Testa, L.; Witzel, I.; Ohtani, S.; et al. Association of tumor mutational burden (TMB) and clinical outcomes with pembrolizumab (pembro) versus chemotherapy (chemo) in patients with metastatic triple-negative breast cancer (mTNBC) from KEYNOTE-119. Am. Soc. Clin. Oncol. 2020, 38, 1013. [Google Scholar] [CrossRef]
- O’Meara, T.A.; Tolaney, S.M. Tumor mutational burden as a predictor of immunotherapy response in breast cancer. Oncotarget 2021, 12, 394. [Google Scholar] [CrossRef]
- Gómez-Miragaya, J.; Díaz-Navarro, A.; Tonda, R.; Beltran, S.; Palomero, L.; Palafox, M.; Dobrolecki, L.E.; Huang, C.; Vasaikar, S.; Zhang, B.; et al. Chromosome 12p amplification in triple-negative/BRCA1-mutated breast cancer associates with emergence of docetaxel resistance and carboplatin sensitivity. Cancer Res. 2019, 79, 4258–4271. [Google Scholar] [CrossRef] [Green Version]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.W.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Davis, A.; McDonald, T.O.; Sei, E.; Shi, X.; Wang, Y.; Tsai, P.-C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 2016, 48, 1119. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell 2018, 173, 879–893. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Huang, Y.; Liu, W.; Meng, W.; Zhao, H.; Yang, Q.; Gu, S.-J.; Xiao, C.-C.; Jia, C.-C.; Zhang, B.; et al. Overexpression of zinc finger protein 687 enhances tumorigenic capability and promotes recurrence of hepatocellular carcinoma. Oncogenesis 2017, 6, e363. [Google Scholar] [CrossRef]
- Pogoda, K.; Niwinska, A.; Murawska, M.; Pien´kowski, T. Analysis of pattern, time and risk factors influencing recurrence in triple-negative breast cancer patients. Med. Oncol. 2013, 30, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, T.-Y.; Wang, C.-Y.; Hung, Y.-H.; Chen, W.-C.; Chen, Y.-L.; Lai, M.-D. Differential expression pattern of THBS1 and THBS2 in lung cancer: Clinical outcome and a systematic-analysis of microarray databases. PLoS ONE 2016, 11, e0161007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.-H.; Yao, L.; Xu, H.-W.; Tang, W.-T.; Fu, J.-H.; Hu, X.-F.; Cui, L.; Xu, X.-M. Identification of MXRA5 as a novel biomarker in colorectal cancer. Oncol. Lett. 2013, 5, 544–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Aberrant Gene States | Pathway | |
|---|---|---|
| 1 | BRCA1/2 mut/del | DNA repair pathway |
| 2 | CDKN1A, SFN, EI24, SERPINE1, DDB2, STEAP3, MDM2 over-exp | TP53 signaling pathway |
| 3 | PIK3CA mut/amp; AKT3 amp/mut; PTEN del/mut; TSC1 del/mut; INPP4B del; TSC1 | PI3K/AKT/mTOR pathway |
| 4 | FGFR1 amp; EGFR amp; IGF1R amp; ERBB2/3/4 mut; BRAF amp/mut; KRAS amp/mut; HRAS mut; DUSP4 del | RAS/RAF/MEK pathway |
| 5 | RB1 del; CDK6 amp; CCND1/2 amp | Cell-cycle checkpoints |
| 6 | JAK2 amp | IL-6/JAK/STAT pathway |
| 7 | Notch1/2/3/4 over-exp, JAG1/2, JAG2, DLL4 | Notch pathway |
| 8 | GLI1, Zeb1/2, Snail1/2, Twist 1/2, FOXM1, FOXC1/2 over-exp | Hedgehog signaling |
| Gene | Pathway |
|---|---|
| NOL6-9p13.3 | Major pathway of rRNA processing in the nucleolus and cytosol; rRNA modification in the nucleus and cytosol |
| FAT3-11q14.3 | Wnt signaling pathway; cadherin signaling pathway |
| AHNAK-11q12.3 | EGFR1 |
| BRCA1-17q21.31 | TP53 regulates transcription of DNA repair genes; processing of DNA double-strand break ends; ATM pathway; presynaptic phase of homologous DNA pairing and strand exchange; homologous DNA pairing and strand exchange; G2/M DNA damage checkpoint; generic transcription pathway |
| CACNA1B-9q34.3 | Presynaptic depolarization and calcium channel opening |
| F5-1q24.2 | Common pathway of fibrin clot formation; regulation of IGF activity by IGFBP; post-translational protein phosphorylation |
| FLG2-1q21.3 | Neutrophil degranulation |
| RNF213-17q25.3 | Antigen processing: ubiquitination and proteasome degradation |
| ABCA9-17q24.2 | ABC transporters in lipid homeostasis; ABC-family proteins mediated transport |
| DNAH5-5p15.2 | Huntington disease |
| LAMA1-18p11.31 | MET activates PTK2 signaling; laminin interactions; L1CAM interactions |
| PLXNA2-1q32.2 | SEMA3A-plexin repulsion signaling by inhibiting integrin adhesion; sema3A PAK-dependent axon repulsion |
| VWF-12p13.31 | GP1b-IX-V activation signaling; integrin alphaIIb beta3 signaling; MAP2K and MAPK activation; signaling by high-kinase activity BRAF mutants |
| DYNC2H1-11q22.3 | Huntington disease; hedgehog ‘off’ state |
| PCNT-21q22.3 | AURKA activation by TPX2; regulation of PLK1 activity at G2/M transition |
| SALL1-16q12.1 | POU5F1 (OCT4), SOX2, NANOG activate genes related to proliferation; transcriptional regulation of pluripotent stem cells; regulation of PTEN gene transcription; PI3K/AKT signaling |
| TG-8q24.22 | Thyroid hormone synthesis; interleukin-6 signaling; constitutive signaling by AKT1 E17K in cancer; IL-6-type cytokine receptor ligand interactions; signaling by EGFR; signaling by ERBB4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibragimova, M.K.; Tsyganov, M.M.; Litviakov, N.V. Molecular-Genetic Portrait of Breast Cancer with Triple Negative Phenotype. Cancers 2021, 13, 5348. https://doi.org/10.3390/cancers13215348
Ibragimova MK, Tsyganov MM, Litviakov NV. Molecular-Genetic Portrait of Breast Cancer with Triple Negative Phenotype. Cancers. 2021; 13(21):5348. https://doi.org/10.3390/cancers13215348
Chicago/Turabian StyleIbragimova, Marina K., Matvey M. Tsyganov, and Nikolai V. Litviakov. 2021. "Molecular-Genetic Portrait of Breast Cancer with Triple Negative Phenotype" Cancers 13, no. 21: 5348. https://doi.org/10.3390/cancers13215348
APA StyleIbragimova, M. K., Tsyganov, M. M., & Litviakov, N. V. (2021). Molecular-Genetic Portrait of Breast Cancer with Triple Negative Phenotype. Cancers, 13(21), 5348. https://doi.org/10.3390/cancers13215348