
Transcriptomics, Epigenetics, and Metabolomics of Primary Aldosteronism




Abstract
:Simple Summary
Abstract
1. Introduction
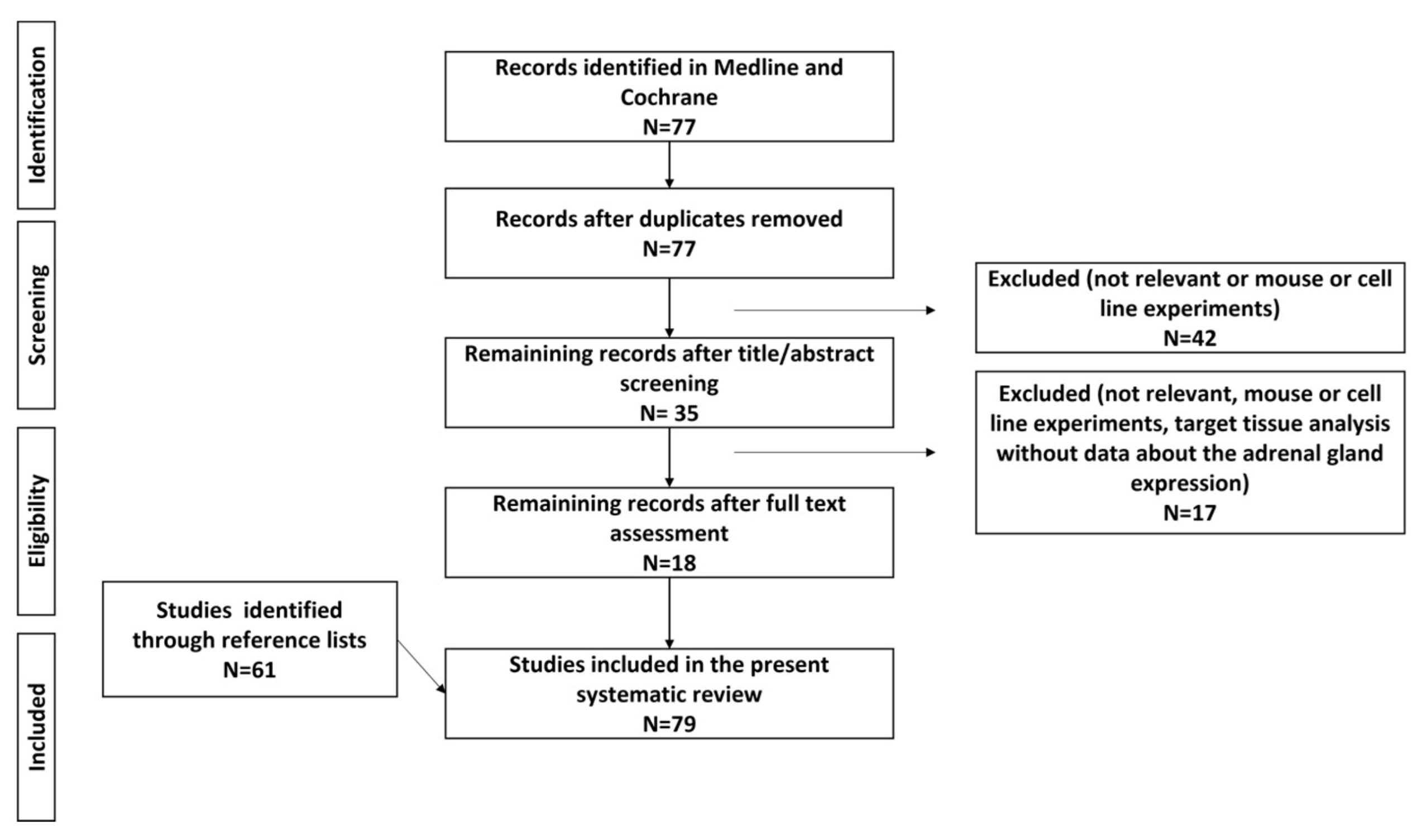
2. Methodology
3. Results
3.1. Clinical and Histological Traits of PA Patients
3.2. Transcriptomics
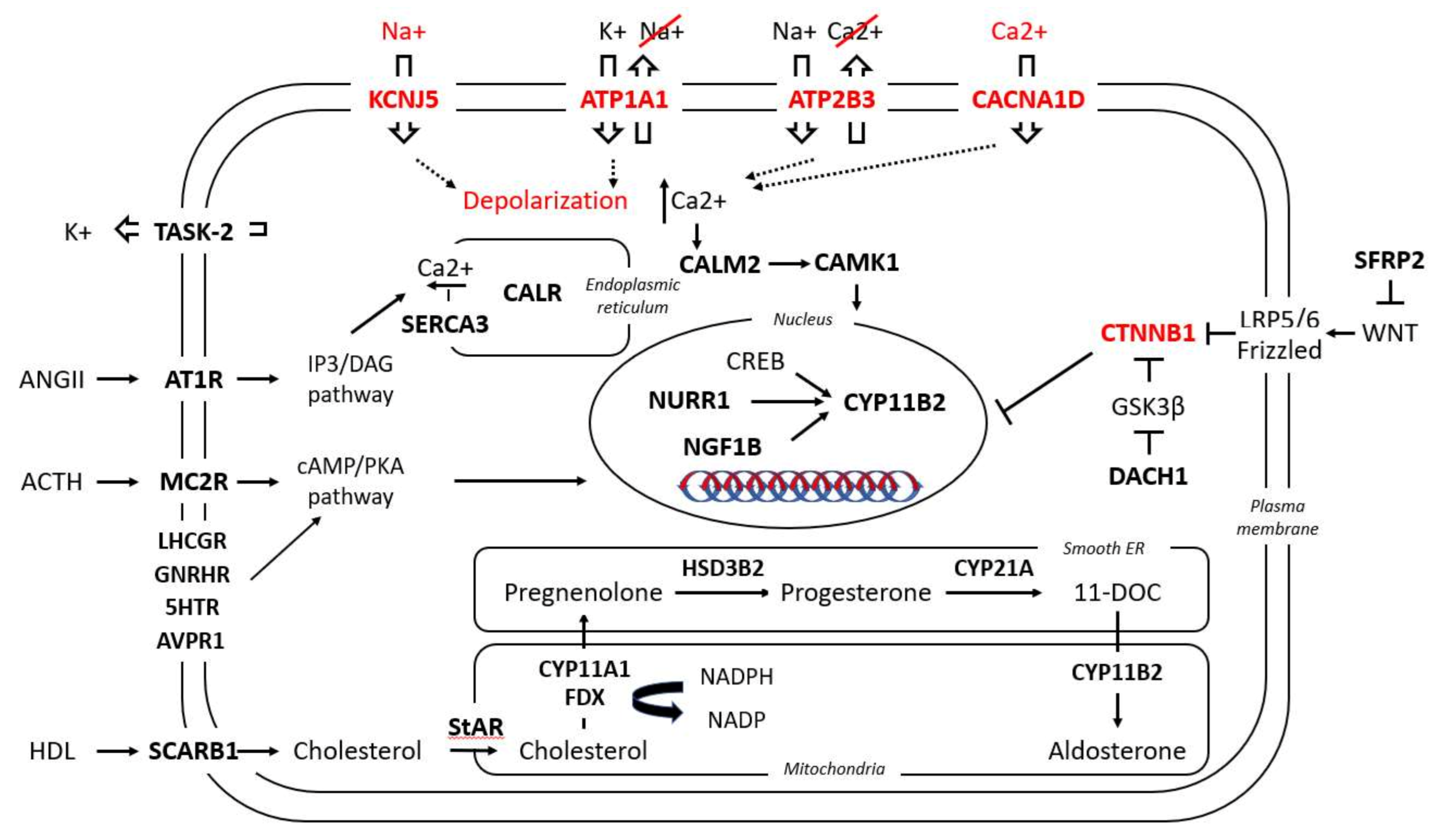
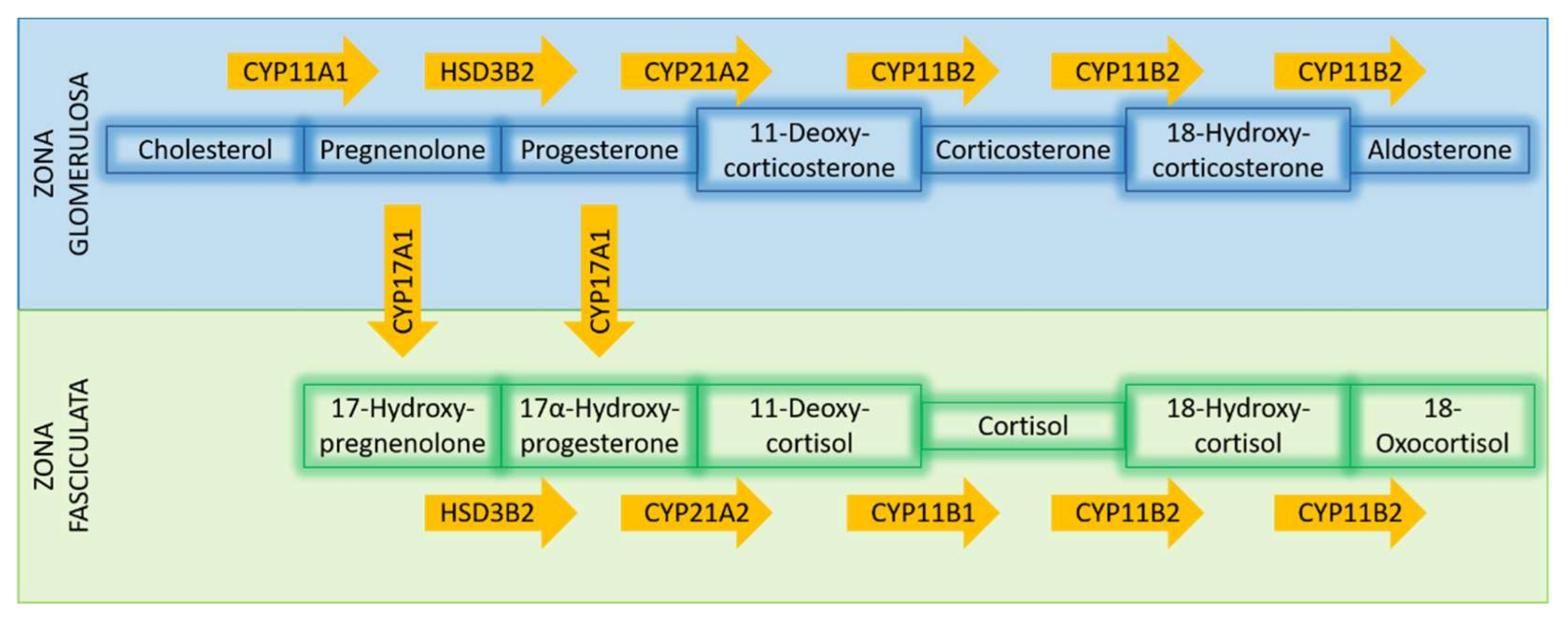
3.2.1. Steroidogenic Enzymes
3.2.2. Nuclear Receptor Transcription Factors
3.2.3. Plasma Membrane Receptors
3.2.4. Ion Channels
3.2.5. Calcium Signaling
3.2.6. G-Protein Coupled Receptors (GPCRs)
3.2.7. Energy
3.2.8. Protein Binding
3.2.9. Cell Growth/Cell Death
3.2.10. Immune Response
3.2.11. DNA Binding/RNA Polymerase
3.3. Epigenetics
3.4. Metabolomics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Funder, J.W.; Carey, R.M.; Mantero, F.; Murad, M.H.; Reincke, M.; Shibata, H.; Stowasser, M.; Young, W.F., Jr. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2016, 101, 1889–1916. [Google Scholar] [CrossRef] [PubMed]
- Monticone, S.; Burrello, J.; Tizzani, D.; Bertello, C.; Viola, A.; Buffolo, F.; Gabetti, L.; Mengozzi, G.; Williams, T.A.; Rabbia, F.; et al. Prevalence and Clinical Manifestations of Primary Aldosteronism Encountered in Primary Care Practice. J. Am. Coll. Cardiol. 2017, 69, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Monticone, S.; D’Ascenzo, F.; Moretti, C.; Williams, T.A.; Veglio, F.; Gaita, F.; Mulatero, P. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2018, 6, 41–50. [Google Scholar] [CrossRef]
- Choi, M.; Scholl, U.I.; Yue, P.; Bjorklund, P.; Zhao, B.; Nelson-Williams, C.; Ji, W.; Cho, Y.; Patel, A.; Men, C.J.; et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011, 331, 768–772. [Google Scholar] [CrossRef] [Green Version]
- Okamura, T.; Nakajima, Y.; Katano-Toki, A.; Horiguchi, K.; Matsumoto, S.; Yoshino, S.; Yamada, E.; Tomaru, T.; Ishii, S.; Saito, T.; et al. Characteristics of Japanese aldosterone-producing adenomas with KCNJ5 mutations. Endocr. J. 2017, 64, 39–47. [Google Scholar] [CrossRef]
- Azizan, E.A.; Murthy, M.; Stowasser, M.; Gordon, R.; Kowalski, B.; Xu, S.; Brown, M.J.; O’Shaughnessy, K.M. Somatic mutations affecting the selectivity filter of KCNJ5 are frequent in 2 large unselected collections of adrenal aldosteronomas. Hypertension 2012, 59, 587–591. [Google Scholar] [CrossRef]
- Boulkroun, S.; Beuschlein, F.; Rossi, G.P.; Golib-Dzib, J.F.; Fischer, E.; Amar, L.; Mulatero, P.; Samson-Couterie, B.; Hahner, S.; Quinkler, M.; et al. Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension 2012, 59, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Beuschlein, F.; Boulkroun, S.; Osswald, A.; Wieland, T.; Nielsen, H.N.; Lichtenauer, U.D.; Penton, D.; Schack, V.R.; Amar, L.; Fischer, E.; et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat. Genet. 2013, 45, 440–444. [Google Scholar] [CrossRef]
- Scholl, U.I.; Goh, G.; Stolting, G.; de Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054. [Google Scholar] [CrossRef]
- Stindl, J.; Tauber, P.; Sterner, C.; Tegtmeier, I.; Warth, R.; Bandulik, S. Pathogenesis of Adrenal Aldosterone-Producing Adenomas Carrying Mutations of the Na(+)/K(+)-ATPase. Endocrinology 2015, 156, 4582–4591. [Google Scholar] [CrossRef]
- Berthon, A.; Drelon, C.; Ragazzon, B.; Boulkroun, S.; Tissier, F.; Amar, L.; Samson-Couterie, B.; Zennaro, M.C.; Plouin, P.F.; Skah, S.; et al. WNT/beta-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum. Mol. Genet. 2014, 23, 889–905. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Azizan, E.A.B.; Cabrera, C.P.; Fernandes-Rosa, F.L.; Boulkroun, S.; Argentesi, G.; Cottrell, E.; Amar, L.; Wu, X.; O’Toole, S.; et al. Somatic mutations of GNA11 and GNAQ in CTNNB1-mutant aldosterone-producing adenomas presenting in puberty, pregnancy or menopause. Nat. Genet. 2021, 53, 1360–1372. [Google Scholar] [CrossRef]
- Dutta, R.K.; Arnesen, T.; Heie, A.; Walz, M.; Alesina, P.; Soderkvist, P.; Gimm, O. A somatic mutation in CLCN2 identified in a sporadic aldosterone-producing adenoma. Eur. J. Endocrinol. 2019, 181, K37–K41. [Google Scholar] [CrossRef]
- Rhayem, Y.; Perez-Rivas, L.G.; Dietz, A.; Bathon, K.; Gebhard, C.; Riester, A.; Mauracher, B.; Gomez-Sanchez, C.; Eisenhofer, G.; Schwarzmayr, T.; et al. PRKACA Somatic Mutations Are Rare Findings in Aldosterone-Producing Adenomas. J. Clin. Endocrinol. Metab. 2016, 101, 3010–3017. [Google Scholar] [CrossRef]
- Nanba, K.; Blinder, A.R.; Rege, J.; Hattangady, N.G.; Else, T.; Liu, C.J.; Tomlins, S.A.; Vats, P.; Kumar-Sinha, C.; Giordano, T.J.; et al. Somatic CACNA1H Mutation as a Cause of Aldosterone-Producing Adenoma. Hypertension 2020, 75, 645–649. [Google Scholar] [CrossRef]
- Lifton, R.P.; Dluhy, R.G.; Powers, M.; Rich, G.M.; Cook, S.; Ulick, S.; Lalouel, J.M. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 1992, 355, 262–265. [Google Scholar] [CrossRef]
- Scholl, U.I.; Stolting, G.; Schewe, J.; Thiel, A.; Tan, H.; Nelson-Williams, C.; Vichot, A.A.; Jin, S.C.; Loring, E.; Untiet, V.; et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat. Genet. 2018, 50, 349–354. [Google Scholar] [CrossRef]
- Monticone, S.; Tetti, M.; Burrello, J.; Buffolo, F.; De Giovanni, R.; Veglio, F.; Williams, T.A.; Mulatero, P. Familial hyperaldosteronism type III. J. Hum. Hypertens. 2017, 31, 776–781. [Google Scholar] [CrossRef]
- Scholl, U.I.; Stolting, G.; Nelson-Williams, C.; Vichot, A.A.; Choi, M.; Loring, E.; Prasad, M.L.; Goh, G.; Carling, T.; Juhlin, C.C.; et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. elife 2015, 4, e06315. [Google Scholar] [CrossRef] [Green Version]
- Daniil, G.; Fernandes-Rosa, F.L.; Chemin, J.; Blesneac, I.; Beltrand, J.; Polak, M.; Jeunemaitre, X.; Boulkroun, S.; Amar, L.; Strom, T.M.; et al. CACNA1H Mutations Are Associated with Different Forms of Primary Aldosteronism. EBioMedicine 2016, 13, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Azizan, E.A.; Lam, B.Y.; Newhouse, S.J.; Zhou, J.; Kuc, R.E.; Clarke, J.; Happerfield, L.; Marker, A.; Hoffman, G.J.; Brown, M.J. Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J. Clin. Endocrinol. Metab. 2012, 97, E819–E829. [Google Scholar] [CrossRef] [Green Version]
- Monticone, S.; Castellano, I.; Versace, K.; Lucatello, B.; Veglio, F.; Gomez-Sanchez, C.E.; Williams, T.A.; Mulatero, P. Immunohistochemical, genetic and clinical characterization of sporadic aldosterone-producing adenomas. Mol. Cell Endocrinol. 2015, 411, 146–154. [Google Scholar] [CrossRef]
- Scholl, U.I.; Healy, J.M.; Thiel, A.; Fonseca, A.L.; Brown, T.C.; Kunstman, J.W.; Horne, M.J.; Dietrich, D.; Riemer, J.; Kucukkoylu, S.; et al. Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin. Endocrinol. 2015, 83, 779–789. [Google Scholar] [CrossRef]
- Fernandes-Rosa, F.L.; Williams, T.A.; Riester, A.; Steichen, O.; Beuschlein, F.; Boulkroun, S.; Strom, T.M.; Monticone, S.; Amar, L.; Meatchi, T.; et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 2014, 64, 354–361. [Google Scholar] [CrossRef]
- Azizan, E.A.; Poulsen, H.; Tuluc, P.; Zhou, J.; Clausen, M.V.; Lieb, A.; Maniero, C.; Garg, S.; Bochukova, E.G.; Zhao, W.; et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat. Genet. 2013, 45, 1055–1060. [Google Scholar] [CrossRef]
- Williams, T.A.; Gomez-Sanchez, C.E.; Rainey, W.E.; Giordano, T.J.; Lam, A.K.; Marker, A.; Mete, O.; Yamazaki, Y.; Zerbini, M.C.N.; Beuschlein, F.; et al. International Histopathology Consensus for Unilateral Primary Aldosteronism. J. Clin. Endocrinol. Metab. 2021, 106, 42–54. [Google Scholar] [CrossRef]
- Boulkroun, S.; Samson-Couterie, B.; Dzib, J.F.; Lefebvre, H.; Louiset, E.; Amar, L.; Plouin, P.F.; Lalli, E.; Jeunemaitre, X.; Benecke, A.; et al. Adrenal cortex remodeling and functional zona glomerulosa hyperplasia in primary aldosteronism. Hypertension 2010, 56, 885–892. [Google Scholar] [CrossRef] [Green Version]
- Nishimoto, K.; Nakagawa, K.; Li, D.; Kosaka, T.; Oya, M.; Mikami, S.; Shibata, H.; Itoh, H.; Mitani, F.; Yamazaki, T.; et al. Adrenocortical zonation in humans under normal and pathological conditions. J. Clin. Endocrinol. Metab. 2010, 95, 2296–2305. [Google Scholar] [CrossRef] [Green Version]
- Omata, K.; Anand, S.K.; Hovelson, D.H.; Liu, C.J.; Yamazaki, Y.; Nakamura, Y.; Ito, S.; Satoh, F.; Sasano, H.; Rainey, W.E.; et al. Aldosterone-Producing Cell Clusters Frequently Harbor Somatic Mutations and Accumulate with Age in Normal Adrenals. J. Endocr. Soc. 2017, 1, 787–799. [Google Scholar] [CrossRef] [Green Version]
- Omata, K.; Tomlins, S.A.; Rainey, W.E. Aldosterone-Producing Cell Clusters in Normal and Pathological States. Horm. Metab. Res. 2017, 49, 951–956. [Google Scholar] [CrossRef]
- Nanba, K.; Vaidya, A.; Williams, G.H.; Zheng, I.; Else, T.; Rainey, W.E. Age-Related Autonomous Aldosteronism. Circulation 2017, 136, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, Y.; Takeo, E.; Shimma, S.; Yokota, M.; Higashi, T.; Seki, T.; Mizuno, Y.; Oya, M.; Kosaka, T.; Omura, M.; et al. Aldosterone and 18-Oxocortisol Coaccumulation in Aldosterone-Producing Lesions. Hypertension 2018, 72, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, K.; Tomlins, S.A.; Kuick, R.; Cani, A.K.; Giordano, T.J.; Hovelson, D.H.; Liu, C.J.; Sanjanwala, A.R.; Edwards, M.A.; Gomez-Sanchez, C.E.; et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc. Natl. Acad. Sci. USA 2015, 112, E4591–E4599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Sanchez, C.E.; Kuppusamy, M.; Reincke, M.; Williams, T.A. Disordered CYP11B2 Expression in Primary Aldosteronism. Horm. Metab. Res. 2017, 49, 957–962. [Google Scholar] [CrossRef] [Green Version]
- Nishimoto, K.; Seki, T.; Kurihara, I.; Yokota, K.; Omura, M.; Nishikawa, T.; Shibata, H.; Kosaka, T.; Oya, M.; Suematsu, M.; et al. Case Report: Nodule Development from Subcapsular Aldosterone-Producing Cell Clusters Causes Hyperaldosteronism. J. Clin. Endocrinol. Metab. 2016, 101, 6–9. [Google Scholar] [CrossRef]
- De Sousa, K.; Abdellatif, A.B.; Giscos-Douriez, I.; Meatchi, T.; Amar, L.; Fernandes-Rosa, F.L.; Boulkroun, S.; Zennaro, M.C. Colocalization of Wnt/beta-catenin and ACTH signaling pathways and paracrine regulation in aldosterone producing adenoma. J. Clin. Endocrinol. Metab. 2021, dgab707. [Google Scholar] [CrossRef]
- De Sousa, K.; Boulkroun, S.; Baron, S.; Nanba, K.; Wack, M.; Rainey, W.E.; Rocha, A.; Giscos-Douriez, I.; Meatchi, T.; Amar, L.; et al. Genetic, Cellular, and Molecular Heterogeneity in Adrenals with Aldosterone-Producing Adenoma. Hypertension 2020, 75, 1034–1044. [Google Scholar] [CrossRef]
- Vouillarmet, J.; Fernandes-Rosa, F.; Graeppi-Dulac, J.; Lantelme, P.; Decaussin-Petrucci, M.; Thivolet, C.; Peix, J.L.; Boulkroun, S.; Clauser, E.; Zennaro, M.C. Aldosterone-Producing Adenoma with a Somatic KCNJ5 Mutation Revealing APC-Dependent Familial Adenomatous Polyposis. J. Clin. Endocrinol. Metab. 2016, 101, 3874–3878. [Google Scholar] [CrossRef]
- Dekkers, T.; ter Meer, M.; Lenders, J.W.; Hermus, A.R.; Schultze Kool, L.; Langenhuijsen, J.F.; Nishimoto, K.; Ogishima, T.; Mukai, K.; Azizan, E.A.; et al. Adrenal nodularity and somatic mutations in primary aldosteronism: One node is the culprit? J. Clin. Endocrinol. Metab. 2014, 99, E1341–E1351. [Google Scholar] [CrossRef] [Green Version]
- Nanba, K.; Chen, A.X.; Omata, K.; Vinco, M.; Giordano, T.J.; Else, T.; Hammer, G.D.; Tomlins, S.A.; Rainey, W.E. Molecular Heterogeneity in Aldosterone-Producing Adenomas. J. Clin. Endocrinol. Metab. 2016, 101, 999–1007. [Google Scholar] [CrossRef]
- Itcho, K.; Oki, K.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Ohno, H.; Kobuke, K.; Nagano, G.; Yoshii, Y.; Baba, R.; Hattori, N.; et al. Endoplasmic Reticulum Chaperone Calmegin Is Upregulated in Aldosterone-Producing Adenoma and Associates with Aldosterone Production. Hypertension 2020, 75, 492–499. [Google Scholar] [CrossRef]
- Omata, K.; Satoh, F.; Morimoto, R.; Ito, S.; Yamazaki, Y.; Nakamura, Y.; Anand, S.K.; Guo, Z.; Stowasser, M.; Sasano, H.; et al. Cellular and Genetic Causes of Idiopathic Hyperaldosteronism. Hypertension 2018, 72, 874–880. [Google Scholar] [CrossRef]
- Hacini, I.; De Sousa, K.; Boulkroun, S.; Meatchi, T.; Amar, L.; Zennaro, M.C.; Fernandes-Rosa, F.L. Somatic mutations in adrenals from patients with primary aldosteronism not cured after adrenalectomy suggest common pathogenic mechanisms between unilateral and bilateral disease. Eur. J. Endocrinol. 2021, 185, 405–412. [Google Scholar] [CrossRef]
- Fernandes-Rosa, F.L.; Amar, L.; Tissier, F.; Bertherat, J.; Meatchi, T.; Zennaro, M.C.; Boulkroun, S. Functional histopathological markers of aldosterone producing adenoma and somatic KCNJ5 mutations. Mol. Cell Endocrinol. 2015, 408, 220–226. [Google Scholar] [CrossRef]
- Plaska, S.W.; Liu, C.J.; Lim, J.S.; Rege, J.; Bick, N.R.; Lerario, A.M.; Hammer, G.D.; Giordano, T.J.; Else, T.; Tomlins, S.A.; et al. Targeted RNAseq of Formalin-Fixed Paraffin-Embedded Tissue to Differentiate among Benign and Malignant Adrenal Cortical Tumors. Horm. Metab. Res. 2020, 52, 607–613. [Google Scholar] [CrossRef]
- Monticone, S.; Hattangady, N.G.; Nishimoto, K.; Mantero, F.; Rubin, B.; Cicala, M.V.; Pezzani, R.; Auchus, R.J.; Ghayee, H.K.; Shibata, H.; et al. Effect of KCNJ5 mutations on gene expression in aldosterone-producing adenomas and adrenocortical cells. J. Clin. Endocrinol. Metab. 2012, 97, E1567–E1572. [Google Scholar] [CrossRef] [Green Version]
- Akerstrom, T.; Willenberg, H.S.; Cupisti, K.; Ip, J.; Backman, S.; Moser, A.; Maharjan, R.; Robinson, B.; Iwen, K.A.; Dralle, H.; et al. Novel somatic mutations and distinct molecular signature in aldosterone-producing adenomas. Endocr. Relat. Cancer 2015, 22, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Bassett, M.H.; Mayhew, B.; Rehman, K.; White, P.C.; Mantero, F.; Arnaldi, G.; Stewart, P.M.; Bujalska, I.; Rainey, W.E. Expression profiles for steroidogenic enzymes in adrenocortical disease. J. Clin. Endocrinol. Metab. 2005, 90, 5446–5455. [Google Scholar] [CrossRef] [Green Version]
- Backman, S.; Akerstrom, T.; Maharjan, R.; Cupisti, K.; Willenberg, H.S.; Hellman, P.; Bjorklund, P. RNA Sequencing Provides Novel Insights into the Transcriptome of Aldosterone Producing Adenomas. Sci. Rep. 2019, 9, 6269. [Google Scholar] [CrossRef] [Green Version]
- Saner-Amigh, K.; Mayhew, B.A.; Mantero, F.; Schiavi, F.; White, P.C.; Rao, C.V.; Rainey, W.E. Elevated expression of luteinizing hormone receptor in aldosterone-producing adenomas. J. Clin. Endocrinol. Metab. 2006, 91, 1136–1142. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Satoh, F.; Morimoto, R.; Nakamura, Y.; Sasano, H.; Auchus, R.J.; Edwards, M.A.; Rainey, W.E. Gene expression profiles in aldosterone-producing adenomas and adjacent adrenal glands. Eur. J. Endocrinol. 2011, 164, 613–619. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.A.; Monticone, S.; Crudo, V.; Warth, R.; Veglio, F.; Mulatero, P. Visinin-like 1 is upregulated in aldosterone-producing adenomas with KCNJ5 mutations and protects from calcium-induced apoptosis. Hypertension 2012, 59, 833–839. [Google Scholar] [CrossRef] [Green Version]
- Assie, G.; Auzan, C.; Gasc, J.M.; Baviera, E.; Balaton, A.; Elalouf, J.M.; Jeunemaitre, X.; Plouin, P.F.; Corvol, P.; Clauser, E. Steroidogenesis in aldosterone-producing adenoma revisited by transcriptome analysis. J. Clin. Endocrinol. Metab. 2005, 90, 6638–6649. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.X.; Yang, X.C.; Gao, Y.X.; Zhuang, M.; Wang, K.P.; Sun, L.J.; Wang, X.S. Expression of aldosterone synthase and adrenocorticotropic hormone receptor in adrenal incidentalomas from normotensive and hypertensive patients: Distinguishing subclinical or atypical primary aldosteronism from adrenal incidentaloma. Int. J. Mol. Med. 2012, 30, 1396–1402. [Google Scholar] [CrossRef]
- El Zein, R.M.; Soria, A.H.; Golib Dzib, J.F.; Rickard, A.J.; Fernandes-Rosa, F.L.; Samson-Couterie, B.; Giscos-Douriez, I.; Rocha, A.; Poglitsch, M.; Gomez-Sanchez, C.E.; et al. Retinoic acid receptor alpha as a novel contributor to adrenal cortex structure and function through interactions with Wnt and Vegfa signalling. Sci. Rep. 2019, 9, 14677. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Zhao, C.; Zhang, Z.; Wang, M.; Zhang, Z.; Yang, A.; Ma, B.; Gu, M.; Cui, R.; Xin, Z.; et al. Transcriptome analysis of primary aldosteronism in adrenal glands and controls. Int. J. Clin. Exp. Pathol. 2017, 10, 10009–10018. [Google Scholar]
- Lenzini, L.; Seccia, T.M.; Aldighieri, E.; Belloni, A.S.; Bernante, P.; Giuliani, L.; Nussdorfer, G.G.; Pessina, A.C.; Rossi, G.P. Heterogeneity of aldosterone-producing adenomas revealed by a whole transcriptome analysis. Hypertension 2007, 50, 1106–1113. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.A.; Monticone, S.; Schack, V.R.; Stindl, J.; Burrello, J.; Buffolo, F.; Annaratone, L.; Castellano, I.; Beuschlein, F.; Reincke, M.; et al. Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension 2014, 63, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Kitamoto, T.; Suematsu, S.; Yamazaki, Y.; Nakamura, Y.; Sasano, H.; Matsuzawa, Y.; Saito, J.; Omura, M.; Nishikawa, T. Clinical and Steroidogenic Characteristics of Aldosterone-Producing Adenomas with ATPase or CACNA1D Gene Mutations. J. Clin. Endocrinol. Metab. 2016, 101, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Fallo, F.; Castellano, I.; Gomez-Sanchez, C.E.; Rhayem, Y.; Pilon, C.; Vicennati, V.; Santini, D.; Maffeis, V.; Fassina, A.; Mulatero, P.; et al. Histopathological and genetic characterization of aldosterone-producing adenomas with concurrent subclinical cortisol hypersecretion: A case series. Endocrine 2017, 58, 503–512. [Google Scholar] [CrossRef]
- Bassett, M.H.; Suzuki, T.; Sasano, H.; De Vries, C.J.; Jimenez, P.T.; Carr, B.R.; Rainey, W.E. The orphan nuclear receptor NGFIB regulates transcription of 3beta-hydroxysteroid dehydrogenase. implications for the control of adrenal functional zonation. J. Biol. Chem. 2004, 279, 37622–37630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Lam, B.; Neogi, S.G.; Yeo, G.S.; Azizan, E.A.; Brown, M.J. Transcriptome Pathway Analysis of Pathological and Physiological Aldosterone-Producing Human Tissues. Hypertension 2016, 68, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Ouyang, J.; Wu, Z.; Shi, T.; Wang, B.; Ma, X.; Li, H.; Wang, S.; Zhang, X. Elementary studies on elevated steroidogenic factor-1 expression in aldosterone-producing adenoma. Urol. Oncol. 2012, 30, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Reincke, M.; Beuschlein, F.; Lalli, E.; Arlt, W.; Vay, S.; Sassone-Corsi, P.; Allolio, B. DAX-1 expression in human adrenocortical neoplasms: Implications for steroidogenesis. J. Clin. Endocrinol. Metab. 1998, 83, 2597–2600. [Google Scholar] [CrossRef]
- Lenzini, L.; Caroccia, B.; Campos, A.G.; Fassina, A.; Belloni, A.S.; Seccia, T.M.; Kuppusamy, M.; Ferraro, S.; Skander, G.; Bader, M.; et al. Lower expression of the TWIK-related acid-sensitive K+ channel 2 (TASK-2) gene is a hallmark of aldosterone-producing adenoma causing human primary aldosteronism. J. Clin. Endocrinol. Metab. 2014, 99, E674–E682. [Google Scholar] [CrossRef] [Green Version]
- Maniero, C.; Scudieri, P.; Haris Shaikh, L.; Zhao, W.; Gurnell, M.; Galietta, L.J.V.; Brown, M.J. ANO4 (Anoctamin 4) Is a Novel Marker of Zona Glomerulosa That Regulates Stimulated Aldosterone Secretion. Hypertension 2019, 74, 1152–1159. [Google Scholar] [CrossRef]
- Kobuke, K.; Oki, K.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Ohno, H.; Itcho, K.; Yoshii, Y.; Yoneda, M.; Hattori, N. Calneuron 1 Increased Ca(2+) in the Endoplasmic Reticulum and Aldosterone Production in Aldosterone-Producing Adenoma. Hypertension 2018, 71, 125–133. [Google Scholar] [CrossRef]
- Oki, K.; Gomez-Sanchez, C.E. The landscape of molecular mechanism for aldosterone production in aldosterone-producing adenoma. Endocr. J. 2020, 67, 989–995. [Google Scholar] [CrossRef]
- Li, X.; Wang, B.; Tang, L.; Zhang, Y.; Chen, L.; Gu, L.; Zhang, F.; Ouyang, J.; Zhang, X. GSTA1 Expression Is Correlated With Aldosterone Level in KCNJ5-Mutated Adrenal Aldosterone-Producing Adenoma. J. Clin. Endocrinol. Metab. 2018, 103, 813–823. [Google Scholar] [CrossRef]
- Ye, P.; Mariniello, B.; Mantero, F.; Shibata, H.; Rainey, W.E. G-protein-coupled receptors in aldosterone-producing adenomas: A potential cause of hyperaldosteronism. J. Endocrinol. 2007, 195, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Itcho, K.; Oki, K.; Kobuke, K.; Yoshii, Y.; Ohno, H.; Yoneda, M.; Hattori, N. Aberrant G protein-receptor expression is associated with DNA methylation in aldosterone-producing adenoma. Mol. Cell Endocrinol. 2018, 461, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.S.; Plaska, S.W.; Rege, J.; Rainey, W.E.; Turcu, A.F. Aldosterone-Regulating Receptors and Aldosterone-Driver Somatic Mutations. Front. Endocrinol. 2021, 12, 644382. [Google Scholar] [CrossRef]
- Rossi, G.P.; Caroccia, B.; Seccia, T.M. Role of estrogen receptors in modulating aldosterone biosynthesis and blood pressure. Steroids 2019, 152, 108486. [Google Scholar] [CrossRef]
- Caroccia, B.; Seccia, T.M.; Campos, A.G.; Gioco, F.; Kuppusamy, M.; Ceolotto, G.; Guerzoni, E.; Simonato, F.; Mareso, S.; Lenzini, L.; et al. GPER-1 and estrogen receptor-beta ligands modulate aldosterone synthesis. Endocrinology 2014, 155, 4296–4304. [Google Scholar] [CrossRef] [Green Version]
- Gong, S.; Tetti, M.; Reincke, M.; Williams, T.A. Primary Aldosteronism: Metabolic Reprogramming and the Pathogenesis of Aldosterone-Producing Adenomas. Cancers 2021, 13, 3716. [Google Scholar] [CrossRef]
- Maniero, C.; Garg, S.; Zhao, W.; Johnson, T.I.; Zhou, J.; Gurnell, M.; Brown, M.J. NEFM (Neurofilament Medium) Polypeptide, a Marker for Zona Glomerulosa Cells in Human Adrenal, Inhibits D1R (Dopamine D1 Receptor)-Mediated Secretion of Aldosterone. Hypertension 2017, 70, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Teo, A.E.; Garg, S.; Johnson, T.I.; Zhao, W.; Zhou, J.; Gomez-Sanchez, C.E.; Gurnell, M.; Brown, M.J. Physiological and Pathological Roles in Human Adrenal of the Glomeruli-Defining Matrix Protein NPNT (Nephronectin). Hypertension 2017, 69, 1207–1216. [Google Scholar] [CrossRef]
- Williams, T.A.; Monticone, S.; Morello, F.; Liew, C.C.; Mengozzi, G.; Pilon, C.; Asioli, S.; Sapino, A.; Veglio, F.; Mulatero, P. Teratocarcinoma-derived growth factor-1 is upregulated in aldosterone-producing adenomas and increases aldosterone secretion and inhibits apoptosis in vitro. Hypertension 2010, 55, 1468–1475. [Google Scholar] [CrossRef]
- Penny, M.K.; Finco, I.; Hammer, G.D. Cell signaling pathways in the adrenal cortex: Links to stem/progenitor biology and neoplasia. Mol. Cell Endocrinol. 2017, 445, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Tissier, F.; Cavard, C.; Groussin, L.; Perlemoine, K.; Fumey, G.; Hagnere, A.M.; Rene-Corail, F.; Jullian, E.; Gicquel, C.; Bertagna, X.; et al. Mutations of beta-catenin in adrenocortical tumors: Activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005, 65, 7622–7627. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Shaikh, L.H.; Neogi, S.G.; McFarlane, I.; Zhao, W.; Figg, N.; Brighton, C.A.; Maniero, C.; Teo, A.E.; Azizan, E.A.; et al. DACH1, a zona glomerulosa selective gene in the human adrenal, activates transforming growth factor-beta signaling and suppresses aldosterone secretion. Hypertension 2015, 65, 1103–1110. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Tetti, M.; Vohra, T.; Adolf, C.; Seissler, J.; Hristov, M.; Belavgeni, A.; Bidlingmaier, M.; Linkermann, A.; Mulatero, P.; et al. BEX1 Is Differentially Expressed in Aldosterone-Producing Adenomas and Protects Human Adrenocortical Cells from Ferroptosis. Hypertension 2021, 77, 1647–1658. [Google Scholar] [CrossRef]
- Howard, B.; Wang, Y.; Xekouki, P.; Faucz, F.R.; Jain, M.; Zhang, L.; Meltzer, P.G.; Stratakis, C.A.; Kebebew, E. Integrated analysis of genome-wide methylation and gene expression shows epigenetic regulation of CYP11B2 in aldosteronomas. J. Clin. Endocrinol. Metab. 2014, 99, E536–E543. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Yoshimoto, T.; Nakabayashi, K.; Tsuchiya, K.; Minami, I.; Bouchi, R.; Izumiyama, H.; Fujii, Y.; Abe, K.; Tayama, C.; et al. Integration of transcriptome and methylome analysis of aldosterone-producing adenomas. Eur. J. Endocrinol. 2015, 173, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Di Dalmazi, G.; Morandi, L.; Rubin, B.; Pilon, C.; Asioli, S.; Vicennati, V.; De Leo, A.; Ambrosi, F.; Santini, D.; Pagotto, U.; et al. DNA Methylation of Steroidogenic Enzymes in Benign Adrenocortical Tumors: New Insights in Aldosterone-Producing Adenomas. J. Clin. Endocrinol. Metab. 2020, 105, e4605–e4615. [Google Scholar] [CrossRef]
- Yoshii, Y.; Oki, K.; Gomez-Sanchez, C.E.; Ohno, H.; Itcho, K.; Kobuke, K.; Yoneda, M. Hypomethylation of CYP11B2 in Aldosterone-Producing Adenoma. Hypertension 2016, 68, 1432–1437. [Google Scholar] [CrossRef] [Green Version]
- Kometani, M.; Yoneda, T.; Demura, M.; Aono, D.; Gondoh, Y.; Karashima, S.; Nishimoto, K.; Yasuda, M.; Horike, S.I.; Takeda, Y. Genetic and epigenetic analyses of aldosterone-producing adenoma with hypercortisolemia. Steroids 2019, 151, 108470. [Google Scholar] [CrossRef]
- Kobuke, K.; Oki, K.; Gomez-Sanchez, C.E.; Ohno, H.; Itcho, K.; Yoshii, Y.; Yoneda, M.; Hattori, N. Purkinje Cell Protein 4 Expression Is Associated with DNA Methylation Status in Aldosterone-Producing Adenoma. J. Clin. Endocrinol. Metab. 2018, 103, 965–971. [Google Scholar] [CrossRef]
- Ettaieb, M.; Kerkhofs, T.; van Engeland, M.; Haak, H. Past, Present and Future of Epigenetics in Adrenocortical Carcinoma. Cancers 2020, 12, 1218. [Google Scholar] [CrossRef]
- Robertson, S.; MacKenzie, S.M.; Alvarez-Madrazo, S.; Diver, L.A.; Lin, J.; Stewart, P.M.; Fraser, R.; Connell, J.M.; Davies, E. MicroRNA-24 is a novel regulator of aldosterone and cortisol production in the human adrenal cortex. Hypertension 2013, 62, 572–578. [Google Scholar] [CrossRef] [Green Version]
- Nakano, Y.; Yoshimoto, T.; Watanabe, R.; Murakami, M.; Fukuda, T.; Saito, K.; Fujii, Y.; Akashi, T.; Tanaka, T.; Yamada, T.; et al. miRNA299 involvement in CYP11B2 expression in aldosterone-producing adenoma. Eur. J. Endocrinol. 2019, 181, 69–78. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Cao, Y.; Su, T.; Jiang, Y.; Jiang, L.; Zhou, W.; Zhang, C.; Wang, W.; Ning, G. Downregulation of miR-375 in aldosterone-producing adenomas promotes tumour cell growth via MTDH. Clin. Endocrinol. 2015, 83, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.Y.; Chang, H.M.; Lin, Y.F.; Chan, C.K.; Chang, C.H.; Chueh, S.J.; Yang, S.Y.; Huang, K.H.; Lin, Y.H.; Wu, V.C.; et al. miRNA-203 Modulates Aldosterone Levels and Cell Proliferation by Targeting Wnt5a in Aldosterone-Producing Adenomas. J. Clin. Endocrinol. Metab. 2018, 103, 3737–3747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decmann, A.; Nyiro, G.; Darvasi, O.; Turai, P.; Bancos, I.; Kaur, R.J.; Pezzani, R.; Iacobone, M.; Kraljevic, I.; Kastelan, D.; et al. Circulating miRNA Expression Profiling in Primary Aldosteronism. Front. Endocrinol. 2019, 10, 739. [Google Scholar] [CrossRef]
- Raman, P.B.; Sharma, D.C.; Dorfman, R.I.; Gabrilove, J.L. Biosynthesis of C-18-oxygenated steroids by an aldosterone-secreting human adrenal tumor. Metabolism of [4-14C] progesterone, [1,2-3H]11-deoxycorticosterone, and [4-14C] pregnenolone. Biochemistry 1965, 4, 1376–1385. [Google Scholar] [CrossRef]
- Gordon, R.D.; Hamlet, S.M.; Tunny, T.J.; Gomez-Sanchez, C.E.; Jayasinghe, L.S. Distinguishing aldosterone-producing adenoma from other forms of hyperaldosteronism and lateralizing the tumour pre-operatively. Clin. Exp. Pharm. Physiol. 1986, 13, 325–328. [Google Scholar] [CrossRef]
- Nakamura, Y.; Satoh, F.; Morimoto, R.; Kudo, M.; Takase, K.; Gomez-Sanchez, C.E.; Honma, S.; Okuyama, M.; Yamashita, K.; Rainey, W.E.; et al. 18-oxocortisol measurement in adrenal vein sampling as a biomarker for subclassifying primary aldosteronism. J. Clin. Endocrinol. Metab. 2011, 96, E1272–E1278. [Google Scholar] [CrossRef] [Green Version]
- Mulatero, P.; di Cella, S.M.; Monticone, S.; Schiavone, D.; Manzo, M.; Mengozzi, G.; Rabbia, F.; Terzolo, M.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E.; et al. 18-hydroxycorticosterone, 18-hydroxycortisol, and 18-oxocortisol in the diagnosis of primary aldosteronism and its subtypes. J. Clin. Endocrinol. Metab. 2012, 97, 881–889. [Google Scholar] [CrossRef] [Green Version]
- Satoh, F.; Morimoto, R.; Ono, Y.; Iwakura, Y.; Omata, K.; Kudo, M.; Takase, K.; Seiji, K.; Sasamoto, H.; Honma, S.; et al. Measurement of peripheral plasma 18-oxocortisol can discriminate unilateral adenoma from bilateral diseases in patients with primary aldosteronism. Hypertension 2015, 65, 1096–1102. [Google Scholar] [CrossRef] [Green Version]
- Eisenhofer, G.; Dekkers, T.; Peitzsch, M.; Dietz, A.S.; Bidlingmaier, M.; Treitl, M.; Williams, T.A.; Bornstein, S.R.; Haase, M.; Rump, L.C.; et al. Mass Spectrometry-Based Adrenal and Peripheral Venous Steroid Profiling for Subtyping Primary Aldosteronism. Clin. Chem. 2016, 62, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Lenders, J.W.M.; Williams, T.A.; Reincke, M.; Gomez-Sanchez, C.E. Diagnosis of endocrine disease: 18-Oxocortisol and 18-hydroxycortisol: Is there clinical utility of these steroids? Eur. J. Endocrinol. 2018, 178, R1–R9. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Rhayem, Y.; Kunzke, T.; Sun, N.; Feuchtinger, A.; Ludwig, P.; Strom, T.M.; Gomez-Sanchez, C.; Knosel, T.; Kirchner, T.; et al. In situ metabolomics of aldosterone-producing adenomas. JCI Insight 2019, 4, e130356. [Google Scholar] [CrossRef]
- Williams, T.A.; Peitzsch, M.; Dietz, A.S.; Dekkers, T.; Bidlingmaier, M.; Riester, A.; Treitl, M.; Rhayem, Y.; Beuschlein, F.; Lenders, J.W.; et al. Genotype-Specific Steroid Profiles Associated with Aldosterone-Producing Adenomas. Hypertension 2016, 67, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Eisenhofer, G.; Duran, C.; Cannistraci, C.V.; Peitzsch, M.; Williams, T.A.; Riester, A.; Burrello, J.; Buffolo, F.; Prejbisz, A.; Beuschlein, F.; et al. Use of Steroid Profiling Combined with Machine Learning for Identification and Subtype Classification in Primary Aldosteronism. JAMA Netw. Open 2020, 3, e2016209. [Google Scholar] [CrossRef]
- Lana, A.; Alexander, K.; Castagna, A.; D’Alessandro, A.; Morandini, F.; Pizzolo, F.; Zorzi, F.; Mulatero, P.; Zolla, L.; Olivieri, O. Urinary Metabolic Signature of Primary Aldosteronism: Gender and Subtype-Specific Alterations. Proteom. Clin. Appl. 2019, 13, e1800049. [Google Scholar] [CrossRef]
- Arlt, W.; Lang, K.; Sitch, A.J.; Dietz, A.S.; Rhayem, Y.; Bancos, I.; Feuchtinger, A.; Chortis, V.; Gilligan, L.C.; Ludwig, P.; et al. Steroid metabolome analysis reveals prevalent glucocorticoid excess in primary aldosteronism. JCI Insight 2017, 2, e93136. [Google Scholar] [CrossRef]
- Erlic, Z.; Reel, P.; Reel, S.; Amar, L.; Pecori, A.; Larsen, C.K.; Tetti, M.; Pamporaki, C.; Prehn, C.; Adamski, J.; et al. Targeted Metabolomics as a Tool in Discriminating Endocrine from Primary Hypertension. J. Clin. Endocrinol. Metab. 2021, 106, 1111–1128. [Google Scholar] [CrossRef]
- Sun, N.; Meyer, L.S.; Feuchtinger, A.; Kunzke, T.; Knosel, T.; Reincke, M.; Walch, A.; Williams, T.A. Mass Spectrometry Imaging Establishes 2 Distinct Metabolic Phenotypes of Aldosterone-Producing Cell Clusters in Primary Aldosteronism. Hypertension 2020, 75, 634–644. [Google Scholar] [CrossRef]
- Swierczynska, M.M.; Betz, M.J.; Colombi, M.; Dazert, E.; Jeno, P.; Moes, S.; Pfaff, C.; Glatz, K.; Reincke, M.; Beuschlein, F.; et al. Proteomic Landscape of Aldosterone-Producing Adenoma. Hypertension 2019, 73, 469–480. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Genes | Description | Trend |
|---|---|---|
| Steroidogenic enzymes | ||
| CYP11B2 | Cytochrome P450 Family 11 Subfamily B Member 2 | ↑(a), ↔ ↑(b) |
| CYP11B1 | Cytochrome P450 Family 11 Subfamily B Member 1 | ↑(b) |
| CYP21A2 | Cytochrome P450 Family 21 Subfamily A Member 2 | ↑(a) |
| HSD3B2 | Hydroxy-Delta-5-Steroid Dehydrogenase, 3 Beta- And Steroid Delta-Isomerase 2 | ↑(a) |
| CYP17A1 | Cytochrome P450 Family 17 Subfamily A Member 1 | ↓ (a), ↑(b) * |
| CYP11A1 | Cytochrome P450 Family 11 Subfamily A Member 1 | ↑(a) |
| AKR1C3 | Aldo-Keto Reductase Family 1 Member C3 | ↓(a) |
| Nuclear receptors/transcription factors | ||
| NR4A2 | Nuclear Receptor Subfamily 4 Group A Member 2 (NURR1) | ↑(a), ↑(b) |
| NR4A1 | Nuclear Receptor Subfamily 4 Group A Member 1 (NGF1B) | ↑(a) |
| NR0B1 | Nuclear Receptor Subfamily 0 Group B Member 1 (DAX1) | ↑(a) * |
| NR5A1 | Nuclear Receptor Subfamily 5 Group A Member 1 (Steroidogenic factor 1 _ SF1) | ↑(a) |
| NR1B1 | Retinoic Acid Receptor Alpha (RARα) | ↓(a) |
| Plasma membrane receptor | ||
| SCARB1 | Scavenger Receptor Class B Member 1 (CD36) | ↑(a) |
| Ion channels | ||
| KCNK1 | Potassium Two Pore Domain Channel Subfamily K Member 1 (TWIK-1) | ↓(b) |
| KCNK5 | Potassium Two Pore Domain Channel Subfamily K Member 5 (TASK-2) | ↓(a) |
| SLC24A3 | Solute Carrier Family 24 Member 3 (sodium calcium exchanger) | ↓(b) |
| ANO4 | Anoctamin 4 (calcium dependent chloride channel) | ↓(a) |
| CACNA1A | Calcium Voltage-Gated Channel Subunit Alpha1 A | ↑(a) |
| CACNA1C | Calcium Voltage-Gated Channel Subunit Alpha1 C | ↑(a) |
| CACNA1E | Calcium Voltage-Gated Channel Subunit Alpha1 E | ↑(a) |
| Calcium signaling | ||
| CALM2 | Calmodulin 2 | ↑(a) |
| CALR | Calreticulin | ↑(a) |
| CAMK1 | Calcium/Calmodulin Dependent Protein Kinase I | ↑(b) |
| CAMK2B | Calcium/Calmodulin Dependent Protein Kinase II Beta | ↓(b) |
| CALN1 | Calneuron 1 | ↑(a) |
| ATP2A3 | ATPase Sarcoplasmic/Endoplasmic Reticulum Ca2+ Transporting 3 (SERCA3) | ↑(a) |
| CLGN | Calmegin | ↑(a) |
| PCP4 | Purkinje Cell Protein 4 | ↑(a) |
| VSNL1 | Visinin Like 1 | ↑(a), ↑(b) |
| GSTA1 | Glutathione S-Transferase Alpha 1 | ↓(a), ↓(b) |
| G-protein coupled receptors | ||
| LHCGR | Luteinizing Hormone/Choriogonadotropin Receptor | ↑(a) ** |
| GNRHR | Gonadotropin Releasing Hormone Receptor | ↑(a) |
| HTR2A | 5-Hydroxytryptamine Receptor 2A | ↑(a), ↑(b) |
| HTR4 | 5-Hydroxytryptamine Receptor 4 | ↑(a) |
| AGTR1 | Angiotensin II Receptor Type 1 (AT1R) | ↑(a), ↑(b) |
| PTGER1 | Prostaglandin E Receptor 1 | ↑(a) |
| GRM3 | Glutamate Metabotropic Receptor 3 | ↑(a) |
| EDNRB | Endothelin Receptor Type B | ↑(a) |
| MC2R | Melanocortin 2 Receptor | ↑(a), ↑(b) |
| AVPR1A | Arginin Vasopressin Receptor 1A | ↓(a) |
| PTGFR | Prostaglandin F Receptor | ↓(a) |
| GPER1 | G-Protein-Coupled Estrogen Receptor 1 | ↑(a) |
| Energy | ||
| FDX1 | Adrenodoxin | ↑(a) |
| POR | Cytochrome P450 Oxidoreductase | ↑(a) |
| CYB5 | Cytochrome B5 Type A | ↑(a) |
| ATAD3C | ATPase Family AAA Domain Containing 3C | ↑(a) |
| ACSS3 | Acyl-CoA Synthetase Short Chain Family Member 3 | ↑(b) |
| - | Genes related to lipid metabolism, glycolysis, and antioxidant systems | ↑(a) |
| Protein binding | ||
| NEFM | Neurofilament Medium Chain | ↓(b) |
| NPNT | Nephronectin | ↓(b) |
| MRAP | Melanocortin 2 Receptor Accessory Protein | ↑(a), ↑(b) |
| PROM1 | Prominin 1 | ↑(a) |
| SFRP2 | Secreted Frizzled Related Protein 2 | ↓(a) |
| Cell growth/cell death | ||
| COPS5 | COP9 Signalosome Subunit 5 (JAB1) | ↑(a) |
| MYC | MYC Proto-Oncogene, BHLH Transcription Factor | ↑(a) |
| IGFBP2 | Insulin Like Growth Factor Binding Protein 2 | ↑(a) |
| CCN3 | Cellular Communication Network Factor 3 (IGFBP9 or NOV) | ↑(a) |
| TDGF1 | Teratocarcinoma-Derived Growth Factor 1 | ↑(a) |
| BID | BH3 Interacting Domain Death Agonist | ↓(b) ** |
| BIRC2 | Baculoviral IAP Repeat Containing 2 | ↓(b) ** |
| BIRC3 | Baculoviral IAP Repeat Containing 3 | ↓(b) ** |
| Immune response | ||
| - | Genes related to inflammatory response, interferon-γ response, and IL-6, JAK/STAT3 signaling | ↓(a) |
| DNA binding/RNA polymerase | ||
| GATA6 | GATA Binding Protein 6 | ↑(a) |
| PRRX1 | Paired Related Homeobox 1 | ↑(a) |
| DACH1 | Dachshund Family Transcription Factor 1 | ↓(a) |
| BEX1 | Brain-Expressed X-Linked 1 | ↑ (a), ↓(b) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spyroglou, A.; Piaditis, G.P.; Kaltsas, G.; Alexandraki, K.I. Transcriptomics, Epigenetics, and Metabolomics of Primary Aldosteronism. Cancers 2021, 13, 5582. https://doi.org/10.3390/cancers13215582
Spyroglou A, Piaditis GP, Kaltsas G, Alexandraki KI. Transcriptomics, Epigenetics, and Metabolomics of Primary Aldosteronism. Cancers. 2021; 13(21):5582. https://doi.org/10.3390/cancers13215582
Chicago/Turabian StyleSpyroglou, Ariadni, George P. Piaditis, Gregory Kaltsas, and Krystallenia I. Alexandraki. 2021. "Transcriptomics, Epigenetics, and Metabolomics of Primary Aldosteronism" Cancers 13, no. 21: 5582. https://doi.org/10.3390/cancers13215582
APA StyleSpyroglou, A., Piaditis, G. P., Kaltsas, G., & Alexandraki, K. I. (2021). Transcriptomics, Epigenetics, and Metabolomics of Primary Aldosteronism. Cancers, 13(21), 5582. https://doi.org/10.3390/cancers13215582