
The Epithelial and Stromal Immune Microenvironment in Gastric Cancer: A Comprehensive Analysis Reveals Prognostic Factors with Digital Cytometry


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Gastric Cancer Bulk Gene Expression Data
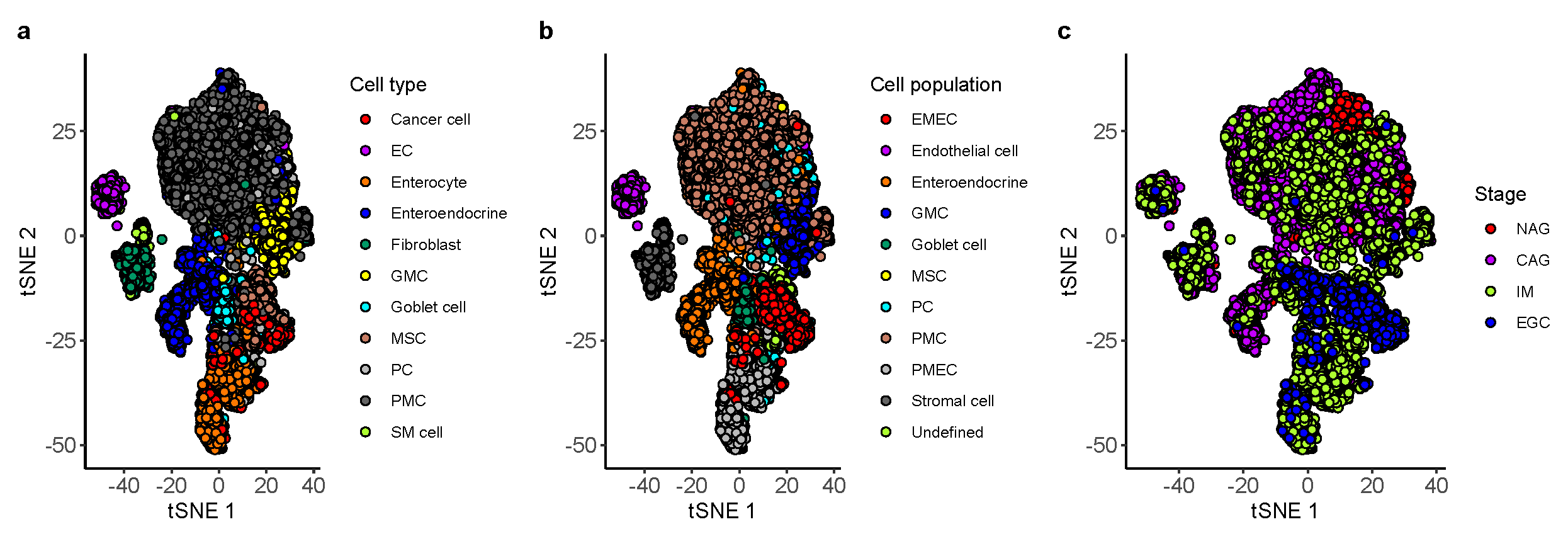
2.2. Identification of Ten Non-Immune Cell Populations from Single-Cell RNA-Seq of Gastric Antral Mucosa Biopsies
2.3. Marker Selection and Signature Matrix Construction
2.4. Deconvolving Bulk Gene Expression Samples
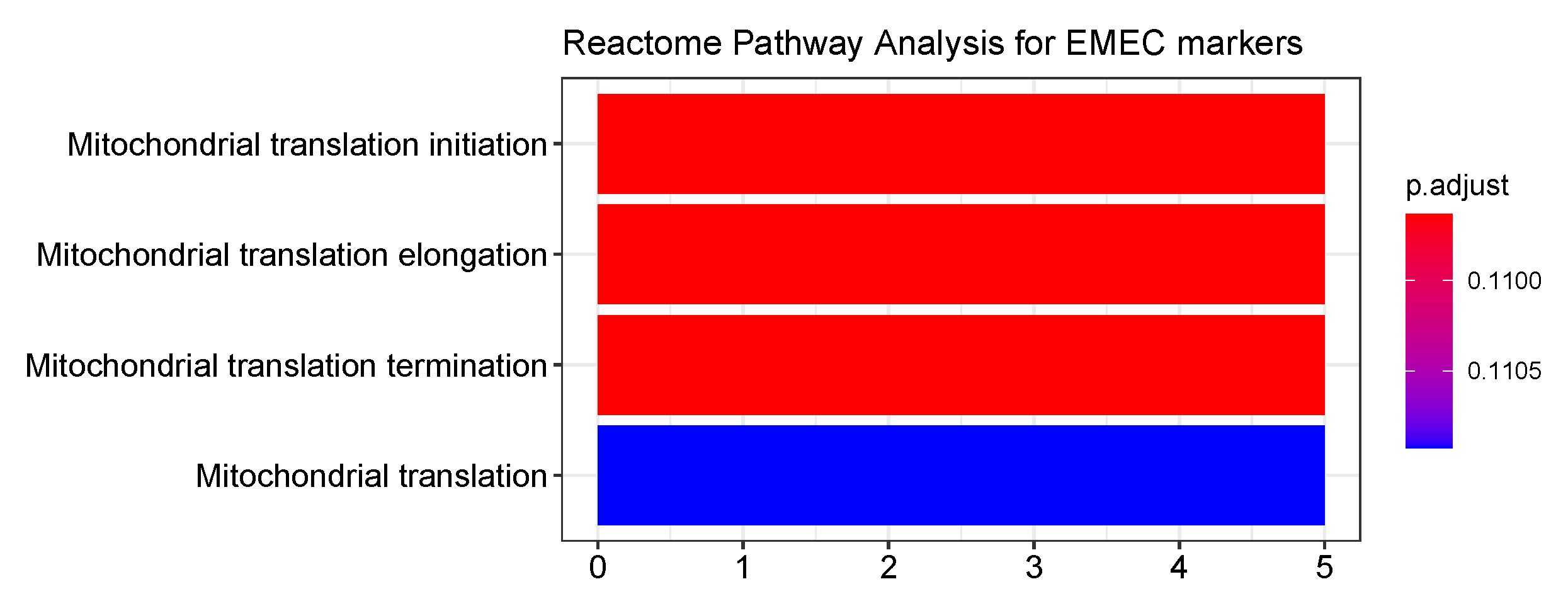
2.5. Go Enrichment Analyses
2.6. Determination of Optimal Stem Score Cutoff
2.7. Tme Subtypes Identification in GC
2.8. Statistical Analysis
3. Results
3.1. Building a Non-Immune Signature Matrix for GC from a Single-Cell Rna-Seq Data Set
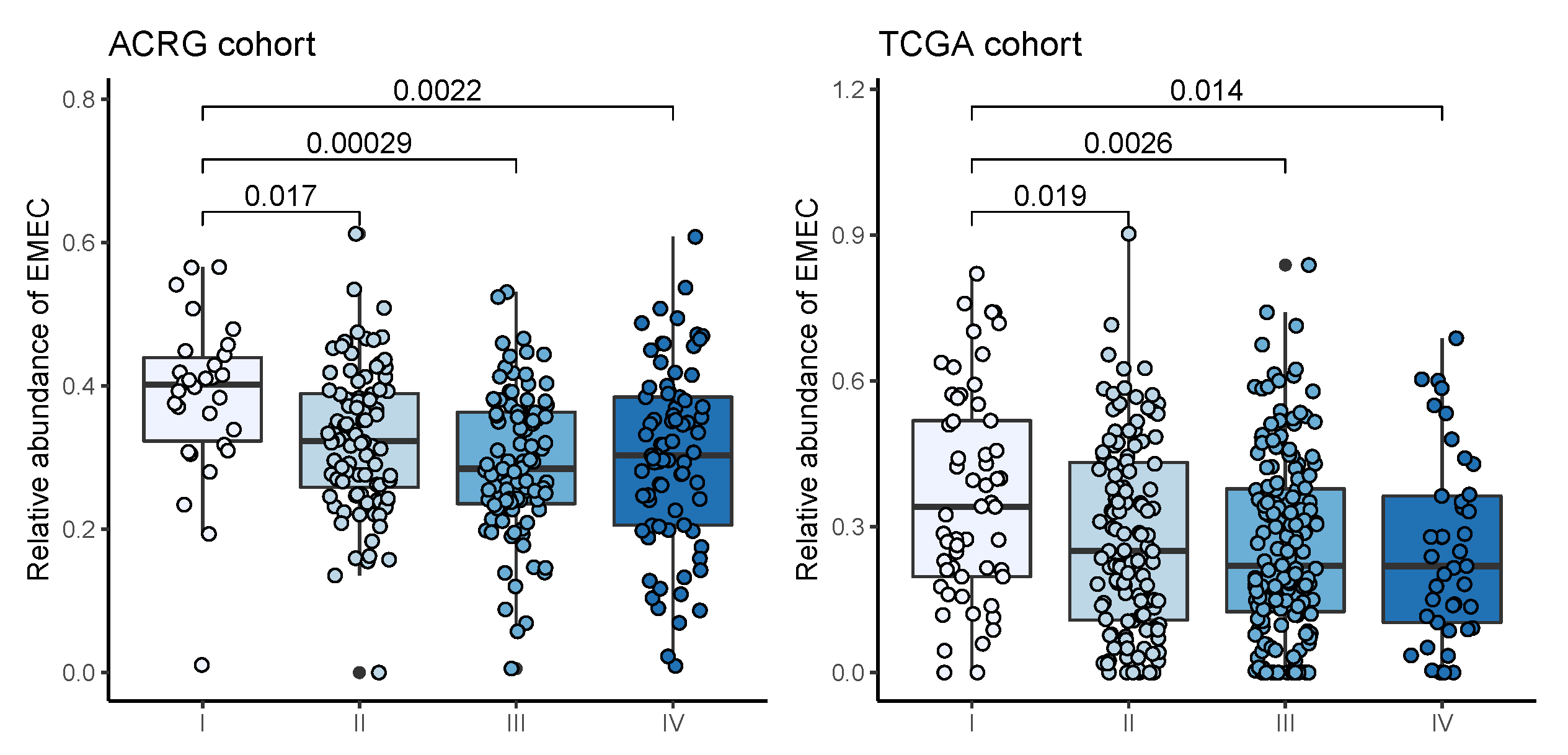
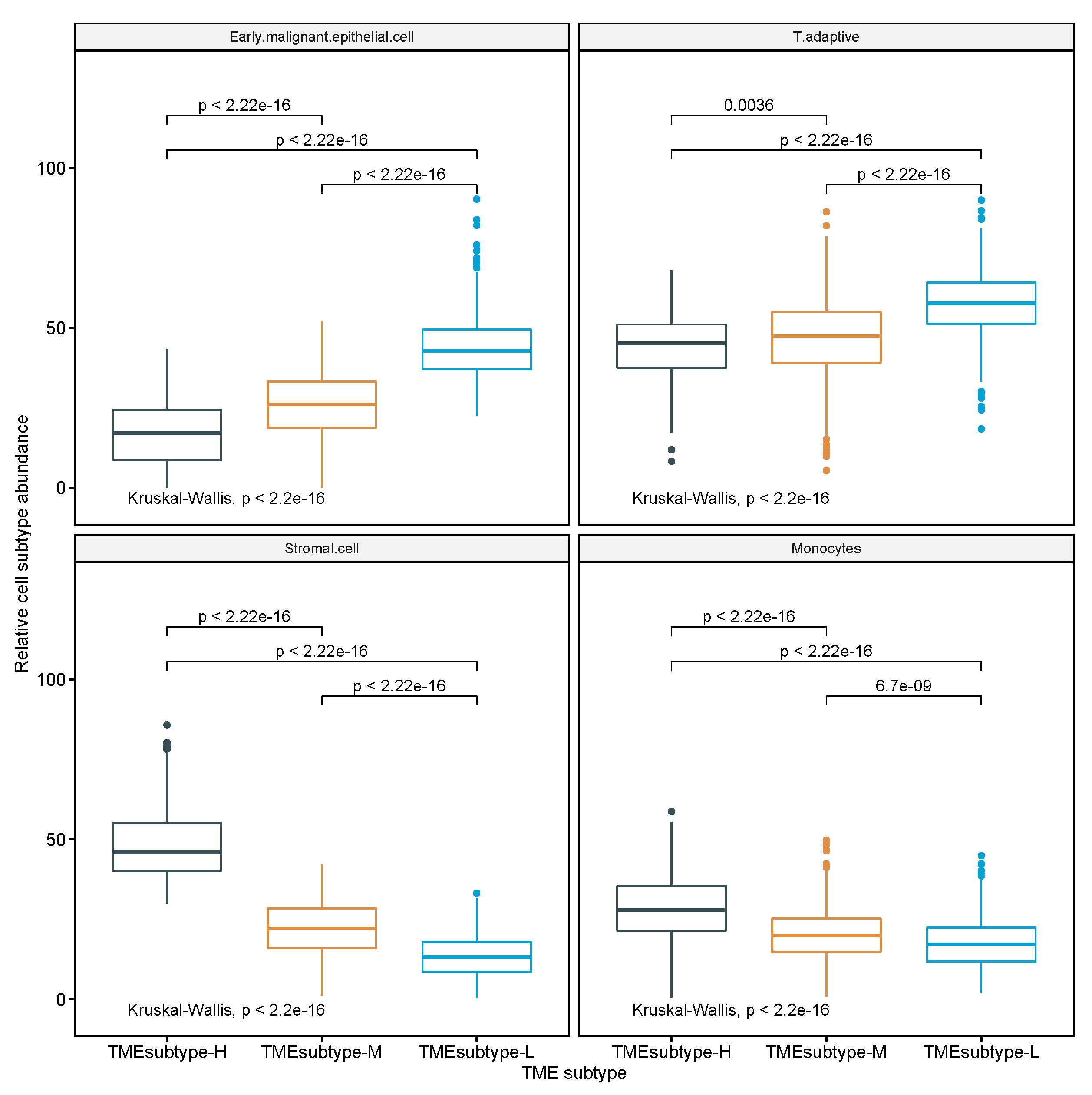
3.2. Dissecting Epithelium-Stroma-Immune Signals from Gastric Cancer Samples
3.3. Correlates of Non-Immune/Immune Factors with Overall Survival
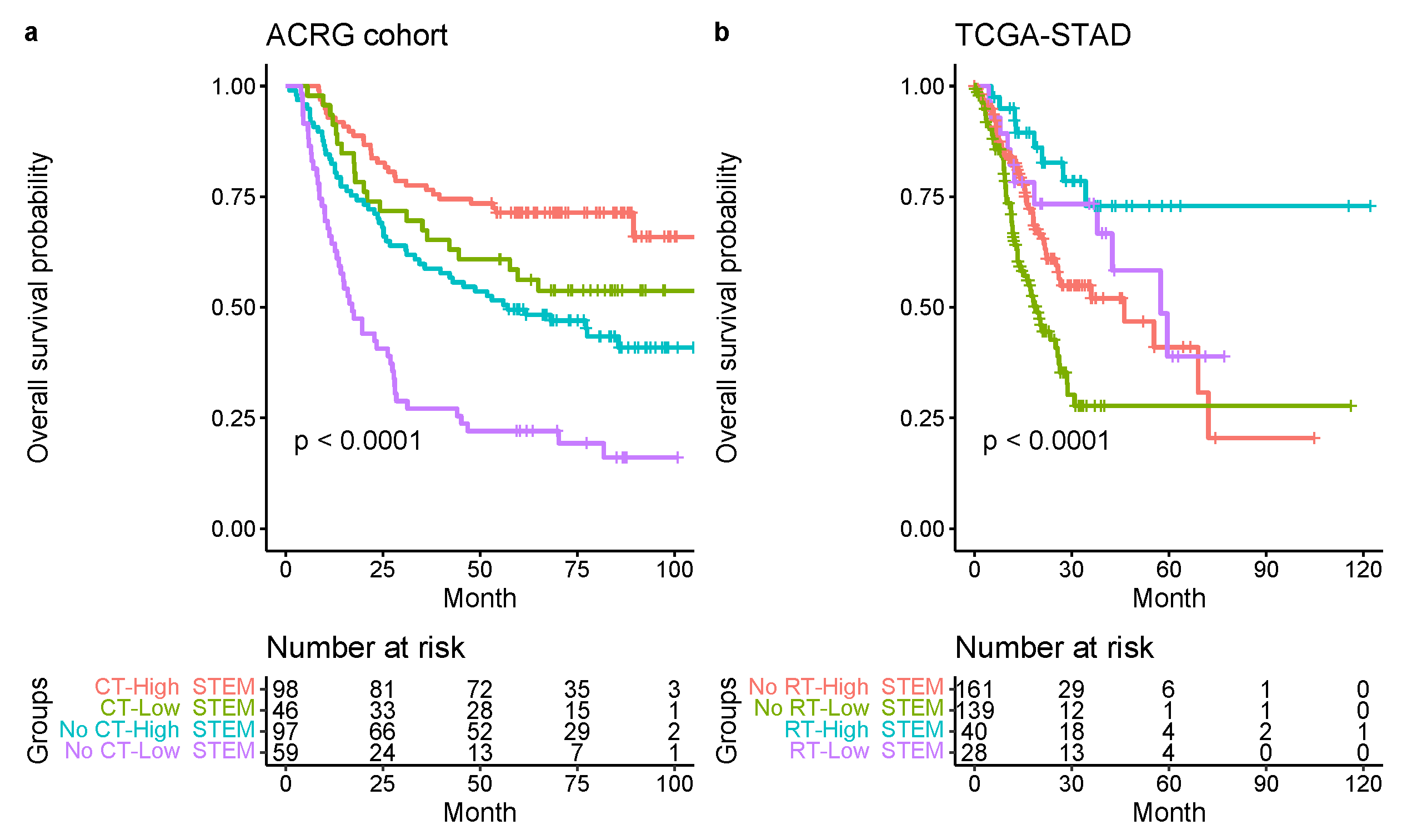
3.4. Increased Stem Score Associated with Superior Survival
3.5. Comparison with Other Reported Molecular Classifications for GC
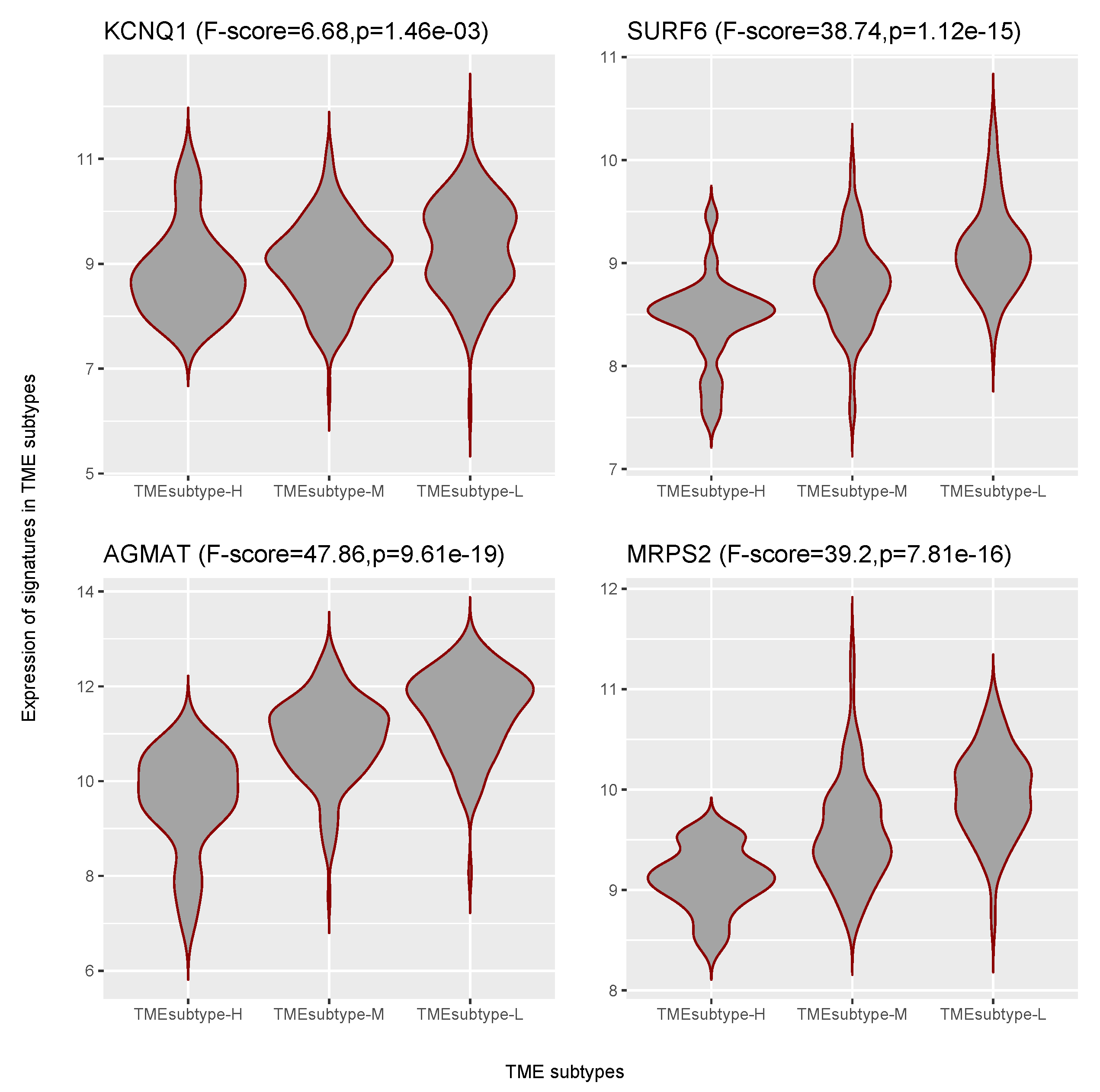
3.6. Identification of GC Prognostic Gene Signatures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| GC | gastric cancer |
| TME | tumor microenvironment |
| EMEC | early malignant epithelial cell |
| PMEC | premalignant epithelial cell |
| STEM score | TME signature score by integrating stromal cells, adaptive T cells, EMEC, and monocytes |
| ACRG | The Asian Cancer Research Group |
| STAD | stomach adenocarcinoma |
| EMT | epithelial-to-mesenchymal transition |
| EBV | Epstein–Barr virus-positivity |
| MSI | microsatellite instability |
| GS | genomically stable |
| CIN | chromosomal instability |
| TCGA | The Cancer Genome Atlas |
| CAF | cancer-associated fibroblasts |
| TIL | tumor-infiltrating lymphocytes |
| scRNAseq | single-cell RNA-sequencing |
| NAG | non-atrophic gastritis |
| CAG | chronic atrophic gastritis |
| IM | intestinal metaplasia |
| EGC | early gastric cancer |
| MSC | metaplastic stem-like cell |
| PC | proliferative cell |
| PMC | Pit mucous cell |
| EC | endothelial cell |
| GMC | antral basal gland mucous cell |
| SM cell | smooth muscle cell |
| TSR | tumor-to-stroma ratio |
| LMR | lymphocyte-to-monocyte ratio |
| TPM | transcripts per kilobase million |
| GO | gene ontology |
| BP | biological process |
| CC | cellular component |
| OS | overall survival |
| HR | hazard ratio |
| CI | confidence interval |
| BH | Benjamini–Hochberg |
| FDR | false discovery rate. |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma: An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Teh, M.; Lee, Y.S. Intestinal and diffuse carcinoma of the stomach among the ethnic and dialect groups in Singapore. Cancer 1987, 60, 921–925. [Google Scholar] [CrossRef]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heppner, G.H.; Miller, B.E. Tumor heterogeneity: Biological implications and therapeutic consequences. Cancer Metastasis Rev. 1983, 2, 5–23. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Aranda, V.; Bardelli, A.; Blanpain, C.; Bock, C.; Borowski, C.; Caldas, C.; Califano, A.; Doherty, M.; Elsner, M.; et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015, 21, 846. [Google Scholar] [CrossRef]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-associated fibroblasts build and secure the tumor microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Wang, R.; Guan, W.; Qiao, M.; Wang, L. Roles of microRNAs in cancer associated fibroblasts of gastric cancer. Pathol.-Res. Pract. 2017, 213, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Shen, J.; Xie, G.; Wu, J.; He, M.; Gao, L.; Zhang, Y.; Yao, X.; Shen, L. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019, 454, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; He, W.; Wu, C.; Tan, Y.; He, Y.; Xu, B.; Chen, L.; Li, Q.; Jiang, J. Scoring system for tumor-infiltrating lymphocytes and its prognostic value for gastric cancer. Front. Immunol. 2019, 10, 71. [Google Scholar] [CrossRef] [Green Version]
- Ling, Z.; Shao, L.; Liu, X.; Cheng, Y.; Yan, C.; Mei, Y.; Ji, F.; Liu, X. Regulatory T cells and plasmacytoid dendritic cells within the tumor microenvironment in gastric cancer are correlated with gastric microbiota dysbiosis: A preliminary study. Front. Immunol. 2019, 10, 533. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Luo, Q.; Wang, W.; Li, J.; Wang, T.; Wang, P.; Chen, L.; Zhang, P.; Chen, H.; Liu, Y.; et al. Tumor-associated macrophages-derived exosomes promote the migration of gastric cancer cells by transfer of functional Apolipoprotein E. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Peng, C.; Liu, J.; Yang, G.; Li, Y. The tumor-stromal ratio as a strong prognosticator for advanced gastric cancer patients: Proposal of a new TSNM staging system. J. Gastroenterol. 2018, 53, 606–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sammarco, G.; Varricchi, G.; Ferraro, V.; Ammendola, M.; De Fazio, M.; Altomare, D.F.; Luposella, M.; Maltese, L.; Currò, G.; Marone, G.; et al. Mast cells, angiogenesis and lymphangiogenesis in human gastric cancer. Int. J. Mol. Sci. 2019, 20, 2106. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Jiang, Y.; Li, G.; Fisher, G.A., Jr.; Li, R. Natural killer cell and stroma abundance are independently prognostic and predict gastric cancer chemotherapy benefit. JCI Insight 2020, 5, e136570. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Park, J.; Susztak, K.; Zhang, N.R.; Li, M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, G.; Lee, B.; Xu, W.; Mustafah, S.; Hwang, Y.Y.; Carre, C.; Burdin, N.; Visan, L.; Ceccarelli, M.; Poidinger, M.; et al. RNA-Seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 2019, 26, 1627–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Malika, C.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust: An R package for determining the relevant number of clusters in a data Set. J. Stat. Softw 2014, 61, 1–36. [Google Scholar]
- Zhang, P.; Yang, M.; Zhang, Y.; Xiao, S.; Lai, X.; Tan, A.; Du, S.; Li, S. Dissecting the single-cell transcriptome network underlying gastric premalignant lesions and early gastric cancer. Cell Rep. 2019, 27, 1934–1947. [Google Scholar] [CrossRef] [Green Version]
- Jakupciak, J.P.; Maragh, S.; Markowitz, M.E.; Greenberg, A.K.; Hoque, M.O.; Maitra, A.; Barker, P.E.; Wagner, P.D.; Rom, W.N.; Srivastava, S.; et al. Performance of mitochondrial DNA mutations detecting early stage cancer. BMC Cancer 2008, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Verschoor, M.L.; Ungard, R.; Harbottle, A.; Jakupciak, J.P.; Parr, R.; Singh, G. Mitochondria and cancer: Past, present, and future. BioMed Res. Int. 2013, 2013, 612369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Li, Y.; Hu, B. Potential role of mitochondria in gastric cancer detection: Fission and glycolysis. Oncol. Lett. 2021, 21, 1–7. [Google Scholar] [CrossRef]
- Camargo, M.C.; Kim, W.H.; Chiaravalli, A.M.; Kim, K.M.; Corvalan, A.H.; Matsuo, K.; Yu, J.; Sung, J.J.; Herrera-Goepfert, R.; Meneses-Gonzalez, F.; et al. Improved survival of gastric cancer with tumour Epstein–Barr virus positivity: An international pooled analysis. Gut 2014, 63, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, Z.; Wang, Y.; Zhang, C.; Liu, Y.; Qu, X. Microsatellite instability and survival in gastric cancer: A systematic review and meta-analysis. Mol. Clin. Oncol. 2015, 3, 699–705. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Mishra, P.; Banerjee, D.; Ben-Baruch, A. Chemokines at the crossroads of tumor-fibroblast interactions that promote malignancy. J. Leukoc. Biol. 2011, 89, 31–39. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Littlepage, L.E.; Egeblad, M.; Werb, Z. Coevolution of cancer and stromal cellular responses. Cancer Cell 2005, 7, 499–500. [Google Scholar] [CrossRef] [Green Version]
- Kemi, N.; Eskuri, M.; Herva, A.; Leppänen, J.; Huhta, H.; Helminen, O.; Saarnio, J.; Karttunen, T.J.; Kauppila, J.H. Tumour-stroma ratio and prognosis in gastric adenocarcinoma. Br. J. Cancer 2018, 119, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, S.M.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.; Ellis, I.O.; Green, A.R. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J. Clin. Oncol. 2011, 29, 1949–1955. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Banerjee, A. Effector, memory, and dysfunctional CD8+ T cell fates in the antitumor immune response. J. Immunol. Res. 2016, 2016, 8941260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A. Progress in human tumour immunology and immunotherapy. Nature 2001, 411, 380–384. [Google Scholar] [CrossRef]
- Zikos, T.; Donnenberg, A.; Landreneau, R.; Luketich, J.; Donnenberg, V. Lung T-cell subset composition at the time of surgical resection is a prognostic indicator in non-small cell lung cancer. Cancer Immunol. Immunother. 2011, 60, 819–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.M.; Huang, J.J.; Xia, Y.; Sun, J.; Huang, Y.; Wang, Y.; Zhu, Y.J.; Li, Y.J.; Zhao, W.; Wei, W.X.; et al. Blood lymphocyte-to-monocyte ratio identifies high-risk patients in diffuse large B-cell lymphoma treated with R-CHOP. PLoS ONE 2012, 7, e41658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stotz, M.; Pichler, M.; Absenger, G.; Szkandera, J.; Arminger, F.; Schaberl-Moser, R.; Samonigg, H.; Stojakovic, T.; Gerger, A. The preoperative lymphocyte to monocyte ratio predicts clinical outcome in patients with stage III colon cancer. Br. J. Cancer 2014, 110, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Rhee, D.K.; Park, S.H.; Jang, Y.K. Molecular signatures associated with transformation and progression to breast cancer in the isogenic MCF10 model. Genomics 2008, 92, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Donahue, T.R.; Tran, L.M.; Hill, R.; Li, Y.; Kovochich, A.; Calvopina, J.H.; Patel, S.G.; Wu, N.; Hindoyan, A.; Farrell, J.J.; et al. Integrative survival-based molecular profiling of human pancreatic cancer. Clin. Cancer Res. 2012, 18, 1352–1363. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Ye, Y.; Dong, L.; Vainionpää, S.; Mustonen, H.; Puolakkainen, P.; Wang, S. Kindlin-2: A novel adhesion protein related to tumor invasion, lymph node metastasis, and patient outcome in gastric cancer. Am. J. Surg. 2012, 203, 222–229. [Google Scholar] [CrossRef]
- Zhao, T.; Guan, L.; Yu, Y.; Pei, X.; Zhan, J.; Han, L.; Tang, Y.; Li, F.; Fang, W.; Zhang, H. Kindlin-2 promotes genome instability in breast cancer cells. Cancer Lett. 2013, 330, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.H.; Roehrl, M.H.; Wang, J.Y. Tissue proteomics reveals differential and compartment-specific expression of the homologs transgelin and transgelin-2 in lung adenocarcinoma and its stroma. J. Proteome Res. 2009, 8, 5610–5618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, H.; Chiyomaru, T.; Enokida, H.; Kawakami, K.; Tatarano, S.; Nishiyama, K.; Nohata, N.; Seki, N.; Nakagawa, M. The tumour-suppressive function of miR-1 and miR-133a targeting TAGLN2 in bladder cancer. Br. J. Cancer 2011, 104, 808–818. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Chen, M. MYLK and MYL9 expression in non-small cell lung cancer identified by bioinformatics analysis of public expression data. Tumor Biol. 2014, 35, 12189–12200. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Zhang, L.; Huang, S.T.; Xu, J.; Zhou, Y.; Yu, X.J.; Luo, R.Z.; Wen, Z.S.; Jia, W.H.; Zheng, M. Expression and prognostic significance of MYL9 in esophageal squamous cell carcinoma. PLoS ONE 2017, 12, e0175280. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bian, S.; Zhou, X.; Cui, Y.; Wang, W.; Wen, L.; Guo, L.; Fu, W.; Tang, F. Single-Cell Multiomics Sequencing Reveals Prevalent Genomic Alterations in Tumor Stromal Cells of Human Colorectal Cancer. Cancer Cell 2020, 38, 818–828. [Google Scholar] [CrossRef]
- Hou, J.Y.; Wang, Y.G.; Ma, S.J.; Yang, B.Y.; Li, Q.P. Identification of a prognostic 5-Gene expression signature for gastric cancer. J. Cancer Res. Clin. Oncol. 2017, 143, 619–629. [Google Scholar] [CrossRef]
- Hagel, C.; Dornblut, C.; Schulz, A.; Wiehl, U.; Friedrich, R.; Huckhagel, T.; Mautner, V.F.; Morrison, H. The putative oncogene CPI-17 is up-regulated in schwannoma. Neuropathol. Appl. Neurobiol. 2016, 42, 664–668. [Google Scholar] [CrossRef]
- Riecken, L.B.; Zoch, A.; Wiehl, U.; Reichert, S.; Scholl, I.; Cui, Y.; Ziemer, M.; Anderegg, U.; Hagel, C.; Morrison, H. CPI-17 drives oncogenic Ras signaling in human melanomas via Ezrin-Radixin-Moesin family proteins. Oncotarget 2016, 7, 78242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, R.; Goto, Y.; Kojima, S.; Enokida, H.; Chiyomaru, T.; Kinoshita, T.; Sakamoto, S.; Fuse, M.; Nakagawa, M.; Naya, Y.; et al. Tumor-suppressive microRNA-29s inhibit cancer cell migration and invasion via targeting LAMC1 in prostate cancer. Int. J. Oncol. 2014, 45, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.C.; Jong, H.S.; Kim, T.Y.; Song, S.H.; Lee, D.S.; Lee, J.W.; Kim, T.Y.; Kim, N.K.; Bang, Y.J. AKAP12/Gravin is inactivated by epigenetic mechanism in human gastric carcinoma and shows growth suppressor activity. Oncogene 2004, 23, 7095–7103. [Google Scholar] [CrossRef] [Green Version]
- Gelman, I.H. Emerging roles for SSeCKS/Gravin/AKAP12 in the control of cell proliferation, cancer malignancy, and barriergenesis. Genes Cancer 2010, 1, 1147–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Sun, Z.; Jiang, S.; Jin, X.; Wang, H. Identification and validation of a two-gene metabolic signature for survival prediction in patients with kidney renal clear cell carcinoma. Aging 2021, 13, 8276. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Lisanti, M.P. Mitochondrial mRNA transcripts predict overall survival, tumor recurrence and progression in serous ovarian cancer: Companion diagnostics for cancer therapy. Oncotarget 2017, 8, 66925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleich, M.; Warth, R. The very small-conductance K+ channel K V LQT1 and epithelial function. Pflügers Arch. 2000, 440, 202–206. [Google Scholar]
- Than, B.L.; Goos, J.; Sarver, A.L.; O’Sullivan, M.G.; Rod, A.; Starr, T.K.; Fijneman, R.J.; Meijer, G.A.; Zhao, L.; Zhang, Y.; et al. The role of KCNQ1 in mouse and human gastrointestinal cancers. Oncogene 2014, 33, 3861–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapetti-Mauss, R.; Bustos, V.; Thomas, W.; McBryan, J.; Harvey, H.; Lajczak, N.; Madden, S.F.; Pellissier, B.; Borgese, F.; Soriani, O.; et al. Bidirectional KCNQ1: β-catenin interaction drives colorectal cancer cell differentiation. Proc. Natl. Acad. Sci. USA 2017, 114, 4159–4164. [Google Scholar] [CrossRef] [Green Version]
- Den Uil, S.H.; Coupé, V.M.; Linnekamp, J.F.; Van Den Broek, E.; Goos, J.A.; Delis-van Diemen, P.M.; Eric, J.; Van Grieken, N.C.; Scott, P.M.; Vermeulen, L.; et al. Loss of KCNQ1 expression in stage II and stage III colon cancer is a strong prognostic factor for disease recurrence. Br. J. Cancer 2016, 115, 1565–1574. [Google Scholar] [CrossRef] [Green Version]
- Kunitomi, H.; Kobayashi, Y.; Wu, R.C.; Takeda, T.; Tominaga, E.; Banno, K.; Aoki, D. LAMC1 is a prognostic factor and a potential therapeutic target in endometrial cancer. J. Gynecol. Oncol. 2020, 31, e11. [Google Scholar] [CrossRef]
- Nohata, N.; Hanazawa, T.; Kikkawa, N.; Sakurai, D.; Sasaki, K.; Chiyomaru, T.; Kawakami, K.; Yoshino, H.; Enokida, H.; Nakagawa, M.; et al. Identification of novel molecular targets regulated by tumor suppressive miR-1/miR-133a in maxillary sinus squamous cell carcinoma. Int. J. Oncol. 2011, 39, 1099–1107. [Google Scholar]
- Cai, J.; Chen, S.; Zhang, W.; Hu, S.; Lu, J.; Xing, J.; Dong, Y. Paeonol reverses paclitaxel resistance in human breast cancer cells by regulating the expression of transgelin 2. Phytomedicine 2014, 21, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Chen, S.; Zhang, W.; Zheng, X.; Hu, S.; Pang, C.; Lu, J.; Xing, J.; Dong, Y. Salvianolic acid A reverses paclitaxel resistance in human breast cancer MCF-7 cells via targeting the expression of transgelin 2 and attenuating PI3 K/Akt pathway. Phytomedicine 2014, 21, 1725–1732. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chen, S.; Yang, Q.; Cai, J.; Zhang, W.; You, H.; Xing, J.; Dong, Y. Salvianolic acid A reverses the paclitaxel resistance and inhibits the migration and invasion abilities of human breast cancer cells by inactivating transgelin 2. Cancer Biol. Ther. 2015, 16, 1407–1414. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Deng, W.; Wang, P.; Yan, Y.; Xie, C.; Cao, X.; Chen, M.; Zhang, C.; Shi, D.; Dong, Y.; et al. Fermitin family member 2 promotes melanoma progression by enhancing the binding of p-α-Pix to Rac1 to activate the MAPK pathway. Oncogene 2021, 40, 5626–5638. [Google Scholar] [CrossRef] [PubMed]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|
| Non-Immune Factors | HR (95% CI for HR) | pValue (FDR-Adjusted) | HR (95% CI for HR) | pValue (FDR-Adjusted) |
| EMEC/Stromal cell | 0.75 (0.65–0.86) | <0.001 (<0.001) | 0.82 (0.71–0.94) | 0.005 (0.022) |
| PC/Stromal cell | 0.80 (0.71–0.90) | <0.001 (<0.001) | 0.85 (0.75–0.96) | 0.011 (0.022) |
| Endothelial cell/Stromal cell | 0.27 (0.08–0.90) | 0.032 (0.064) | 0.44 (0.13–1.47) | 0.182 (0.273) |
| Stromal cell/Endothelial cell | 1.04 (1.00–1.08) | 0.081 (0.122) | 1.07 (1.02–1.13) | 0.009 (0.022) |
| PC/Endothelial cell | 0.98 (0.96–1.01) | 0.150 (0.167) | 0.99 (0.97–1.02) | 0.463 (0.463) |
| EMEC/Endothelial cell | 0.99 (0.97–1.01) | 0.167 (0.167) | 0.99 (0.97–1.01) | 0.415 (0.463) |
| Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|
| Immune Factors | HR (95% CI for HR) | pValue (FDR-Adjusted) | HR (95% CI for HR) | pValue (FDR-Adjusted) |
| T adaptive/Monocytes | 0.86 (0.78–0.96) | 0.004 (0.056) | 0.88 (0.80–0.97) | 0.010 (0.140) |
| Monocytes/T adaptive | 2.54 (1.24–5.21) | 0.011 (0.077) | 2.61 (1.11–6.15) | 0.028 (0.196) |
| Monocytes/B adaptive | 1.14 (1.00–1.29) | 0.048 (0.224) | 1.08 (0.95–1.22) | 0.239 (0.837) |
| B adaptive/Monocytes | 0.80 (0.61–1.04) | 0.097 (0.291) | 0.78 (0.59–1.03) | 0.078 (0.364) |
| T Innate/T adaptive | 2.76 (0.81–9.39) | 0.104 (0.291) | 1.60 (0.41–6.21) | 0.500 (0.875) |
| Granulocytes/Monocytes | 0.79 (0.50–1.24) | 0.304 (0.596) | 0.90 (0.63–1.29) | 0.582 (0.905) |
| B adaptive/T Innate | 0.99 (0.96–1.02) | 0.347 (0.596) | 0.99 (0.97–1.01) | 0.354 (0.875) |
| T Innate/B adaptive | 1.16 (0.85–1.59) | 0.360 (0.596) | 1.00 (0.72–1.37) | 0.978 (0.997) |
| B adaptive/T adaptive | 1.44 (0.61–3.43) | 0.405 (0.596) | 1.43 (0.54–3.78) | 0.471 (0.875) |
| Granulocytes/B adaptive | 0.87 (0.63–1.22) | 0.426 (0.596) | 1.05 (0.76–1.45) | 0.781 (0.953) |
| Granulocytes/T Innate | 0.96 (0.86–1.09) | 0.553 (0.698) | 1.00 (0.99–1.01) | 0.997 (0.997) |
| T Innate/Monocytes | 0.89 (0.57–1.38) | 0.598 (0.698) | 0.82 (0.52–1.30) | 0.393 (0.875) |
| T adaptive/B adaptive | 0.99 (0.93–1.05) | 0.742 (0.749) | 0.99 (0.93–1.06) | 0.817 (0.953) |
| Granulocytes/T adaptive | 0.85 (0.32–2.29) | 0.749 (0.749) | 1.17 (0.46–2.98) | 0.741 (0.953) |
| Variable | HR (95% CI for HR) | p Value |
|---|---|---|
| STEM score | 0.90 (0.84–0.97) | 0.003 |
| Age | 1.02 (1.00–1.03) | 0.019 |
| Gender (Male vs. Female) | 1.21 (0.85–1.71) | 0.283 |
| Stage (II vs. I) | 1.78 (0.68–4.63) | 0.239 |
| Stage (III vs. I) | 3.58 (1.40–9.15) | 0.008 |
| Stage (IV vs. I) | 8.49 (3.36–21.47) | <0.001 |
| Lauren (Intestinal vs. Diffuse/Mixed) | 0.75 (0.53–1.07) | 0.111 |
| Chemotherapy (Yes vs. No) | 0.45 (0.31–0.64) | <0.001 |
| TCGA-STAD | GSE15459 | GSE84437 | ||||
|---|---|---|---|---|---|---|
| Variable | HR (95% CI) | pValue | HR (95% CI) | pValue | HR (95% CI) | pValue |
| STEM score | 0.94 (0.91–0.97) | 0.001 | 0.89 (0.82–0.97) | 0.010 | 0.86 (0.80–0.92) | <0.001 |
| Age | 1.02 (1.00–1.04) | 0.011 | 1.01 (1.00–1.03) | 0.128 | 1.02 (1.01–1.03) | <0.001 |
| Gender (Male vs. Female) | 1.09 (0.76–1.57) | 0.626 | 0.74 (0.47–1.19) | 0.213 | 1.31 (0.96–1.77) | 0.086 |
| Stage (II vs. I) | 1.43 (0.70–2.92) | 0.321 | 2.22 (0.68–7.22) | 0.186 | ||
| Stage (III vs. I) | 2.64 (1.34–5.21) | 0.005 | 7.96 (2.80–22.61) | <0.001 | ||
| Stage (IV vs. I) | 4.15 (1.89–9.08) | <0.001 | 23.28 (7.92–68.46) | <0.001 | ||
| Lauren (Intestinal vs. Diffuse/Mixed) | 1.25 (0.80–1.95) | 0.322 | ||||
| Radiationtherapy (Yes vs. No) | 0.41 (0.25–0.69) | 0.001 | ||||
| Cell Population | Gene Symbol | Gene Name | HR (95% CI) | DE |
|---|---|---|---|---|
| Stromal cell | FERMT2 | fermitin family member 2 | 1.49 (1.35–1.65) | Up |
| Stromal cell | SGCE | sarcoglycan epsilon | 1.50 (1.35–1.67) | Up |
| Stromal cell | PPP1R14A | protein phosphatase 1 regulatory inhibitor subunit 14A | 1.38 (1.27–1.5) | Up |
| Stromal cell | LAMC1 | laminin subunit gamma 1 | 1.83 (1.56–2.16) | Up |
| Stromal cell | MYL9 | myosin light chain 9 | 1.30 (1.21–1.40) | Up |
| Stromal cell | TPM2 | tropomyosin 2 | 1.34 (1.24–1.46) | Up |
| Stromal cell | TAGLN | transgelin | 1.33 (1.23–1.44) | Up |
| Stromal cell | AKAP12 | A–kinase anchoring protein 12 | 1.40 (1.27–1.54) | Up |
| EMEC | KCNQ1 | potassium voltage–gated channel subfamily Q member 1 | 0.73 (0.65–0.82) | Down |
| EMEC | SURF6 | surfeit 6 | 0.57 (0.45–0.72) | Down |
| EMEC | AGMAT | agmatinase | 0.79 (0.70–0.88) | Down |
| EMEC | MRPS2 | mitochondrial ribosomal protein S2 | 0.60 (0.48–0.76) | Down |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, W.; Wang, G.; Cooper, G.R.; Jiang, Y.; Zhou, X. The Epithelial and Stromal Immune Microenvironment in Gastric Cancer: A Comprehensive Analysis Reveals Prognostic Factors with Digital Cytometry. Cancers 2021, 13, 5382. https://doi.org/10.3390/cancers13215382
Shen W, Wang G, Cooper GR, Jiang Y, Zhou X. The Epithelial and Stromal Immune Microenvironment in Gastric Cancer: A Comprehensive Analysis Reveals Prognostic Factors with Digital Cytometry. Cancers. 2021; 13(21):5382. https://doi.org/10.3390/cancers13215382
Chicago/Turabian StyleShen, Wenjun, Guoyun Wang, Georgia R. Cooper, Yuming Jiang, and Xin Zhou. 2021. "The Epithelial and Stromal Immune Microenvironment in Gastric Cancer: A Comprehensive Analysis Reveals Prognostic Factors with Digital Cytometry" Cancers 13, no. 21: 5382. https://doi.org/10.3390/cancers13215382
APA StyleShen, W., Wang, G., Cooper, G. R., Jiang, Y., & Zhou, X. (2021). The Epithelial and Stromal Immune Microenvironment in Gastric Cancer: A Comprehensive Analysis Reveals Prognostic Factors with Digital Cytometry. Cancers, 13(21), 5382. https://doi.org/10.3390/cancers13215382