
Biology and Treatment Advances in Cutaneous Squamous Cell Carcinoma


{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Epidemiology
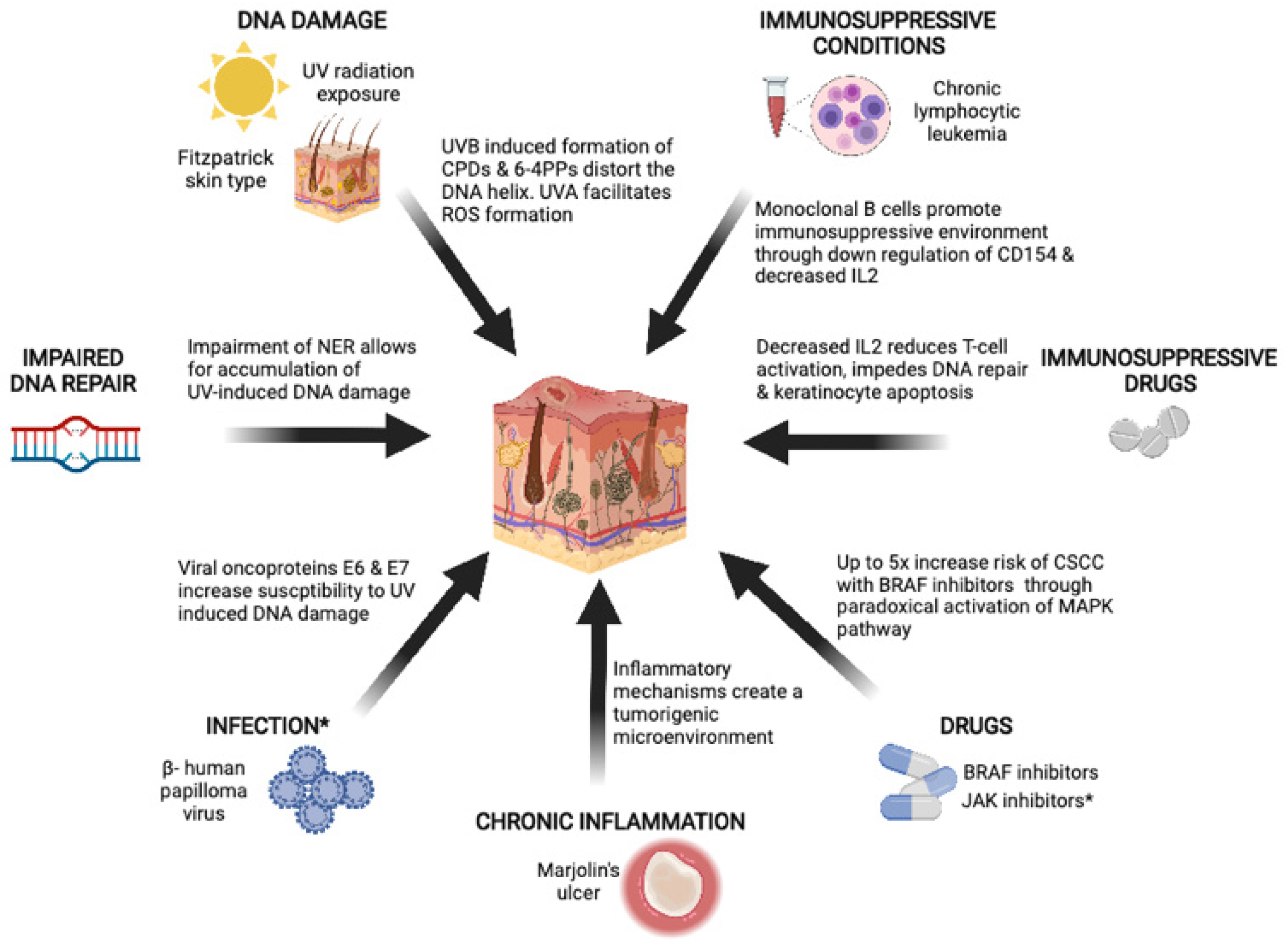
3. Risk Factors
3.1. Ultraviolet Radiation
3.2. Immunosuppression
3.2.1. Chronic Lymphocytic Leukemia
3.2.2. Drugs
Immunosuppressive Drugs
BRAF Inhibitors
JAK1/2 Inhibitors
3.3. Marjolin’s Ulcers
3.4. Environmental Exposure
3.5. Inherited Bone Marrow Failure Sydromes (IBMFSs)
3.6. Beta Human Papillomavirus
4. Biology and Pathogenesis
5. Tumor Mutation Burden
6. Tumor Mutational Signatures
7. Treatment Advances
7.1. Localized Resectable High-Risk Disease
7.1.1. Post-Operative Chemoradiotherapy
7.1.2. Neo/Adjuvant Immunotherapy
7.2. Unresectable Locally Advanced or Metastatic Disease
7.2.1. Immunotherapy
7.2.2. EGFR Pathway Inhibition
7.2.3. Other Approaches
8. Therapeutic Options for Immunocompromised Patients
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Brantsch, K.D.; Meisner, C.; Schönfisch, B.; Trilling, B.; Wehner-Caroli, J.; Röcken, M.; Breuninger, H. Analysis of risk factors determining prognosis of cutaneous squamous cell carcinoma: A prospective study. Lancet Oncol. 2008, 9, 713–720. [Google Scholar] [CrossRef]
- Brougham, N.D.; Dennett, E.R.; Cameron, R.; Tan, S.T. The incidence of metastasis from cutaneous squamous cell carcinoma and the impact of its risk factors. J. Surg. Oncol. 2012, 106, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br. J. Dermatol. 2012, 166, 1069–1080. [Google Scholar] [CrossRef]
- Pandeya, N.; Olsen, C.M.; Whiteman, D.C. The incidence and multiplicity rates of keratinocyte cancers in Australia. Med. J. Aust. 2017, 207, 339–343. [Google Scholar] [CrossRef]
- Goon, P.K.C.; Greenberg, D.C.; Igali, L.; Levell, N.J. Predicted cases of U.K. skin squamous cell carcinoma and basal cell carcinoma in 2020 and 2025: Horizon planning for National Health Service dermatology and dermatopathology. Br. J. Dermatol. 2017, 176, 1351–1353. [Google Scholar] [CrossRef] [PubMed]
- Karia, P.S.; Han, J.; Schmults, C.D. Cutaneous squamous cell carcinoma: Estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J. Am. Acad. Dermatol. 2013, 68, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Ronconi, G.; Piccinni, C.; Dondi, L.; Calabria, S.; Pedrini, A.; Esposito, I.; Ascierto, P.A.; Naldi, L.; Martini, N. Identification of cases and estimate of direct costs of unresectable and advanced cutaneous squamous cell carcinoma: Real-world data from a large Italian database. Br. J. Dermatol. 2020, 183, 172–174. [Google Scholar] [CrossRef]
- Foote, J.A.; Harris, R.B.; Giuliano, A.R.; Roe, D.J.; Moon, T.E.; Cartmel, B.; Alberts, D.S. Predictors for cutaneous basal-and squamous cell carcinoma among actinically damaged adults. Int. J. Cancer 2001, 95, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, B.K.; Kricker, A. The epidemiology of UV induced skin cancer. J. Photochem. Photobiol. B Biol. 2001, 63, 8–18. [Google Scholar] [CrossRef]
- Hall, H.I.; May, D.S.; Lew, R.A.; Koh, H.K.; Nadel, M. Sun protection behaviors of the US white population. Prev. Med. 1997, 26, 401–407. [Google Scholar] [CrossRef]
- McCarthy, E.M.; Ethridge, K.P.; Wagner Jr, R. Beach holiday sunburn: The sunscreen paradox and gender differences. Cutis 1999, 64, 37–42. [Google Scholar]
- Thomas-Ahner, J.M.; Wulff, B.C.; Tober, K.L.; Kusewitt, D.F.; Riggenbach, J.A.; Oberyszyn, T.M. Gender Differences in UVB-Induced Skin Carcinogenesis, Inflammation, and DNA Damage. Cancer Res. 2007, 67, 3468–3474. [Google Scholar] [CrossRef] [Green Version]
- Rosso, S.; Zanetti, R.; Martinez, C.; Tormo, M.J.; Schraub, S.; Sancho-Garnier, H.; Franceschi, S.; Gafà, L.; Perea, E.; Navarro, C.; et al. The multicentre south European study ‘Helios’. II: Different sun exposure patterns in the aetiology of basal cell and squamous cell carcinomas of the skin. Br. J. Cancer 1996, 73, 1447–1454. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.P.; Häder, D.P. UV-induced DNA damage and repair: A review. Photochem. Photobiol. Sci. 2002, 1, 225–236. [Google Scholar] [CrossRef]
- Benjamin, C.L.; Ananthaswamy, H.N. p53 and the pathogenesis of skin cancer. Toxicol. Appl. Pharm. 2007, 224, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benavides, F.; Oberyszyn, T.M.; VanBuskirk, A.M.; Reeve, V.E.; Kusewitt, D.F. The hairless mouse in skin research. J. Dermatol. Sci. 2009, 53, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, T.B. The validity and practicality of sun-reactive skin types I through VI. Arch. Dermatol. 1988, 124, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Staples, M.P.; Elwood, M.; Burton, R.C.; Williams, J.L.; Marks, R.; Giles, G.G. Non-melanoma skin cancer in Australia: The 2002 national survey and trends since 1985. Med. J. Aust. 2006, 184, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Harwood, C.A.; Mesher, D.; McGregor, J.M.; Mitchell, L.; Leedham-Green, M.; Raftery, M.; Cerio, R.; Leigh, I.M.; Sasieni, P.; Proby, C.M. A Surveillance Model for Skin Cancer in Organ Transplant Recipients: A 22-Year Prospective Study in an Ethnically Diverse Population. Am. J. Transplant. 2013, 13, 119–129. [Google Scholar] [CrossRef]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Hober, C.; Fredeau, L.; Pham-Ledard, A.; Boubaya, M.; Herms, F.; Celerier, P.; Aubin, F.; Beneton, N.; Dinulescu, M.; Jannic, A.; et al. Cemiplimab for Locally Advanced and Metastatic Cutaneous Squamous Cell Carcinomas: Real-Life Experience from the French CAREPI Study Group. Cancers 2021, 13, 3547. [Google Scholar] [CrossRef]
- Brewer, J.D.; Shanafelt, T.D.; Khezri, F.; Seda, I.M.S.; Zubair, A.S.; Baum, C.L.; Arpey, C.J.; Cerhan, J.R.; Call, T.G.; Roenigk, R.K. Increased incidence and recurrence rates of nonmelanoma skin cancer in patients with non-Hodgkin lymphoma: A Rochester Epidemiology Project population-based study in Minnesota. J. Am. Acad. Dermatol. 2015, 72, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Levi, F.; Randimbison, L.; Te, V.C.; La Vecchia, C. Non-Hodgkin’s lymphomas, chronic lymphocytic leukaemias and skin cancers. Br. J. Cancer 1996, 74, 1847–1850. [Google Scholar] [CrossRef] [Green Version]
- Que, S.K.T.; Zwald, F.O.; Schmults, C.D. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J. Am. Acad. Dermatol. 2018, 78, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrany, K.; Weenig, R.H.; Lee, K.K.; Pittelkow, M.R.; Otley, C.C. Increased metastasis and mortality from cutaneous squamous cell carcinoma in patients with chronic lymphocytic leukemia. J. Am. Acad. Dermatol. 2005, 53, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Royle, J.A.; Baade, P.D.; Joske, D.; Girschik, J.; Fritschi, L. Second cancer incidence and cancer mortality among chronic lymphocytic leukaemia patients: A population-based study. Br. J. Cancer 2011, 105, 1076–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinstern, G.; Rishi, A.; Achenbach, S.J.; Rabe, K.G.; Kay, N.E.; Shanafelt, T.D.; Ding, W.; Leis, J.F.; Norman, A.D.; Call, T.G.; et al. Delineation of clinical and biological factors associated with cutaneous squamous cell carcinoma among patients with chronic lymphocytic leukemia. J. Am. Acad. Dermatol. 2020, 83, 1581–1589. [Google Scholar] [CrossRef]
- Sarvaria, A.; Madrigal, J.A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell. Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantwell, M.; Hua, T.; Pappas, J.; Kipps, T.J. Acquired CD40-ligand deficiency in chronic lymphocytic leukemia. Nat. Med. 1997, 3, 984–989. [Google Scholar] [CrossRef]
- Lindqvist, C.A.; Christiansson, L.H.; Simonsson, B.; Enblad, G.; Olsson-Strömberg, U.; Loskog, A.S.I. T regulatory cells control T-cell proliferation partly by the release of soluble CD25 in patients with B-cell malignancies. Immunology 2010, 131, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Chikuma, S.; Terawaki, S.; Hayashi, T.; Nabeshima, R.; Yoshida, T.; Shibayama, S.; Okazaki, T.; Honjo, T. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J. Immunol. 2009, 182, 6682–6689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riches, J.C.; Ramsay, A.G.; Gribben, J.G. Immune reconstitution in chronic lymphocytic leukemia. Curr. Hematol. Malig. Rep. 2012, 7, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Besson, C.; Moore, A.; Wu, W.; Camp, N.J.; Vajdic, C.; Morton, L.M.; Smedby, K.E.; de Sanjose, S.; Shanafelt, T.D.; Brewer, J.; et al. Common Genetic Polymorphisms Contribute to the Association between Non-Melanoma Skin Cancer and Chronic Lymphocytic Leukemia. Blood 2017, 130, 1454. [Google Scholar] [CrossRef]
- Veness, M.J.; Quinn, D.I.; Ong, C.S.; Keogh, A.M.; Macdonald, P.S.; Cooper, S.G.; Morgan, G.W. Aggressive cutaneous malignancies following cardiothoracic transplantation: The Australian experience. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1999, 85, 1758–1764. [Google Scholar] [CrossRef]
- Jensen, P.; Hansen, S.; Møller, B.; Leivestad, T.; Pfeffer, P.; Geiran, O.; Fauchald, P.; Simonsen, S. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J. Am. Acad. Dermatol. 1999, 40, 177–186. [Google Scholar] [CrossRef]
- Garrett, G.L.; Lowenstein, S.E.; Singer, J.P.; He, S.Y.; Arron, S.T. Trends of skin cancer mortality after transplantation in the United States: 1987 to 2013. J. Am. Acad. Dermatol. 2016, 75, 106–112. [Google Scholar] [CrossRef]
- Wu, X.; Nguyen, B.-C.; Dziunycz, P.; Chang, S.; Brooks, Y.; Lefort, K.; Hofbauer, G.F.L.; Dotto, G.P. Opposing roles for calcineurin and ATF3 in squamous skin cancer. Nature 2010, 465, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Yarosh, D.B.; Pena, A.V.; Nay, S.L.; Canning, M.T.; Brown, D.A. Calcineurin Inhibitors Decrease DNA Repair and Apoptosis in Human Keratinocytes Following Ultraviolet B Irradiation. J. Investig. Dermatol. 2005, 125, 1020–1025. [Google Scholar] [CrossRef] [Green Version]
- Walsh, S.B.; Xu, J.; Xu, H.; Kurundkar, A.R.; Maheshwari, A.; Grizzle, W.E.; Timares, L.; Huang, C.C.; Kopelovich, L.; Elmets, C.A.; et al. Cyclosporine a mediates pathogenesis of aggressive cutaneous squamous cell carcinoma by augmenting epithelial-mesenchymal transition: Role of TGFβ signaling pathway. Mol. Carcinog. 2011, 50, 516–527. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.C.; Kahan, B.D. Immunosuppressive agents in organ transplantation: Past, present, and future. Semin. Nephrol. 2000, 20, 108–125. [Google Scholar] [PubMed]
- Coghill, A.E.; Johnson, L.G.; Berg, D.; Resler, A.J.; Leca, N.; Madeleine, M.M. Immunosuppressive Medications and Squamous Cell Skin Carcinoma: Nested Case-Control Study Within the Skin Cancer after Organ Transplant (SCOT) Cohort. Am. J. Transplant. 2016, 16, 565–573. [Google Scholar] [CrossRef]
- Salgo, R.; Gossmann, J.; Schöfer, H.; Kachel, H.; Kuck, J.; Geiger, H.; Kaufmann, R.; Scheuermann, E. Switch to a sirolimus-based immunosuppression in long-term renal transplant recipients: Reduced rate of (pre-) malignancies and nonmelanoma skin cancer in a prospective, randomized, assessor-blinded, controlled clinical trial. Am. J. Transplant. 2010, 10, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.G.; Canter, J.A.; Shi, M.; Milne, G.L.; Morrow, J.D.; Sligh, J.E. Cyclosporine A suppresses keratinocyte cell death through MPTP inhibition in a model for skin cancer in organ transplant recipients. Mitochondrion 2010, 10, 94–101. [Google Scholar] [CrossRef]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Hackstein, H.; Taner, T.; Zahorchak, A.F.; Morelli, A.E.; Logar, A.J.; Gessner, A.; Thomson, A.W. Rapamycin inhibits IL-4—Induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood 2003, 101, 4457–4463. [Google Scholar] [CrossRef]
- de Gruijl, F.R.; Koehl, G.E.; Voskamp, P.; Strik, A.; Rebel, H.G.; Gaumann, A.; de Fijter, J.W.; Tensen, C.P.; Bavinck, J.N.B.; Geissler, E.K. Early and late effects of the immunosuppressants rapamycin and mycophenolate mofetil on UV carcinogenesis. Int. J. Cancer 2010, 127, 796–804. [Google Scholar] [CrossRef]
- Wulff, B.C.; Kusewitt, D.F.; VanBuskirk, A.M.; Thomas-Ahner, J.M.; Duncan, F.J.; Oberyszyn, T.M. Sirolimus reduces the incidence and progression of UVB-induced skin cancer in SKH mice even with co-administration of cyclosporine A. J. Investig. Dermatol. 2008, 128, 2467–2473. [Google Scholar] [CrossRef] [Green Version]
- Schaper-Gerhardt, K.; Walter, A.; Schmitz-Rode, C.; Satzger, I.; Gutzmer, R. The mTOR-inhibitor Sirolimus decreases the cyclosporine-induced expression of the oncogene ATF3 in human keratinocytes. J. Dermatol. Sci. 2018, 92, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; In, S.G.; Park, O.J.; Won, C.H.; Lee, M.W.; Choi, J.H.; Kim, C.W.; Kim, S.E.; Moon, K.C.; Chang, S. Increased expression of activating transcription factor 3 is related to the biologic behavior of cutaneous squamous cell carcinomas. Hum. Pathol. 2011, 42, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Euvrard, S.; Kanitakis, J.; Decullier, E.; Butnaru, A.C.; Lefrançois, N.; Boissonnat, P.; Sebbag, L.; Garnier, J.-L.; Pouteil-Noble, C.; Cahen, R. Subsequent skin cancers in kidney and heart transplant recipients after the first squamous cell carcinoma. Transplantation 2006, 81, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Alberú, J.; Pascoe, M.D.; Campistol, J.M.; Schena, F.P.; del Carmen Rial, M.; Polinsky, M.; Neylan, J.F.; Korth-Bradley, J.; Goldberg-Alberts, R.; Maller, E.S. Lower malignancy rates in renal allograft recipients converted to sirolimus-based, calcineurin inhibitor-free immunotherapy: 24-month results from the CONVERT trial. Transplantation 2011, 92, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.; Thomsen, H.F.; Engebjerg, M.C.; Olesen, A.B.; Friis, S.; Karagas, M.R.; Sørensen, H.T. Use of oral glucocorticoids and risk of skin cancer and non-Hodgkin’s lymphoma: A population-based case–control study. Br. J. Cancer 2009, 100, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Baibergenova, A.T.; Weinstock, M.A.; Group, V.T. Oral prednisone use and risk of keratinocyte carcinoma in non-transplant population. The VATTC trial. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1109–1115. [Google Scholar] [CrossRef]
- Sørensen, H.T.; Mellemkjær, L.; Nielsen, G.L.; Baron, J.A.; Olsen, J.H.; Karagas, M.R. Skin Cancers and Non-Hodgkin Lymphoma Among Users of Systemic Glucocorticoids: A Population-Based Cohort Study. JNCI J. Natl. Cancer Inst. 2004, 96, 709–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortina, A.B.; Piaserico, S.; Caforio, A.L.P.; Abeni, D.; Alaibac, M.; Angelini, A.; Iliceto, S.; Peserico, A. Immunosuppressive Level and Other Risk Factors for Basal Cell Carcinoma and Squamous Cell Carcinoma in Heart Transplant Recipients. Arch. Dermatol. 2004, 140, 1079–1085. [Google Scholar] [CrossRef]
- Chen, P.; Chen, F.; Zhou, B. Systematic review and meta-analysis of prevalence of dermatological toxicities associated with vemurafenib treatment in patients with melanoma. Clin. Exp. Dermatol. 2019, 44, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Anforth, R.; Fernandez-Peñas, P.; Long, G.V. Cutaneous toxicities of RAF inhibitors. Lancet. Oncol. 2013, 14, e11–e18. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbe, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.F.; Grob, J.J.; et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 3205–3211. [Google Scholar] [CrossRef]
- Gibney, G.T.; Messina, J.L.; Fedorenko, I.V.; Sondak, V.K.; Smalley, K.S.M. Paradoxical oncogenesis—The long-term effects of BRAF inhibition in melanoma. Nat. Rev. Clin. Oncol. 2013, 10, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anforth, R.; Menzies, A.; Byth, K.; Carlos, G.; Chou, S.; Sharma, R.; Scolyer, R.A.; Kefford, R.; Long, G.V.; Fernandez-Peñas, P. Factors influencing the development of cutaneous squamous cell carcinoma in patients on BRAF inhibitor therapy. J. Am. Acad. Dermatol. 2015, 72, 809–815.e801. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Carlos, G.; Anforth, R.; Clements, A.; Menzies, A.M.; Carlino, M.S.; Chou, S.; Fernandez-Peñas, P. Cutaneous Toxic Effects of BRAF Inhibitors Alone and in Combination with MEK Inhibitors for Metastatic Melanoma. JAMA Derm. 2015, 151, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- March-Rodriguez, Á.; Bellosillo, B.; Álvarez-Larrán, A.; Besses, C.; Pujol, R.M.; Toll, A. Rapidly Growing and Aggressive Cutaneous Squamous Cell Carcinomas in a Patient Treated with Ruxolitinib. Ann. Derm. 2019, 31, 204–208. [Google Scholar] [CrossRef]
- Aboul-Fettouh, N.; Nijhawan, R.I. Aggressive squamous cell carcinoma in a patient on the Janus kinase inhibitor ruxolitinib. JAAD Case Rep. 2018, 4, 455–457. [Google Scholar] [CrossRef]
- Aleisa, A.I.; Plante, J.G.; Hsia, L.-L.B. A case of aggressive squamous cell carcinoma with lymphovascular invasion during treatment with the Janus kinase inhibitor tofacitinib. JAAD Case Rep. 2020, 6, 727–730. [Google Scholar] [CrossRef]
- Harrison, C.N.; Vannucchi, A.M.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, F.; Jones, M.M.; Sun, K.; McQuitty, M.; et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016, 30, 1701–1707. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Abikhair Burgo, M.; Roudiani, N.; Chen, J.; Santana, A.L.; Doudican, N.; Proby, C.; Felsen, D.; Carucci, J.A. Ruxolitinib inhibits cyclosporine-induced proliferation of cutaneous squamous cell carcinoma. JCI Insight 2020, 3, e120750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr-Valentic, M.A.; Samimi, K.; Rohlen, B.H.; Agarwal, J.P.; Rockwell, W.B. Marjolin’s Ulcer: Modern Analysis of an Ancient Problem. Plast. Reconstr. Surg. 2009, 123, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Xiang, F.; Song, H.-P.; Huang, Y.-S. Clinical features and treatment of 140 cases of Marjolin’s ulcer at a major burn center in southwest China. Exp. Med. 2019, 17, 3403–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadir, A.R. Burn scar neoplasm. Ann. Burn. Fire Disasters 2007, 20, 185–188. [Google Scholar]
- Das, K.K.; Chakaraborty, A.; Rahman, A.; Khandkar, S. Incidences of malignancy in chronic burn scar ulcers: Experience from Bangladesh. Burns 2015, 41, 1315–1321. [Google Scholar] [CrossRef]
- Yu, N.; Long, X.; Lujan-Hernandez, J.R.; Hassan, K.Z.; Bai, M.; Wang, Y.; Wang, X.; Zhao, R. Marjolin’s ulcer: A preventable malignancy arising from scars. World J. Surg. Oncol. 2013, 11, 313. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Yu, H.S.; Liao, W.T.; Chai, C.Y. Arsenic carcinogenesis in the skin. J. Biomed. Sci. 2006, 13, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishikawa, M.; Koyama, K.; Iseki, M.; Kobuke, T.; Yonehara, S.; Soda, M.; Ron, E.; Tokunaga, M.; Preston, D.L.; Mabuchi, K.; et al. Histologic characteristics of skin cancer in Hiroshima and Nagasaki: Background incidence and radiation effects. Int. J. Cancer 2005, 117, 363–369. [Google Scholar] [CrossRef]
- Yoshinaga, S.; Hauptmann, M.; Sigurdson, A.J.; Doody, M.M.; Freedman, D.M.; Alexander, B.H.; Linet, M.S.; Ron, E.; Mabuchi, K. Nonmelanoma skin cancer in relation to ionizing radiation exposure among U.S. radiologic technologists. Int. J. Cancer 2005, 115, 828–834. [Google Scholar] [CrossRef]
- Lichter, M.D.; Karagas, M.R.; Mott, L.A.; Spencer, S.K.; Stukel, T.A.; Greenberg, E.R. Therapeutic ionizing radiation and the incidence of basal cell carcinoma and squamous cell carcinoma. The New Hampshire Skin Cancer Study Group. Arch. Dermatol. 2000, 136, 1007–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenehjem, J.S.; Robsahm, T.E.; Bråtveit, M.; Samuelsen, S.O.; Kirkeleit, J.; Grimsrud, T.K. Aromatic hydrocarbons and risk of skin cancer by anatomical site in 25 000 male offshore petroleum workers. Am. J. Ind. Med. 2017, 60, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Pukkala, E.; Martinsen, J.I.; Weiderpass, E.; Kjaerheim, K.; Lynge, E.; Tryggvadottir, L.; Sparén, P.; Demers, P.A. Cancer incidence among firefighters: 45 years of follow-up in five Nordic countries. Occup. Environ. Med. 2014, 71, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Alter, B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Cancer in dyskeratosis congenita. Blood 2009, 113, 6549–6557. [Google Scholar] [CrossRef] [Green Version]
- Alter, B.P.; Giri, N.; Savage, S.A.; Peters, J.A.; Loud, J.T.; Leathwood, L.; Carr, A.G.; Greene, M.H.; Rosenberg, P.S. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br. J. Haematol. 2010, 150, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Risitano, A.M.; Marotta, S.; Calzone, R.; Grimaldi, F.; Zatterale, A. Twenty years of the Italian Fanconi Anemia Registry: Where we stand and what remains to be learned. Haematologica 2016, 101, 319–327. [Google Scholar] [CrossRef] [Green Version]
- de Koning, M.N.C.; Weissenborn, S.J.; Abeni, D.; Bouwes Bavinck, J.N.; Euvrard, S.; Green, A.C.; Harwood, C.A.; Naldi, L.; Neale, R.; Nindl, I.; et al. Prevalence and associated factors of betapapillomavirus infections in individuals without cutaneous squamous cell carcinoma. J. Gen. Virol. 2009, 90, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, A.; Forslund, O.; Ekberg, H.; Sterner, G.; Hansson, B.G. The ubiquity and impressive genomic diversity of human skin papillomaviruses suggest a commensalic nature of these viruses. J. Virol. 2000, 74, 11636–11641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neale, R.E.; Weissenborn, S.; Abeni, D.; Bavinck, J.N.B.; Euvrard, S.; Feltkamp, M.C.W.; Green, A.C.; Harwood, C.; de Koning, M.; Naldi, L.; et al. Human Papillomavirus Load in Eyebrow Hair Follicles and Risk of Cutaneous Squamous Cell Carcinoma. Cancer Epidemiol. Biomark. Prev. 2013, 22, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Bouwes Bavinck, J.N.; Feltkamp, M.C.W.; Green, A.C.; Fiocco, M.; Euvrard, S.; Harwood, C.A.; Nasir, S.; Thomson, J.; Proby, C.M.; Naldi, L.; et al. Human papillomavirus and posttransplantation cutaneous squamous cell carcinoma: A multicenter, prospective cohort study. Am. J. Transplant. 2018, 18, 1220–1230. [Google Scholar] [CrossRef]
- Chahoud, J.; Semaan, A.; Chen, Y.; Cao, M.; Rieber, A.G.; Rady, P.; Tyring, S.K. Association Between β-Genus Human Papillomavirus and Cutaneous Squamous Cell Carcinoma in Immunocompetent Individuals—A Meta-analysis. JAMA Dermatol. 2016, 152, 1354–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagas, M.R.; Nelson, H.H.; Sehr, P.; Waterboer, T.; Stukel, T.A.; Andrew, A.; Green, A.C.; Bouwes Bavinck, J.N.; Perry, A.; Spencer, S.; et al. Human Papillomavirus Infection and Incidence of Squamous Cell and Basal Cell Carcinomas of the Skin. JNCI J. Natl. Cancer Inst. 2006, 98, 389–395. [Google Scholar] [CrossRef]
- Andersson, K.; Waterboer, T.; Kirnbauer, R.; Slupetzky, K.; Iftner, T.; de Villiers, E.-M.; Forslund, O.; Pawlita, M.; Dillner, J. Seroreactivity to Cutaneous Human Papillomaviruses among Patients with Nonmelanoma Skin Cancer or Benign Skin Lesions. Cancer Epidemiol. Biomark. Prev. 2008, 17, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannacone, M.R.; Gheit, T.; Waterboer, T.; Giuliano, A.R.; Messina, J.L.; Fenske, N.A.; Cherpelis, B.S.; Sondak, V.K.; Roetzheim, R.G.; Ferrer-Gil, S.; et al. Case–Control Study of Cutaneous Human Papillomavirus Infection in Basal Cell Carcinoma of the Skin. J. Investig. Dermatol. 2013, 133, 1512–1520. [Google Scholar] [CrossRef] [Green Version]
- Howley, P.M.; Pfister, H.J. Beta genus papillomaviruses and skin cancer. Virology 2015, 479–480, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Weissenborn, S.J.; Nindl, I.; Purdie, K.; Harwood, C.; Proby, C.; Breuer, J.; Majewski, S.; Pfister, H.; Wieland, U. Human papillomavirus-DNA loads in actinic keratoses exceed those in non-melanoma skin cancers. J. Investig. Dermatol. 2005, 125, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Conforti, C.; Paolini, F.; Venuti, A.; Dianzani, C.; Zalaudek, I. The detection rate of human papillomavirus in well-differentiated squamous cell carcinoma and keratoacanthoma: Is there new evidence for a viral pathogenesis of keratoacanthoma? Br. J. Dermatol. 2019, 181, 1309–1311. [Google Scholar] [CrossRef]
- Viarisio, D.; Decker, K.M.; Aengeneyndt, B.; Flechtenmacher, C.; Gissmann, L.; Tommasino, M. Human papillomavirus type 38 E6 and E7 act as tumour promoters during chemically induced skin carcinogenesis. J. Gen. Virol. 2013, 94, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 2012, 8, e1002807. [Google Scholar] [CrossRef] [Green Version]
- Cornet, I.; Bouvard, V.; Campo, M.S.; Thomas, M.; Banks, L.; Gissmann, L.; Lamartine, J.; Sylla, B.S.; Accardi, R.; Tommasino, M. Comparative Analysis of Transforming Properties of E6 and E7 from Different Beta Human Papillomavirus Types. J. Virol. 2012, 86, 2366–2370. [Google Scholar] [CrossRef] [Green Version]
- Caldeira, S.; Zehbe, I.; Accardi, R.; Malanchi, I.; Dong, W.; Giarrè, M.; de Villiers, E.-M.; Filotico, R.; Boukamp, P.; Tommasino, M. The E6 and E7 Proteins of the Cutaneous Human Papillomavirus Type 38 Display Transforming Properties. J. Virol. 2003, 77, 2195–2206. [Google Scholar] [CrossRef] [Green Version]
- Brimer, N.; Lyons, C.; Wallberg, A.E.; Vande Pol, S.B. Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene 2012, 31, 4639–4646. [Google Scholar] [CrossRef] [Green Version]
- Meyers, J.M.; Uberoi, A.; Grace, M.; Lambert, P.F.; Munger, K. Cutaneous HPV8 and MmuPV1 E6 Proteins Target the NOTCH and TGF-β Tumor Suppressors to Inhibit Differentiation and Sustain Keratinocyte Proliferation. PLoS Pathog. 2017, 13, e1006171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.J.A.; White, E.A.; Sowa, M.E.; Harper, J.W.; Aster, J.C.; Howley, P.M. Cutaneous β-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1473–E1480. [Google Scholar] [CrossRef] [Green Version]
- Rozenblatt-Rosen, O.; Deo, R.C.; Padi, M.; Adelmant, G.; Calderwood, M.A.; Rolland, T.; Grace, M.; Dricot, A.; Askenazi, M.; Tavares, M.; et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 2012, 487, 491–495. [Google Scholar] [CrossRef]
- Olivero, C.; Lanfredini, S.; Borgogna, C.; Gariglio, M.; Patel, G.K. HPV-Induced Field Cancerisation: Transformation of Adult Tissue Stem Cell Into Cancer Stem Cell. Front. Microbiol. 2018, 9, 546. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Harris, C.C. p53: Traffic cop at the crossroads of DNA repair and recombination. Nat. Rev. Mol. Cell Biol. 2005, 6, 44–55. [Google Scholar] [CrossRef]
- Kraemer, K.H.; Tamura, D.; Khan, S.G.; Digiovanna, J.J. Burning issues in the diagnosis of xeroderma pigmentosum. Br. J. Dermatol. 2013, 169, 1176. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, H.; English, D.; Randell, P.L.; Nakazawa, K.; Martel, N.; Armstrong, B.K.; Yamasaki, H. UV and skin cancer: Specific p53 gene mutation in normal skin as a biologically relevant exposure measurement. Proc. Natl. Acad. Sci. USA 1994, 91, 360–364. [Google Scholar] [CrossRef] [Green Version]
- Jonason, A.S.; Kunala, S.; Price, G.J.; Restifo, R.J.; Spinelli, H.M.; Persing, J.A.; Leffell, D.J.; Tarone, R.E.; Brash, D.E. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA 1996, 93, 14025–14029. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.P.; Hedrum, A.; Pontén, F.; Nistér, M.; Ahmadian, A.; Lundeberg, J.; Uhlén, M.; Pontén, J. Human epidermal cancer and accompanying precursors have identical p53 mutations different from p53 mutations in adjacent areas of clonally expanded non-neoplastic keratinocytes. Oncogene 1996, 12, 765–773. [Google Scholar]
- Ziegler, A.; Jonason, A.S.; Leffell, D.J.; Simon, J.A.; Sharma, H.W.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D.E. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Ho, C.; Wang, N.J.; Liao, W.; Jakkula, L.R.; Collisson, E.A.; Pons, J.; Chan, S.-W.; Lam, E.T.; Chu, C.; et al. Temporal Dissection of Tumorigenesis in Primary Cancers. Cancer Discov. 2011, 1, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; den Breems, N.Y.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 Mutations Occur Early during Cutaneous Squamous Cell Carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef] [Green Version]
- Di Nardo, L.; Pellegrini, C.; Di Stefani, A.; Del Regno, L.; Sollena, P.; Piccerillo, A.; Longo, C.; Garbe, C.; Fargnoli, M.C.; Peris, K. Molecular genetics of cutaneous squamous cell carcinoma: Perspective for treatment strategies. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 932–941. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972. [Google Scholar] [PubMed]
- Olivier, M.; Langerød, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; Bièche, I.; et al. The clinical value of somatic TP53 gene mutations in 1794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef] [Green Version]
- Berg, R.J.; van Kranen, H.J.; Rebel, H.G.; de Vries, A.; van Vloten, W.A.; Van Kreijl, C.F.; van der Leun, J.C.; de Gruijl, F.R. Early p53 alterations in mouse skin carcinogenesis by UVB radiation: Immunohistochemical detection of mutant p53 protein in clusters of preneoplastic epidermal cells. Proc. Natl. Acad. Sci. USA 1996, 93, 274–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Tron, V.; Ho, V. Induction of squamous cell carcinoma in p53-deficient mice after ultraviolet irradiation. J. Investig. Dermatol. 1998, 110, 72–75. [Google Scholar] [CrossRef] [Green Version]
- Mudgil, A.V.; Segal, N.; Andriani, F.; Wang, Y.; Fusenig, N.E.; Garlick, J.A. Ultraviolet B irradiation induces expansion of intraepithelial tumor cells in a tissue model of early cancer progression. J. Investig. Dermatol. 2003, 121, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef] [Green Version]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch Signaling: Cell Fate Control and Signal Integration in Development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, F.M.; Estrach, S.; Ambler, C.A. Epidermal Notch signalling: Differentiation, cancer and adhesion. Curr. Opin. Cell Biol. 2008, 20, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Nowell, C.S.; Radtke, F. Notch as a tumour suppressor. Nat. Rev. Cancer 2017, 17, 145–159. [Google Scholar] [CrossRef]
- Balint, K.; Xiao, M.; Pinnix, C.C.; Soma, A.; Veres, I.; Juhasz, I.; Brown, E.J.; Capobianco, A.J.; Herlyn, M.; Liu, Z.J. Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J. Clin. Investig. 2005, 115, 3166–3176. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.t.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.-c.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, K.; Mandinova, A.; Ostano, P.; Kolev, V.; Calpini, V.; Kolfschoten, I.; Devgan, V.; Lieb, J.; Raffoul, W.; Hohl, D.; et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev 2007, 21, 562–577. [Google Scholar] [CrossRef] [Green Version]
- Proweller, A.; Tu, L.; Lepore, J.J.; Cheng, L.; Lu, M.M.; Seykora, J.; Millar, S.E.; Pear, W.S.; Parmacek, M.S. Impaired Notch Signaling Promotes De novo Squamous Cell Carcinoma Formation. Cancer Res. 2006, 66, 7438–7444. [Google Scholar] [CrossRef] [Green Version]
- Demehri, S.; Turkoz, A.; Kopan, R. Epidermal Notch1 Loss Promotes Skin Tumorigenesis by Impacting the Stromal Microenvironment. Cancer Cell 2009, 16, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Extance, A. Alzheimer’s failure raises questions about disease-modifying strategies. Nat. Rev. Drug Discov. 2010, 9, 749–751. [Google Scholar] [CrossRef]
- Brown, V.L.; Harwood, C.A.; Crook, T.; Cronin, J.G.; Kelsell, D.P.; Proby, C.M. p16INK4a and p14ARF Tumor Suppressor Genes Are Commonly Inactivated in Cutaneous Squamous Cell Carcinoma. J. Investig. Dermatol. 2004, 122, 1284–1292. [Google Scholar] [CrossRef] [Green Version]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arcy, M.E.; Pfeiffer, R.M.; Rivera, D.R.; Hess, G.P.; Cahoon, E.K.; Arron, S.T.; Brownell, I.; Cowen, E.W.; Israni, A.K.; Triplette, M.A.; et al. Voriconazole and the Risk of Keratinocyte Carcinomas among Lung Transplant Recipients in the United States. JAMA Dermatol. 2020, 156, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Dajee, M.; Lazarov, M.; Zhang, J.Y.; Cai, T.; Green, C.L.; Russell, A.J.; Marinkovich, M.P.; Tao, S.; Lin, Q.; Kubo, Y.; et al. NF-κB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature 2003, 421, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Ridky, T.W.; Chow, J.M.; Wong, D.J.; Khavari, P.A. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat. Med. 2010, 16, 1450–1455. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, B.D.; Tessman, I. Identification of the altered bases in mutated single-stranded DNA: II. In vivo mutagenesis by 5-bromodeoxyuridine and 2-aminopurine. J. Mol. Biol. 1964, 9, 364–371. [Google Scholar] [CrossRef]
- Setlow, R.; Carrier, W. Pyrimidine dimers in ultraviolet-irradiated DNA’s. J. Mol. Biol. 1966, 17, 237–254. [Google Scholar] [CrossRef]
- Cho, R.J.; Alexandrov, L.B.; den Breems, N.Y.; Atanasova, V.S.; Farshchian, M.; Purdom, E.; Nguyen, T.N.; Coarfa, C.; Rajapakshe, K.; Prisco, M.; et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2018, 10, eaas9668. [Google Scholar] [CrossRef] [Green Version]
- Haisma, M.S.; Plaat, B.E.C.; Bijl, H.P.; Roodenburg, J.L.N.; Diercks, G.F.H.; Romeijn, T.R.; Terra, J.B. Multivariate analysis of potential risk factors for lymph node metastasis in patients with cutaneous squamous cell carcinoma of the head and neck. J. Am. Acad. Dermatol. 2016, 75, 722–730. [Google Scholar] [CrossRef]
- Koyfman, S.A.; Cooper, J.S.; Beitler, J.J.; Busse, P.M.; Jones, C.U.; McDonald, M.W.; Quon, H.; Ridge, J.A.; Saba, N.F.; Salama, J.K.; et al. ACR Appropriateness Criteria(®) Aggressive Nonmelanomatous Skin Cancer of the Head and Neck. Head Neck 2016, 38, 175–182. [Google Scholar] [CrossRef] [Green Version]
- National Comprehensive Cancer Network. Squamous Cell Skin Cancer. (Version 1). 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/squamous.pdf (accessed on 5 April 2021).
- Cooper, J.S.; Pajak, T.F.; Forastiere, A.A.; Jacobs, J.; Campbell, B.H.; Saxman, S.B.; Kish, J.A.; Kim, H.E.; Cmelak, A.J.; Rotman, M. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2004, 350, 1937–1944. [Google Scholar] [CrossRef] [Green Version]
- Bernier, J.; Domenge, C.; Ozsahin, M.; Matuszewska, K.; Lefèbvre, J.-L.; Greiner, R.H.; Giralt, J.; Maingon, P.; Rolland, F.; Bolla, M. Postoperative irradiation with or without concomitant chemotherapy for locally advanced head and neck cancer. N. Engl. J. Med. 2004, 350, 1945–1952. [Google Scholar] [CrossRef] [Green Version]
- Denis, F.; Garaud, P.; Bardet, E.; Alfonsi, M.; Sire, C.; Germain, T.; Bergerot, P.; Rhein, B.; Tortochaux, J.; Calais, G. Final results of the 94-01 French Head and Neck Oncology and Radiotherapy Group randomized trial comparing radiotherapy alone with concomitant radiochemotherapy in advanced-stage oropharynx carcinoma. J. Clin. Oncol. 2004, 22, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, J.; Sire, C.; Graff, P.; Grégoire, V.; Maingon, P.; Calais, G.; Gery, B.; Martin, L.; Alfonsi, M.; Desprez, P.; et al. Concomitant chemoradiotherapy versus acceleration of radiotherapy with or without concomitant chemotherapy in locally advanced head and neck carcinoma (GORTEC 99-02): An open-label phase 3 randomised trial. Lancet Oncol. 2012, 13, 145–153. [Google Scholar] [CrossRef]
- Porceddu, S.V.; Bressel, M.; Poulsen, M.G.; Stoneley, A.; Veness, M.J.; Kenny, L.M.; Wratten, C.; Corry, J.; Cooper, S.; Fogarty, G.B.; et al. Postoperative Concurrent Chemoradiotherapy Versus Postoperative Radiotherapy in High-Risk Cutaneous Squamous Cell Carcinoma of the Head and Neck: The Randomized Phase III TROG 05.01 Trial. J. Clin. Oncol. 2018, 36, 1275–1283. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Blake, S.J.; Yong, M.C.; Harjunpää, H.; Ngiow, S.F.; Takeda, K.; Young, A.; O’Donnell, J.S.; Allen, S.; Smyth, M.J.; et al. Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease. Cancer Discov 2016, 6, 1382–1399. [Google Scholar] [CrossRef] [Green Version]
- Ferrarotto, R.; Amit, M.; Nagarajan, P.; Rubin, M.L.; Yuan, Y.; Bell, D.; El-Naggar, A.K.; Johnson, J.M.; Morrison, W.H.; Rosenthal, D.I.; et al. Pilot Phase II Trial of Neoadjuvant Immunotherapy in Locoregionally Advanced, Resectable Cutaneous Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Neoadjuvant Atezolizumab in Surgically Resectable Advanced Cutaneous Squamous Cell Carcinoma. U.S. National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04710498 (accessed on 15 June 2021).
- Neo-adjuvant Nivolumab or Nivolumab With Ipilimumab in Advanced Cutaneous Squamous Cell Carcinoma Prior to Surgery (MATISSE)—NCT04620200. U.S. National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04620200 (accessed on 1 August 2021).
- Rischin, D.; Fury, M.G.; Lowy, I.; Stankevich, E.; Han, H.; Porceddu, S. A phase III, randomized, double-blind study of adjuvant cemiplimab versus placebo post-surgery and radiation therapy (RT) in patients (pts) with high-risk cutaneous squamous cell carcinoma (CSCC). J. Clin. Oncol. 2020, 38, TPS10084. [Google Scholar] [CrossRef]
- Geiger, J.L.; Daniels, G.A.; Cohen, E.E.W.; Ge, J.Y.; Gumuscu, B.; Swaby, R.F.; Chang, A.L.S. KEYNOTE-630: Phase 3 study of adjuvant pembrolizumab versus placebo in patients with high-risk, locally advanced cutaneous squamous cell carcinoma. J. Clin. Oncol. 2019, 37, TPS9597. [Google Scholar] [CrossRef]
- Jarkowski, A., 3rd; Hare, R.; Loud, P.; Skitzki, J.J.; Kane, J.M., 3rd; May, K.S.; Zeitouni, N.C.; Nestico, J.; Vona, K.L.; Groman, A.; et al. Systemic Therapy in Advanced Cutaneous Squamous Cell Carcinoma (CSCC): The Roswell Park Experience and a Review of the Literature. Am. J. Clin. Oncol. 2016, 39, 545–548. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Rischin, D.; Khushalani, N.I.; Schmults, C.D.; Guminski, A.D.; Chang, A.L.S.; Lewis, K.D.; Lim, A.M.L.; Hernandez-Aya, L.F.; Hughes, B.G.M.; Schadendorf, D.; et al. Phase II study of cemiplimab in patients (pts) with advanced cutaneous squamous cell carcinoma (CSCC): Longer follow-up. J. Clin. Oncol. 2020, 38, 10018. [Google Scholar] [CrossRef]
- Rischin, D.; Migden, M.R.; Lim, A.M.; Schmults, C.D.; Khushalani, N.I.; Hughes, B.G.M.; Schadendorf, D.; Dunn, L.A.; Hernandez-Aya, L.; Chang, A.L.S.; et al. Phase 2 study of cemiplimab in patients with metastatic cutaneous squamous cell carcinoma: Primary analysis of fixed-dosing, long-term outcome of weight-based dosing. J. Immunother. Cancer 2020, 8, e000775. [Google Scholar] [CrossRef]
- Rischin, D.; Khushalani, N.I.; Schmults, C.D.; Guminski, A.; Chang, A.L.S.; Lewis, K.D.; Lim, A.M.; Hernandez-Aya, L.; Hughes, B.G.M.; Schadendorf, D.; et al. Integrated analysis of a phase 2 study of cemiplimab in advanced cutaneous squamous cell carcinoma: Extended follow-up of outcomes and quality of life analysis. J. Immunother. Cancer 2021, 9, e002757. [Google Scholar] [CrossRef] [PubMed]
- Grob, J.J.; Gonzalez, R.; Basset-Seguin, N.; Vornicova, O.; Schachter, J.; Joshi, A.; Meyer, N.; Grange, F.; Piulats, J.M.; Bauman, J.R.; et al. Pembrolizumab Monotherapy for Recurrent or Metastatic Cutaneous Squamous Cell Carcinoma: A Single-Arm Phase II Trial (KEYNOTE-629). J. Clin. Oncol. 2020, 38, 2916–2925. [Google Scholar] [CrossRef] [PubMed]
- Maubec, E.; Boubaya, M.; Petrow, P.; Beylot-Barry, M.; Basset-Seguin, N.; Deschamps, L.; Grob, J.-J.; Dréno, B.; Scheer-Senyarich, I.; Bloch-Queyrat, C.; et al. Phase II Study of Pembrolizumab As First-Line, Single-Drug Therapy for Patients With Unresectable Cutaneous Squamous Cell Carcinomas. J. Clin. Oncol. 2020, 38, 3051–3061. [Google Scholar] [CrossRef] [PubMed]
- Munhoz, R.R.; Camargo, V.P.D.; Marta, G.N.; Martins, J.C.; Nardo, M.; Barbosa, C.C.; Souza, C.E.d.; Barbosa, I.; Ricci, H.; Mattos, M.R.d.; et al. CA209-9JC: A phase II study of first-line nivolumab (NIVO) in patients (pts) with locally advanced or metastatic cutaneous squamous cell carcinoma. J. Clin. Oncol. 2020, 38, 10044. [Google Scholar] [CrossRef]
- The UNSCARRed Study: UNresctable Squamous Cell Carcinoma Treated With Avelumab and Radical Radiotherapy (UNSCARRed). U.S. National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03737721 (accessed on 14 July 2021).
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination with Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [Green Version]
- Long, G.; Dummer, R.; Johnson, D.; Michielin, O.; Martin-Algarra, S.; Treichel, S.; Chan, E.; Diede, S.; Ribas, A. 429 Long-term analysis of MASTERKEY-265 phase 1b trial of talimogene laherparepvec (T-VEC) plus pembrolizumab in patients with unresectable stage IIIB-IVM1c melanoma. J. Immunother. Cancer 2020, 8, A261. [Google Scholar] [CrossRef]
- Study Evaluating Cemiplimab Alone and Combined With RP1 in Treating Advanced Squamous Skin Cancer (CERPASS) U.S National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04050436 (accessed on 2 September 2021).
- Talimogene Laherparepvec and Panitumumab for the Treatment of Locally Advanced or Metastatic Squamous Cell Carcinoma of the Skin U.S National Library of Medicine. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04163952 (accessed on 2 September 2021).
- Avelumab with or without Cetuximab in Treating Patients with Advanced Skin Squamous Cell Cancer. U.S. National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03944941 (accessed on 14 July 2021).
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Maubec, E.; Duvillard, P.; Velasco, V.; Crickx, B.; Avril, M.F. Immunohistochemical analysis of EGFR and HER-2 in patients with metastatic squamous cell carcinoma of the skin. Anticancer Res. 2005, 25, 1205–1210. [Google Scholar]
- Shimizu, T.; Izumi, H.; Oga, A.; Furumoto, H.; Murakami, T.; Ofuji, R.; Muto, M.; Sasaki, K. Epidermal Growth Factor Receptor Overexpression and Genetic Aberrations in Metastatic Squamous-Cell Carcinoma of the Skin. Dermatology 2001, 202, 203–206. [Google Scholar] [CrossRef]
- Maubec, E.; Petrow, P.; Scheer-Senyarich, I.; Duvillard, P.; Lacroix, L.; Gelly, J.; Certain, A.; Duval, X.; Crickx, B.; Buffard, V.; et al. Phase II study of cetuximab as first-line single-drug therapy in patients with unresectable squamous cell carcinoma of the skin. J. Clin. Oncol. 2011, 29, 3419–3426. [Google Scholar] [CrossRef]
- Foote, M.C.; McGrath, M.; Guminski, A.; Hughes, B.G.M.; Meakin, J.; Thomson, D.; Zarate, D.; Simpson, F.; Porceddu, S.V. Phase II study of single-agent panitumumab in patients with incurable cutaneous squamous cell carcinoma. Ann. Oncol. 2014, 25, 2047–2052. [Google Scholar] [CrossRef] [PubMed]
- Reigneau, M.; Robert, C.; Routier, E.; Mamelle, G.; Moya-Plana, A.; Tomasic, G.; Mateus, C. Efficacy of neoadjuvant cetuximab alone or with platinum salt for the treatment of unresectable advanced nonmetastatic cutaneous squamous cell carcinomas. Br. J. Dermatol. 2015, 173, 527–534. [Google Scholar] [CrossRef]
- William, W.N., Jr.; Feng, L.; Ferrarotto, R.; Ginsberg, L.; Kies, M.; Lippman, S.; Glisson, B.; Kim, E.S. Gefitinib for patients with incurable cutaneous squamous cell carcinoma: A single-arm phase II clinical trial. J. Am. Acad. Dermatol. 2017, 77, 1110–1113.e1112. [Google Scholar] [CrossRef]
- Gold, K.A.; Kies, M.S.; William, W.N.; Johnson, F.M.; Lee, J.J.; Glisson, B.S. Erlotinib in the treatment of recurrent or metastatic cutaneous squamous cell carcinoma: A single-arm phase 2 clinical trial. Cancer 2018, 124, 2169–2173. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, S.; Perrone, F.; Miceli, R.; Ascierto, P.A.; Locati, L.D.; Bergamini, C.; Granata, R.; Alfieri, S.; Resteghini, C.; Galbiati, D.; et al. Efficacy and safety of single-agent pan-human epidermal growth factor receptor (HER) inhibitor dacomitinib in locally advanced unresectable or metastatic skin squamous cell cancer. Eur. J. Cancer 2018, 97, 7–15. [Google Scholar] [CrossRef]
- Dunn, L.; Ho, A.L.; Eng, J.; Michel, L.S.; Fetten, J.V.; Warner, E.; Kriplani, A.; Zhi, W.I.; Ng, K.K.; Haque, S.; et al. A phase I/Ib study of lenvatinib and cetuximab in patients with recurrent/metastatic (R/M) head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2020, 38, 6541. [Google Scholar] [CrossRef]
- Adelmann, C.H.; Truong, K.A.; Liang, R.J.; Bansal, V.; Gandee, L.; Saporito, R.C.; Lee, W.; Du, L.; Nicholas, C.; Napoli, M.; et al. MEK Is a Therapeutic and Chemopreventative Target in Squamous Cell Carcinoma. J. Investig. Dermatol. 2016, 136, 1920–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobimetinib and Atezolizumab in Treating Participants with Advanced or Refractory Rare Tumors. U.S. National Library of Medicine. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03108131 (accessed on 2 September 2021).
- Ding, L.T.; Zhao, P.; Yang, M.L.; Lv, G.Z.; Zhao, T.L. GDC-0084 inhibits cutaneous squamous cell carcinoma cell growth. Biochem. Biophys. Res. Commun. 2018, 503, 1941–1948. [Google Scholar] [CrossRef]
- Zou, Y.; Ge, M.; Wang, X. Targeting PI3K-AKT-mTOR by LY3023414 inhibits human skin squamous cell carcinoma cell growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2017, 490, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Varghese, A.M.; Hyman, D.M.; Bauer, T.M.; Pant, S.; Callies, S.; Lin, J.; Martinez, R.; Wickremsinhe, E.; Fink, A.; et al. A First-in-Human Phase 1 Study of LY3023414, an Oral PI3K/mTOR Dual Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 3253–3262. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; De Groot, J.F.; Battiste, J.D.; Goldlust, S.A.; Garner, J.S.; Simpson, J.A.; Kijlstra, J.; Olivero, A.; Cloughesy, T.F. Escalation portion of phase II study to evaluate the safety, pharmacokinetics, and clinical activity of the PI3K/mTOR inhibitor paxalisib (GDC-0084) in glioblastoma (GBM) with unmethylated O6-methylguanine-methyltransferase (MGMT) promotor status. J. Clin. Oncol. 2020, 38, 2550. [Google Scholar] [CrossRef]
- Manohar, S.; Thongprayoon, C.; Cheungpasitporn, W.; Markovic, S.N.; Herrmann, S.M. Systematic Review of the Safety of Immune Checkpoint Inhibitors among Kidney Transplant Patients. Kidney Int. Rep. 2019, 5, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Lai, H.-C.; Lin, J.-F.; Hwang, T.I.S.; Liu, Y.-F.; Yang, A.-H.; Wu, C.-K. Programmed Cell Death 1 (PD-1) Inhibitors in Renal Transplant Patients with Advanced Cancer: A Double-Edged Sword? Int. J. Mol. Sci. 2019, 20, 2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacrolimus, Nivolumab, and Ipilimumab in Treating Kidney Transplant Recipients with Selected Unresectable or Metastatic Cancers. U.S National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03816332 (accessed on 2 July 2021).
- Cemiplimab in AlloSCT/SOT Recipients with CSCC (CONTRAC) U.S. National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04339062 (accessed on 2 July 2021).
- Cemiplimab-rwlc for Unresectable Locally Recurrent and/or Metastatic CSCC U.S National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04242173 (accessed on 2 July 2021).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thai, A.A.; Lim, A.M.; Solomon, B.J.; Rischin, D. Biology and Treatment Advances in Cutaneous Squamous Cell Carcinoma. Cancers 2021, 13, 5645. https://doi.org/10.3390/cancers13225645
Thai AA, Lim AM, Solomon BJ, Rischin D. Biology and Treatment Advances in Cutaneous Squamous Cell Carcinoma. Cancers. 2021; 13(22):5645. https://doi.org/10.3390/cancers13225645
Chicago/Turabian StyleThai, Alesha A., Annette M. Lim, Benjamin J. Solomon, and Danny Rischin. 2021. "Biology and Treatment Advances in Cutaneous Squamous Cell Carcinoma" Cancers 13, no. 22: 5645. https://doi.org/10.3390/cancers13225645
APA StyleThai, A. A., Lim, A. M., Solomon, B. J., & Rischin, D. (2021). Biology and Treatment Advances in Cutaneous Squamous Cell Carcinoma. Cancers, 13(22), 5645. https://doi.org/10.3390/cancers13225645