
Pimavanserin: A Novel Autophagy Modulator for Pancreatic Cancer Treatment



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cell Culture
2.3. Cytotoxicity Assay
2.4. Colony Formation Assay
2.5. Acridine Orange Assay Using Flow Cytometry
2.6. Acridine Orange Assay Using Confocal Microscopy
2.7. LysoTracker Assay Using Confocal Microscopy
2.8. Electron Microscopy
2.9. Cancer Genomic Data Analysis
2.10. Western Blotting
2.11. Immunofluorescence
2.12. Annexin-V/APC Assay
2.13. LC3B Silencing, LYN-1604 and Bafilomycin Treatment
2.14. Subcutaneous Implantation of Pancreatic Tumor Xenografts
2.15. Immunohistochemistry (IHC)
2.16. Statistical Analysis
3. Results
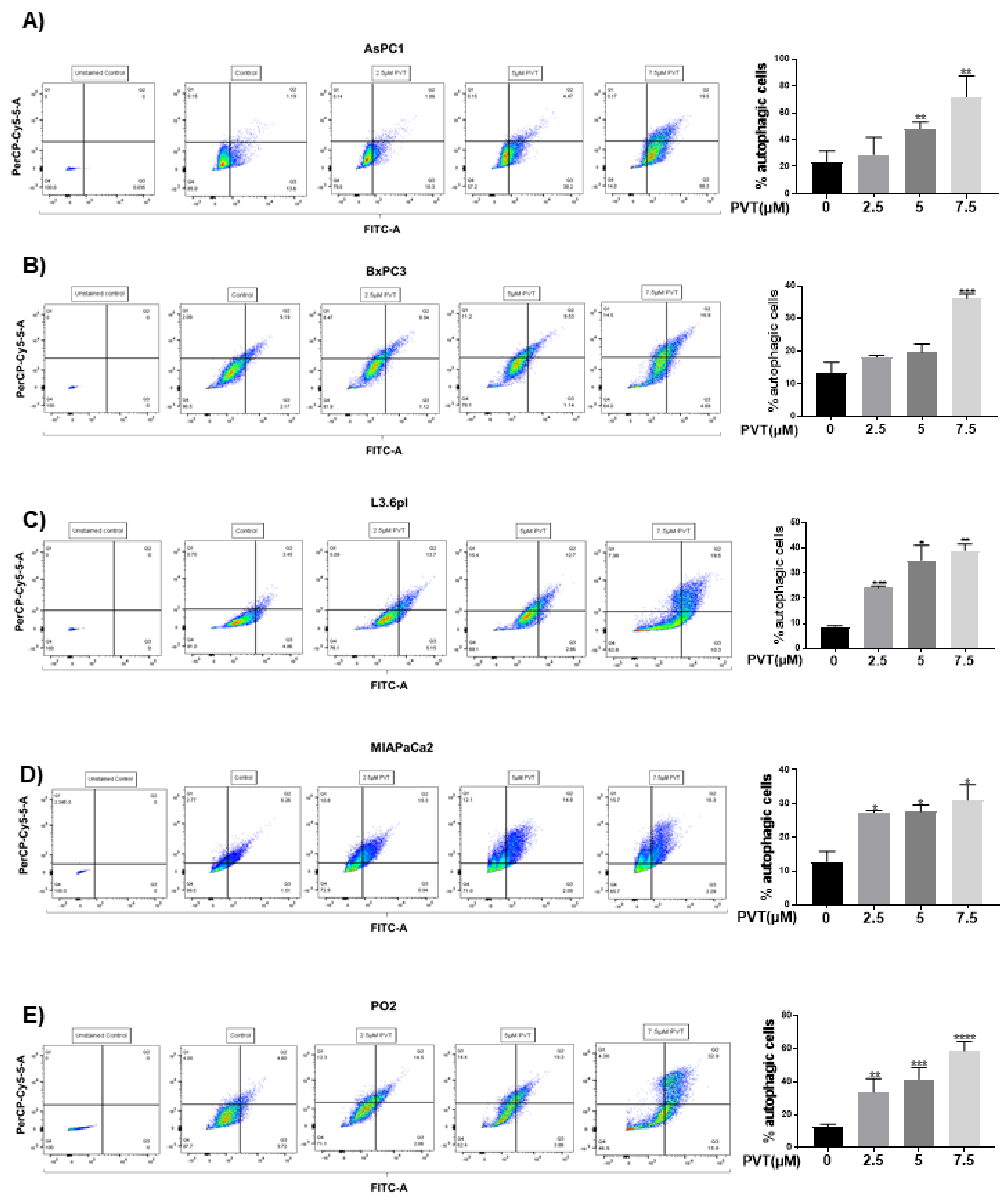
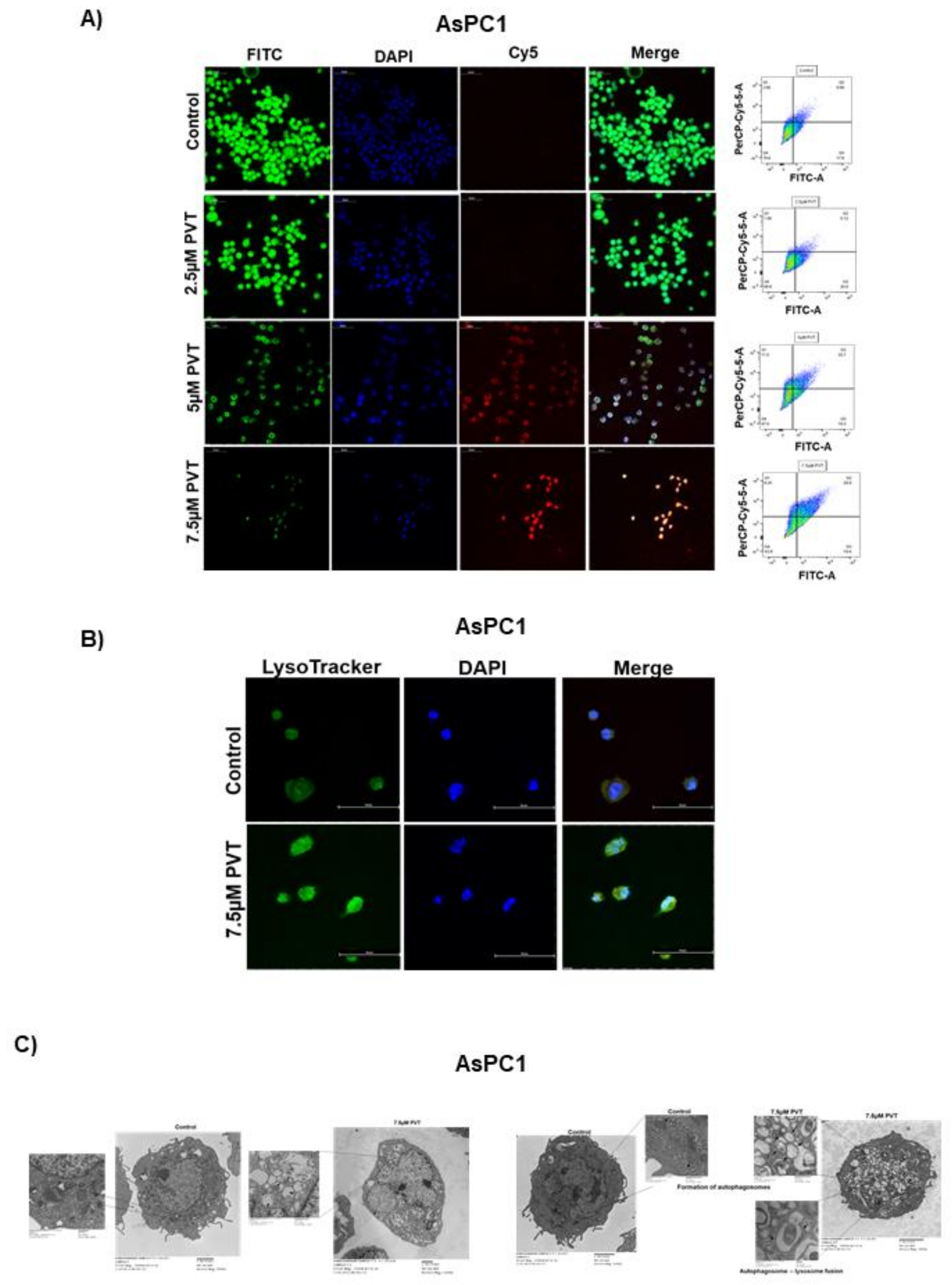
3.1. Induction of Autophagy by PVT
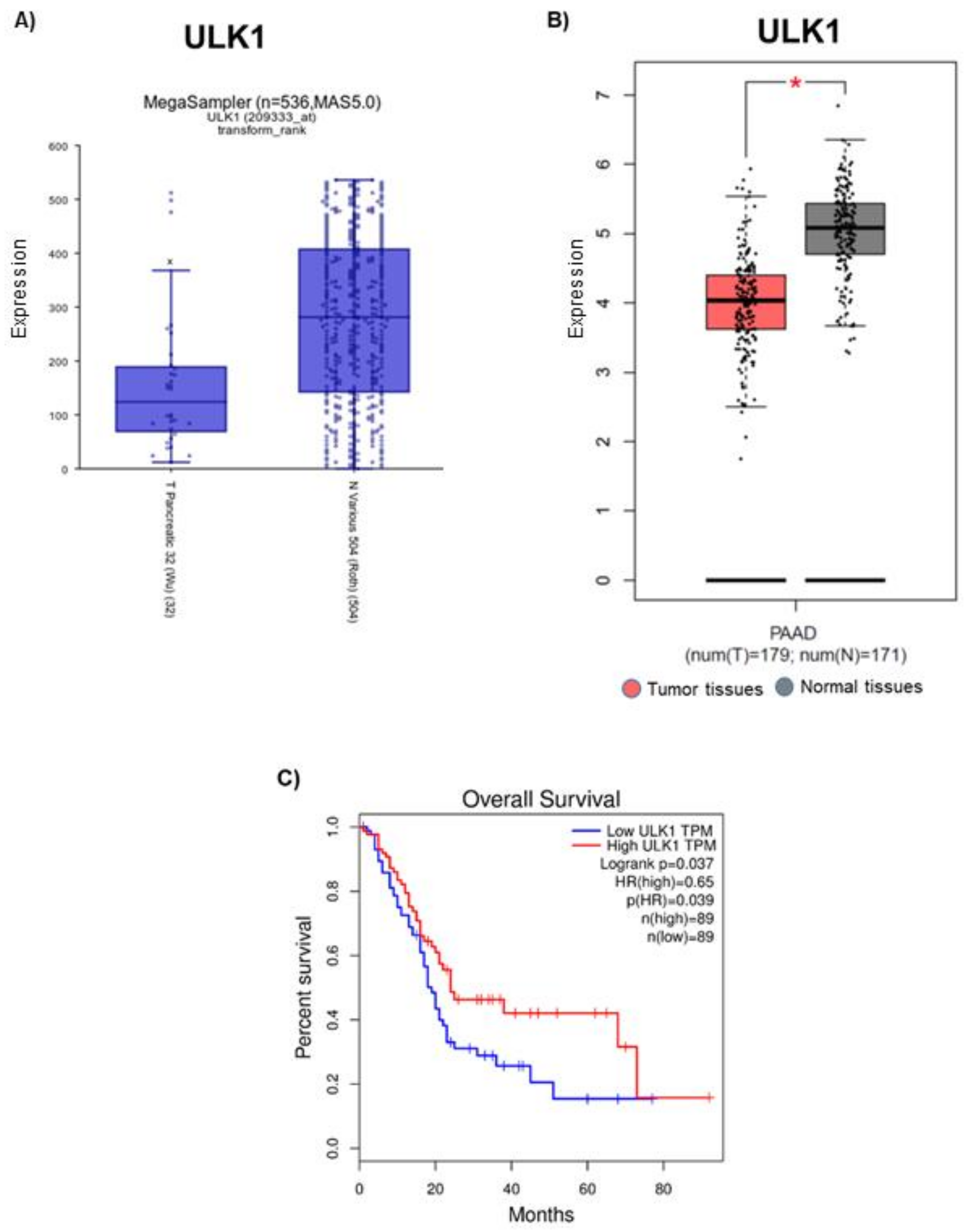
3.2. ULK1 Is Downregulated in Pancreatic Tumor Tissues
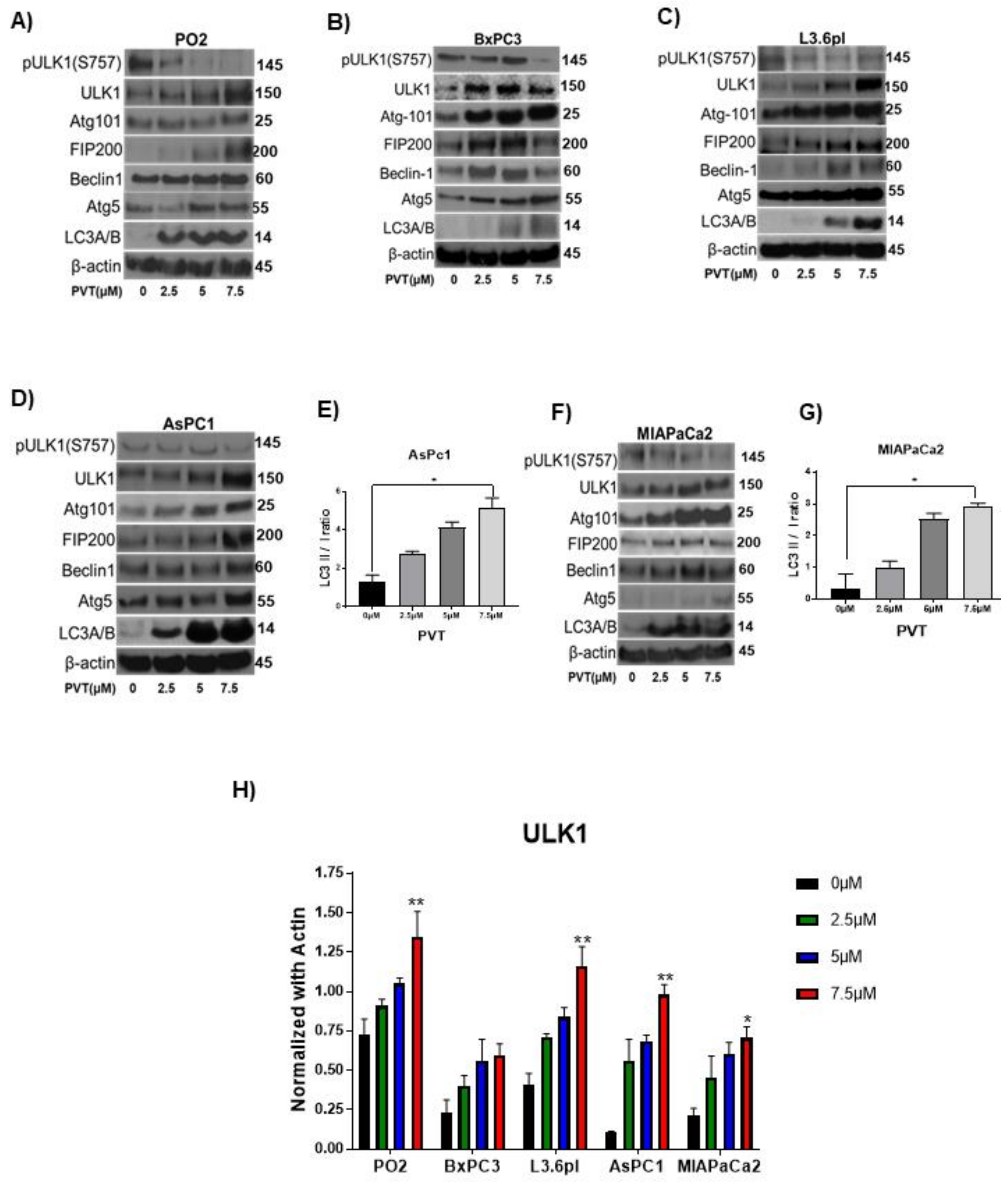
3.3. PVT Induces ULK1-Mediated Autophagy in PDAC Cells
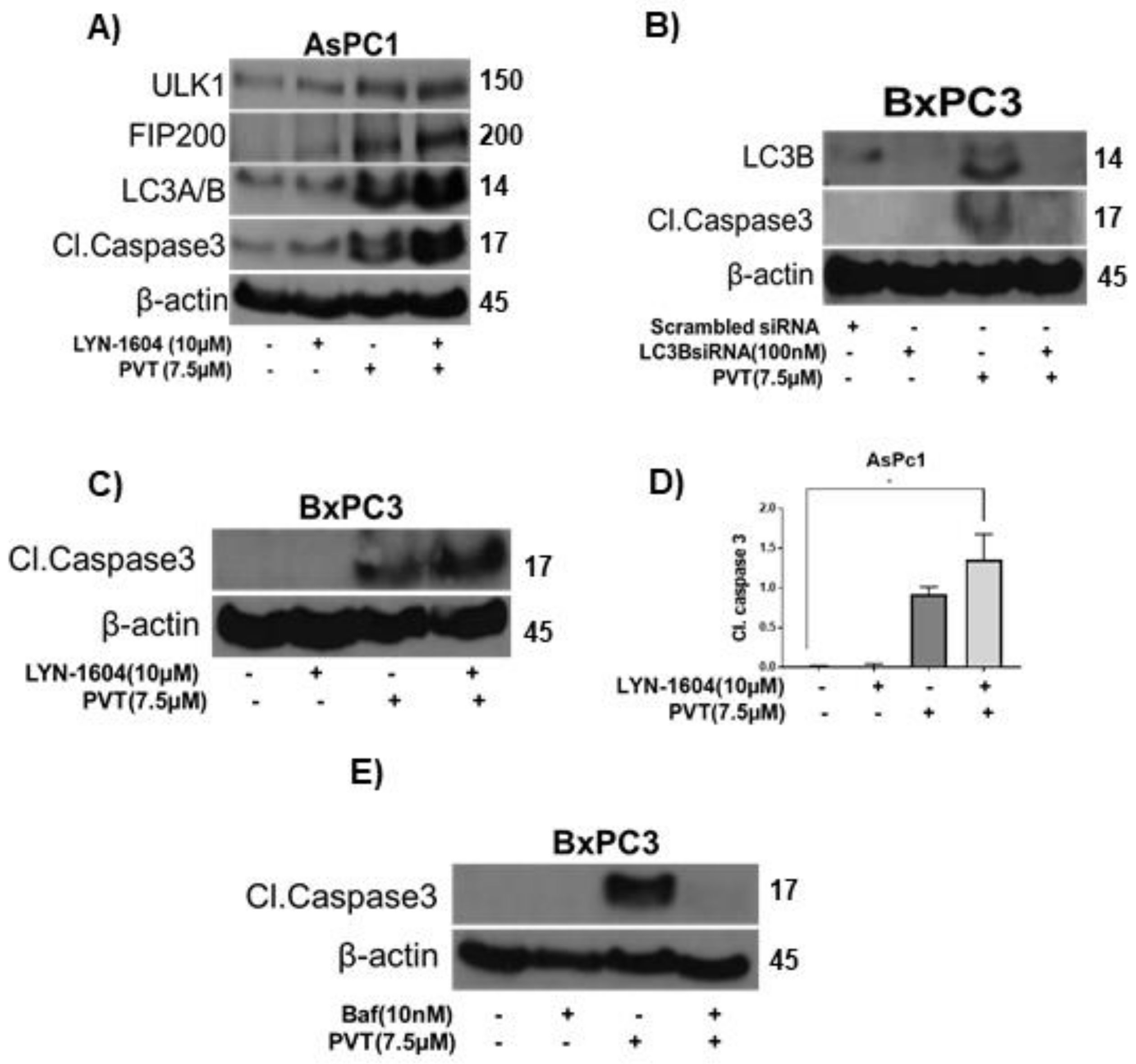
3.4. ULK1 Agonist LYN-1604 Potentiates the Autophagic Effects of PVT
3.5. Autophagy Inhibition by Bafilomycin Abrogates the Autophagic Effects of PVT
3.6. PVT Inhibits Cells Proliferation and Induces Apoptotic Mode of Cell Death in PDAC Cells
3.7. Modulating Autophagy Abrogates or Enhances the Apoptotic Effects of PVT
3.8. PVT Suppresses the Growth of BxPC3 Pancreatic Tumors by Inducing Autophagy-Mediated Apoptosis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [Green Version]
- Pietrocola, F.; Izzo, V.; Niso-Santano, M.; Vacchelli, E.; Galluzzi, L.; Maiuri, M.C.; Kroemer, G. Regulation of autophagy by stress-responsive transcription factors. Semin. Cancer Biol. 2013, 23, 310–322. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Delou, J.; Biasoli, D.; Borges, H.L. The Complex Link between Apoptosis and Autophagy: A Promising New Role for RB. An. Acad. Bras. Cienc. 2016, 88, 2257–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, R.; Ré, A.L.; Archange, C.; Ropolo, A.; Papademetrio, D.L.; Gonzalez, C.D.; Alvarez, E.M.; Iovanna, J.L.; Vaccaro, M.I. Gemcitabine Induces the VMP1—Mediated Autophagy Pathway to Promote Apoptotic Death in Human Pancreatic Cancer Cells. Pancreatology 2010, 10, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Srivastava, S.K. Penfluridol suppresses pancreatic tumor growth by autophagy-mediated apoptosis. Sci. Rep. 2016, 6, 26165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vucicevic, L.; Marjanovic, M.M.; Harhaji-Trajkovic, L.; Maric, N.; Trajkovic, V. Mechanisms and therapeutic significance of autophagy modulation by antipsychotic drugs. Cell Stress 2018, 2, 282–291. [Google Scholar] [CrossRef]
- Ranjan, A.; Kaushik, I.; Srivastava, S.K. Pimozide Suppresses the Growth of Brain Tumors by Targeting STAT3-Mediated Autophagy. Cells 2020, 9, 2141. [Google Scholar] [CrossRef]
- Ramachandran, S.; Srivastava, S.K. Repurposing Pimavanserin, an Anti-Parkinson Drug for Pancreatic Cancer Therapy. Mol. Ther. Oncolytics 2020, 19, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Pi, C.; Wang, G. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed. Pharmacother. 2018, 103, 699–707. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, L.; Qian, X.; Zhou, X.; Yan, Y.; Zhou, J.; Ge, W.; Albahde, M.; Wang, W. GSK343 induces autophagy and downregulates the AKT/mTOR signaling pathway in pancreatic cancer cells. Exp. Ther. Med. 2019, 18, 2608–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, R.; Meng, Q.; Lu, D.; Liu, X.; Wang, Y.; Hao, J. Mitofusin2 Induces Cell Autophagy of Pancreatic Cancer through Inhibiting the PI3K/Akt/mTOR Signaling Pathway. Oxidative Med. Cell. Longev. 2018, 2018, 2798070. [Google Scholar] [CrossRef] [PubMed]
- Koster, J. R2: Genomic Analysis and Visualizatin Platform. Available online: https://hgserver1.amc.nl/cgi-bin/r2/main.cgi (accessed on 14 October 2021).
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Dupont, W.D.; Plummer, W.D., Jr. Power and sample size calculations: A review and computer program. Control. Clin. Trials 1990, 11, 116–128. [Google Scholar] [CrossRef]
- Thomé, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of acridine orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [Green Version]
- Kandala, P.K.; Srivastava, S.K. Regulation of macroautophagy in ovarian cancer cells in vitro and in vivo by controlling Glucose regulatory protein 78 and AMPK. Oncotarget 2012, 3, 435–449. [Google Scholar] [CrossRef]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [Green Version]
- Gillen, S.; Schuster, T.; Büschenfelde, C.M.Z.; Friess, H.; Kleeff, J. Preoperative/Neoadjuvant Therapy in Pancreatic Cancer: A Systematic Review and Meta-Analysis of Response and Resection Percentages. PLoS Med. 2010, 7, e1000267. [Google Scholar] [CrossRef] [Green Version]
- Derle, A.; De Santis, M.C.; Gozzelino, L.; Ratto, E.; Martini, M. The role of metabolic adaptation to nutrient stress in pancreatic cancer. Cell Stress 2018, 2, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gukovskaya, A.S.; Gukovsky, I.; Algül, H.; Habtezion, A. Autophagy, Inflammation, and Immune Dysfunction in the Pathogenesis of Pancreatitis. Gastroenterology 2017, 153, 1212–1226. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy Is Required for Glucose Homeostasis and Lung Tumor Maintenance. Cancer Discov. 2014, 4, 914–927. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Zhang, H.; Meng, L.; Song, H.; Zhou, Q.; Qu, C.; Zhao, P.; Li, Q.; Zou, C.; Liu, X.; et al. Hypoxia-induced acetylation of PAK1 enhances autophagy and promotes brain tumorigenesis via phosphorylating ATG5. Autophagy 2020, 17, 723–742. [Google Scholar] [CrossRef] [Green Version]
- Mah, L.Y.; Ryan, K.M. Autophagy and Cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008821. [Google Scholar] [CrossRef] [Green Version]
- Marsh, T.; Tolani, B.; Debnath, J. The pleiotropic functions of autophagy in metastasis. J. Cell Sci. 2021, 134, jcs247056. [Google Scholar] [CrossRef]
- Kisen, G.; Tessitore, L.; Costelli, P.; Gordon, P.B.; Schwarze, P.E.; Baccino, F.M.; Seglen, P.O. Reduced autophagic activity in primary rat hepatocellular carcinoma and ascites hepatoma cells. Carcinogenesis 1993, 14, 2501–2505. [Google Scholar] [CrossRef]
- Zhang, L.; Fu, L.; Zhang, S.; Zhang, J.; Zhao, Y.; Zheng, Y.; He, G.; Yang, S.; Ouyang, L.; Liu, B. Discovery of a small molecule targeting ULK1-modulated cell death of triple negative breast cancer in vitro and in vivo. Chem. Sci. 2017, 8, 2687–2701. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.J.; Nagaraju, G.P.; Nagalingam, A.; Muniraj, N.; Kuppusamy, P.; Walker, A.; Woo, J.; Győrffy, B.; Gabrielson, E.; Saxena, N.K.; et al. ADIPOQ/adiponectin induces cytotoxic autophagy in breast cancer cells through STK11/LKB1-mediated activation of the AMPK-ULK1 axis. Autophagy 2017, 13, 1386–1403. [Google Scholar] [CrossRef]
- Aryal, P.; Kim, K.; Park, P.-H.; Ham, S.; Cho, J.; Song, K. Baicalein induces autophagic cell death through AMPK/ULK1 activation and downregulation of mTORC1 complex components in human cancer cells. FEBS J. 2014, 281, 4644–4658. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, H.; Chen, X.; Chen, J.; Li, X.; Qiao, G.; Tian, Y.; Yuan, R.; Su, S.; Liu, X.; et al. Aqueous extract of clove inhibits tumor growth by inducing autophagy through AMPK/ULK pathway. Phytother. Res. 2019, 33, 1794–1804. [Google Scholar] [CrossRef]
- Kulkarni, Y.M.; Kaushik, V.; Azad, N.; Wright, C.; Rojanasakul, Y.; O'Doherty, G.; Iyer, A.K.V. Autophagy-Induced Apoptosis in Lung Cancer Cells by a Novel Digitoxin Analog. J. Cell. Physiol. 2016, 231, 817–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Liu, Y.; Li, X. The interaction mechanism between autophagy and apoptosis in colon cancer. Transl. Oncol. 2020, 13, 100871. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhu, L.; Zhao, Y.; Jiang, Y.; Chen, L.; Yu, Y.; Ouyang, L. Fluoxetine induces autophagic cell death via eEF2K-AMPK-mTOR-ULK complex axis in triple negative breast cancer. Cell Prolif. 2018, 51, e12402. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, L.; Zhang, L.; Fu, L.; Liu, B. A small-molecule activator induces ULK1-modulating autophagy-associated cell death in triple negative breast cancer. Autophagy 2017, 13, 777–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Tang, D. Autophagy in pancreatic cancer pathogenesis and treatment. Am. J. Cancer Res. 2012, 2, 383–396. [Google Scholar] [PubMed]
- Bhutia, S.K.; Mukhopadhyay, S.; Sinha, N.; Das, D.N.; Panda, P.K.; Patra, S.K.; Maiti, T.K.; Mandal, M.; Dent, P.; Wang, X.-Y.; et al. Autophagy: Cancer’s friend or foe? Adv. Cancer Res. 2013, 118, 61–95. [Google Scholar] [CrossRef]
- Byun, S.; Lee, E.; Lee, K.W. Therapeutic Implications of Autophagy Inducers in Immunological Disorders, Infection, and Cancer. Int. J. Mol. Sci. 2017, 18, 1959. [Google Scholar] [CrossRef] [PubMed]
- Merenlender-Wagner, A.; Shemer, Z.; Touloumi, O.; Lagoudaki, R.; Giladi, E.; Andrieux, A.; Grigoriadis, N.C.; Gozes, I. New horizons in schizophrenia treatment: Autophagy protection is coupled with behavioral improvements in a mouse model of schizophrenia. Autophagy 2014, 10, 2324–2332. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Miller, A.M.; Woesner, M.E. Autophagy and Schizophrenia: A Closer Look at How Dysregulation of Neuronal Cell Homeostasis Influences the Pathogenesis of Schizophrenia. Einstein J. Biol. Med. 2016, 31, 34–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balic, A.; Sørensen, M.D.; Trabulo, S.M.; Sainz, B.; Cioffi, M.; Vieira, C.R.; Miranda-Lorenzo, I.; Hidalgo, M.; Kleeff, J.; Erkan, M.; et al. Chloroquine Targets Pancreatic Cancer Stem Cells via Inhibition of CXCR4 and Hedgehog Signaling. Mol. Cancer Ther. 2014, 13, 1758–1771. [Google Scholar] [CrossRef] [Green Version]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2020, 124, 333–344. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12 (Suppl. 2), 1528–1534. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Ichim, G.; Green, D.R. Die another way—Non-apoptotic mechanisms of cell death. J. Cell Sci. 2014, 127, 2135–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 2018, 3, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramachandran, S.; Kaushik, I.S.; Srivastava, S.K. Pimavanserin: A Novel Autophagy Modulator for Pancreatic Cancer Treatment. Cancers 2021, 13, 5661. https://doi.org/10.3390/cancers13225661
Ramachandran S, Kaushik IS, Srivastava SK. Pimavanserin: A Novel Autophagy Modulator for Pancreatic Cancer Treatment. Cancers. 2021; 13(22):5661. https://doi.org/10.3390/cancers13225661
Chicago/Turabian StyleRamachandran, Sharavan, Itishree S. Kaushik, and Sanjay K. Srivastava. 2021. "Pimavanserin: A Novel Autophagy Modulator for Pancreatic Cancer Treatment" Cancers 13, no. 22: 5661. https://doi.org/10.3390/cancers13225661
APA StyleRamachandran, S., Kaushik, I. S., & Srivastava, S. K. (2021). Pimavanserin: A Novel Autophagy Modulator for Pancreatic Cancer Treatment. Cancers, 13(22), 5661. https://doi.org/10.3390/cancers13225661