
An HLA-A*11:01-Binding Neoantigen from Mutated NPM1 as Target for TCR Gene Therapy in AML

 ,
,  , , ,
, , ,  , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Collection and Culture
2.2. Peptide and Peptide-HLA Tetramer Production
2.3. Peptide-HLA Class I Binding Assays
2.4. Isolation of dNPM1-Specific T-Cells
2.5. Transduction of Cell Lines
2.6. Generation of TCR-Transduced T-Cells for In Vitro Assays
2.7. Antibodies and Flow Cytometry Experiments
2.8. T-Cell Assays
2.9. In Vivo Experiments
3. Results
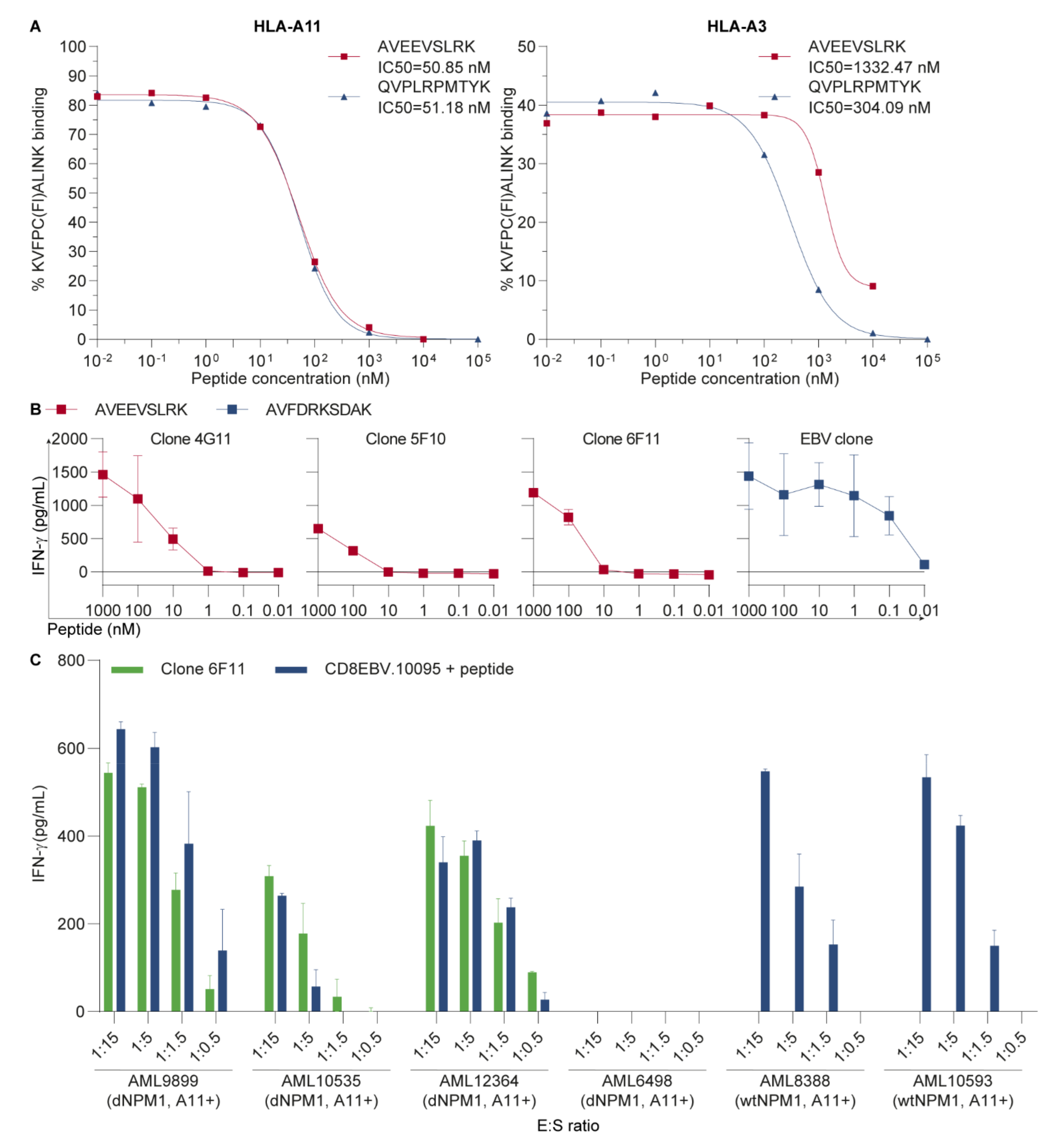
3.1. AVEEVSLRK Is an HLA-A11-Binding Neoantigen on Primary AML
3.2. The HLA-A11-Restricted TCR for dNPM1 Specifically Targets Primary AML
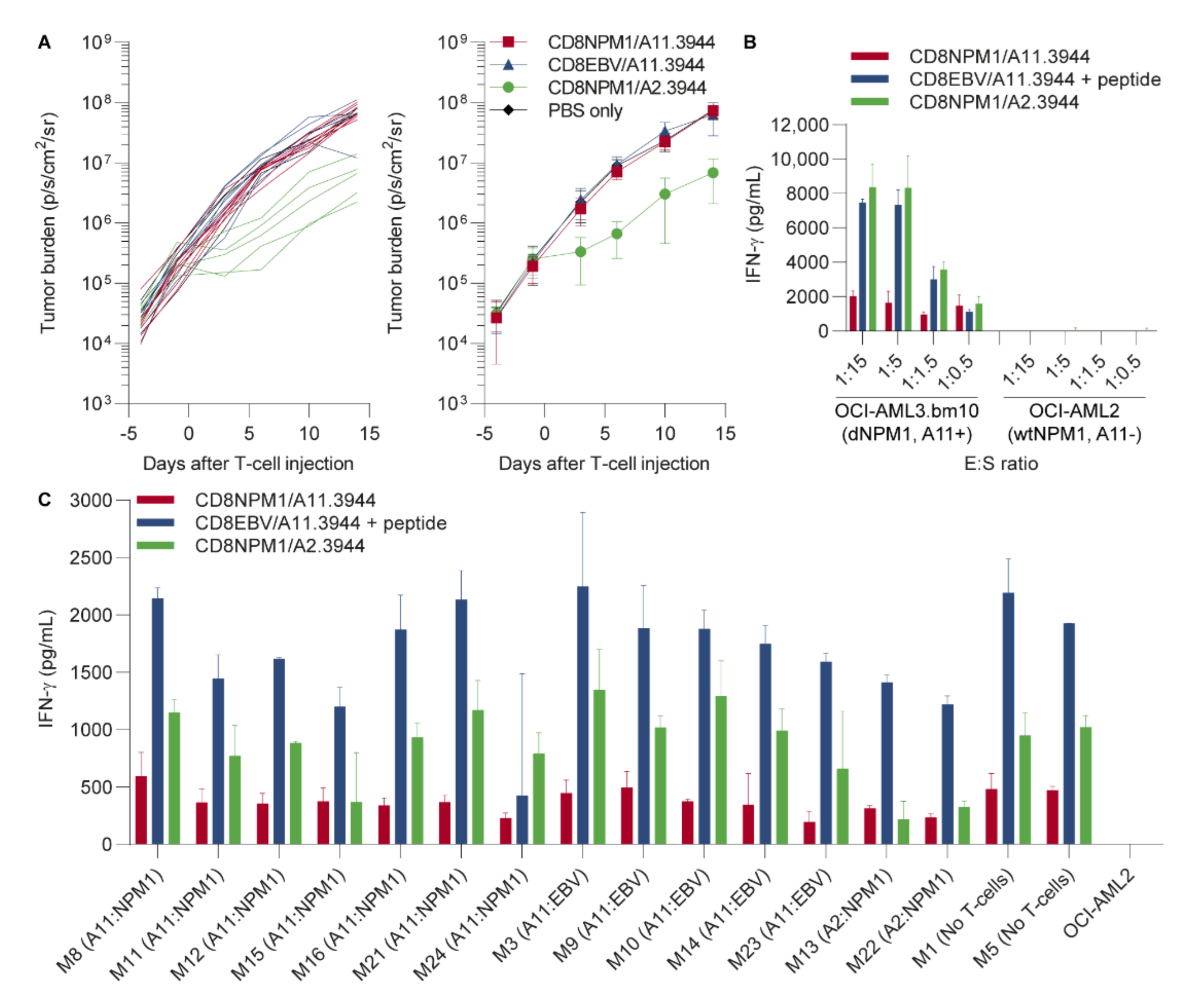
3.3. The HLA-A11-Restricted TCR for dNPM1 Fails to Target AML in Mice
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Thiede, C.; Koch, S.; Creutzig, E.; Steudel, C.; Illmer, T.; Schaich, M.; Ehninger, G. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood 2006, 107, 4011–4020. [Google Scholar] [CrossRef]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef]
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. NPM1-mutated acute myeloid leukemia: From bench to bedside. Blood 2020, 136, 1707–1721. [Google Scholar] [CrossRef] [PubMed]
- Cocciardi, S.; Dolnik, A.; Kapp-Schwoerer, S.; Rücker, F.G.; Lux, S.; Blätte, T.J.; Skambraks, S.; Krönke, J.; Heidel, F.H.; Schnöder, T.M.; et al. Clonal evolution patterns in acute myeloid leukemia with NPM1 mutation. Nat. Commun. 2019, 10, 2031. [Google Scholar] [CrossRef]
- Höllein, A.; Meggendorfer, M.; Dicker, F.; Jeromin, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T. NPM1 mutated AML can relapse with wild-type NPM1: Persistent clonal hematopoiesis can drive relapse. Blood Adv. 2018, 2, 3118–3125. [Google Scholar] [CrossRef]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Bullinger, L.; Teleanu, V.; Tschürtz, F.; Gaidzik, V.I.; Kühn, M.W.; Rücker, F.G.; Holzmann, K.; Paschka, P.; Kapp-Schwörer, S.; et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood 2013, 122, 100–108. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, D.I.; Reijmers, R.M.; Honders, M.W.; Hagedoorn, R.S.; de Jong, R.C.; Kester, M.G.; van der Steen, D.M.; de Ru, A.H.; Kweekel, C.; Bijen, H.M.; et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J. Clin. Investig. 2019, 129, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.; Olsson, N.; Wagar, L.E.; Medeiros, B.C.; Meyer, E.; Czerwinski, D.; Khodadoust, M.S.; Zhang, L.; Schultz, L.; Davis, M.M.; et al. Acute myeloid leukemia immunopeptidome reveals HLA presentation of mutated nucleophosmin. PLoS ONE 2019, 14, e0219547. [Google Scholar] [CrossRef]
- Miller, G.; Shope, T.; Lisco, H.; Stitt, D.; Lipman, M. Epstein-Barr virus: Transformation, cytopathic changes, and viral antigens in squirrel monkey and marmoset leukocytes. Proc. Natl. Acad. Sci. USA 1972, 69, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Kremer, A.N.; van der Meijden, E.D.; Honders, M.W.; Goeman, J.J.; Wiertz, E.J.; Falkenburg, J.H.; Griffioen, M. Endogenous HLA class II epitopes that are immunogenic in vivo show distinct behavior toward HLA-DM and its natural inhibitor HLA-DO. Blood 2012, 120, 3246–3255. [Google Scholar] [CrossRef] [PubMed]
- Garboczi, D.N.; Hung, D.T.; Wiley, D.C. HLA-A2-peptide complexes: Refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc. Natl. Acad. Sci. USA 1992, 89, 3429–3433. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.L.; Geluk, A.; Toebes, M.; Ottenhoff, T.H.; Drijfhout, J.W. A novel, highly efficient peptide-HLA class I binding assay using unfolded heavy chain molecules: Identification of HIV-1 derived peptides that bind to HLA-A*0201 and HLA-A*0301. J. Immunol. Methods 1997, 205, 201–209. [Google Scholar] [CrossRef]
- van Bergen, C.A.; van Luxemburg-Heijs, S.A.; de Wreede, L.C.; Eefting, M.; von dem Borne, P.A.; van Balen, P.; Heemskerk, M.H.; Mulder, A.; Claas, F.H.; Navarrete, M.A.; et al. Selective graft-versus-leukemia depends on magnitude and diversity of the alloreactive T cell response. J. Clin. Investig. 2017, 127, 517–529. [Google Scholar] [CrossRef]
- Linnemann, C.; Heemskerk, B.; Kvistborg, P.; Kluin, R.J.; Bolotin, D.A.; Chen, X.; Bresser, K.; Nieuwland, M.; Schotte, R.; Michels, S.; et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat. Med. 2013, 19, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Parham, P.; Brodsky, F.M. Partial purification and some properties of BB7.2. A cytotoxic monoclonal antibody with specificity for HLA-A2 and a variant of HLA-A28. Hum. Immunol. 1981, 3, 277–299. [Google Scholar] [CrossRef]
- Dustin, M.L. The immunological synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, R.E.; Zwinderman, K.H.; Kluin-Nelemans, H.C.; van Luxemburg-Heijs, S.A.; Willemze, R.; Falkenburg, J.H. Expression and induction of costimulatory and adhesion molecules on acute myeloid leukemic cells: Implications for adoptive immunotherapy. Exp. Hematol. 2000, 28, 161–168. [Google Scholar] [CrossRef]
- Costello, R.T.; Mallet, F.; Sainty, D.; Maraninchi, D.; Gastaut, J.A.; Olive, D. Regulation of CD80/B7-1 and CD86/B7-2 molecule expression in human primary acute myeloid leukemia and their role in allogenic immune recognition. Eur. J. Immunol. 1998, 28, 90–103. [Google Scholar] [CrossRef]
- Villalobos, I.B.; Takahashi, Y.; Akatsuka, Y.; Muramatsu, H.; Nishio, N.; Hama, A.; Yagasaki, H.; Saji, H.; Kato, M.; Ogawa, S.; et al. Relapse of leukemia with loss of mismatched HLA resulting from uniparental disomy after haploidentical hematopoietic stem cell transplantation. Blood 2010, 115, 3158–3161. [Google Scholar] [CrossRef] [PubMed]
- Vago, L.; Perna, S.K.; Zanussi, M.; Mazzi, B.; Barlassina, C.; Stanghellini, M.T.; Perrelli, N.F.; Cosentino, C.; Torri, F.; Angius, A.; et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N. Engl. J. Med. 2009, 361, 478–488. [Google Scholar] [CrossRef]
- Brouwer, R.E.; van der Heiden, P.; Schreuder, G.M.; Mulder, A.; Datema, G.; Anholts, J.D.; Willemze, R.; Claas, F.H.; Falkenburg, J.H. Loss or downregulation of HLA class I expression at the allelic level in acute leukemia is infrequent but functionally relevant, and can be restored by interferon. Hum. Immunol. 2002, 63, 200–210. [Google Scholar] [CrossRef]
- Karpanen, T.; Olweus, J. The Potential of Donor T-Cell Repertoires in Neoantigen-Targeted Cancer Immunotherapy. Front. Immunol. 2017, 8, 1718. [Google Scholar] [CrossRef] [PubMed]
- Falkenburg, J.H.; Jedema, I. Allo-reactive T cells for the treatment of hematological malignancies. Mol. Oncol. 2015, 9, 1894–1903. [Google Scholar] [CrossRef]
- Kunert, A.; Obenaus, M.; Lamers, C.H.J.; Blankenstein, T.; Debets, R. T-cell Receptors for Clinical Therapy: In Vitro Assessment of Toxicity Risk. Clin. Cancer Res. 2017, 23, 6012–6020. [Google Scholar] [CrossRef]
- Holler, P.D.; Holman, P.O.; Shusta, E.V.; O’Herrin, S.; Wittrup, K.D.; Kranz, D.M. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc. Natl. Acad. Sci. USA 2000, 97, 5387–5392. [Google Scholar] [CrossRef]
- Li, Y.; Moysey, R.; Molloy, P.E.; Vuidepot, A.L.; Mahon, T.; Baston, E.; Dunn, S.; Liddy, N.; Jacob, J.; Jakobsen, B.K.; et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005, 23, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Chervin, A.S.; Aggen, D.H.; Raseman, J.M.; Kranz, D.M. Engineering higher affinity T cell receptors using a T cell display system. J. Immunol. Methods 2008, 339, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Malecek, K.; Zhong, S.; McGary, K.; Yu, C.; Huang, K.; Johnson, L.A.; Rosenberg, S.A.; Krogsgaard, M. Engineering improved T cell receptors using an alanine-scan guided T cell display selection system. J. Immunol. Methods 2013, 392, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Haidar, J.N.; Pierce, B.; Yu, Y.; Tong, W.; Li, M.; Weng, Z. Structure-based design of a T-cell receptor leads to nearly 100-fold improvement in binding affinity for pepMHC. Proteins 2009, 74, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Malecek, K.; Grigoryan, A.; Zhong, S.; Gu, W.J.; Johnson, L.A.; Rosenberg, S.A.; Cardozo, T.; Krogsgaard, M. Specific increase in potency via structure-based design of a TCR. J. Immunol. 2014, 193, 2587–2599. [Google Scholar] [CrossRef] [PubMed]
- Bassan, D.; Gozlan, Y.M.; Sharbi-Yunger, A.; Tzehoval, E.; Eisenbach, L. Optimizing T-cell receptor avidity with somatic hypermutation. Int. J. Cancer 2019, 145, 2816–2826. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Mohammed, F.; Reijmers, R.M.; Woolston, A.; Stauss, T.; Kennedy, A.; Stirling, D.; Holler, A.; Green, L.; Jones, D.; et al. Framework engineering to produce dominant T cell receptors with enhanced antigen-specific function. Nat. Commun. 2019, 10, 4451. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; June, C.H. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 2017, 27, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Tahk, S.; Lohner, A.; Hänel, G.; Maiser, A.; Hauke, M.; Patel, L.; Rothe, M.; Josenhans, C.; Leonhardt, H.; et al. Fusion of Bacterial Flagellin to a Dendritic Cell-Targeting αCD40 Antibody Construct Coupled With Viral or Leukemia-Specific Antigens Enhances Dendritic Cell Maturation and Activates Peptide-Responsive T Cells. Front. Immunol. 2020, 11, 602802. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Lee, D.I.; Koutsoumpli, G.; Reijmers, R.M.; Honders, M.W.; de Jong, R.C.M.; Remst, D.F.G.; Wachsmann, T.L.A.; Hagedoorn, R.S.; Franken, K.L.M.C.; Kester, M.G.D.; et al. An HLA-A*11:01-Binding Neoantigen from Mutated NPM1 as Target for TCR Gene Therapy in AML. Cancers 2021, 13, 5390. https://doi.org/10.3390/cancers13215390
van der Lee DI, Koutsoumpli G, Reijmers RM, Honders MW, de Jong RCM, Remst DFG, Wachsmann TLA, Hagedoorn RS, Franken KLMC, Kester MGD, et al. An HLA-A*11:01-Binding Neoantigen from Mutated NPM1 as Target for TCR Gene Therapy in AML. Cancers. 2021; 13(21):5390. https://doi.org/10.3390/cancers13215390
Chicago/Turabian Stylevan der Lee, Dyantha I., Georgia Koutsoumpli, Rogier M. Reijmers, M. Willy Honders, Rob C. M. de Jong, Dennis F. G. Remst, Tassilo L. A. Wachsmann, Renate S. Hagedoorn, Kees L. M. C. Franken, Michel G. D. Kester, and et al. 2021. "An HLA-A*11:01-Binding Neoantigen from Mutated NPM1 as Target for TCR Gene Therapy in AML" Cancers 13, no. 21: 5390. https://doi.org/10.3390/cancers13215390
APA Stylevan der Lee, D. I., Koutsoumpli, G., Reijmers, R. M., Honders, M. W., de Jong, R. C. M., Remst, D. F. G., Wachsmann, T. L. A., Hagedoorn, R. S., Franken, K. L. M. C., Kester, M. G. D., Harber, K. J., Roelofsen, L. M., Schouten, A. M., Mulder, A., Drijfhout, J. W., Veelken, H., van Veelen, P. A., Heemskerk, M. H. M., Falkenburg, J. H. F., & Griffioen, M. (2021). An HLA-A*11:01-Binding Neoantigen from Mutated NPM1 as Target for TCR Gene Therapy in AML. Cancers, 13(21), 5390. https://doi.org/10.3390/cancers13215390