
miRGalaxy: Galaxy-Based Framework for Interactive Analysis of microRNA and isomiR Sequencing Data


 and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
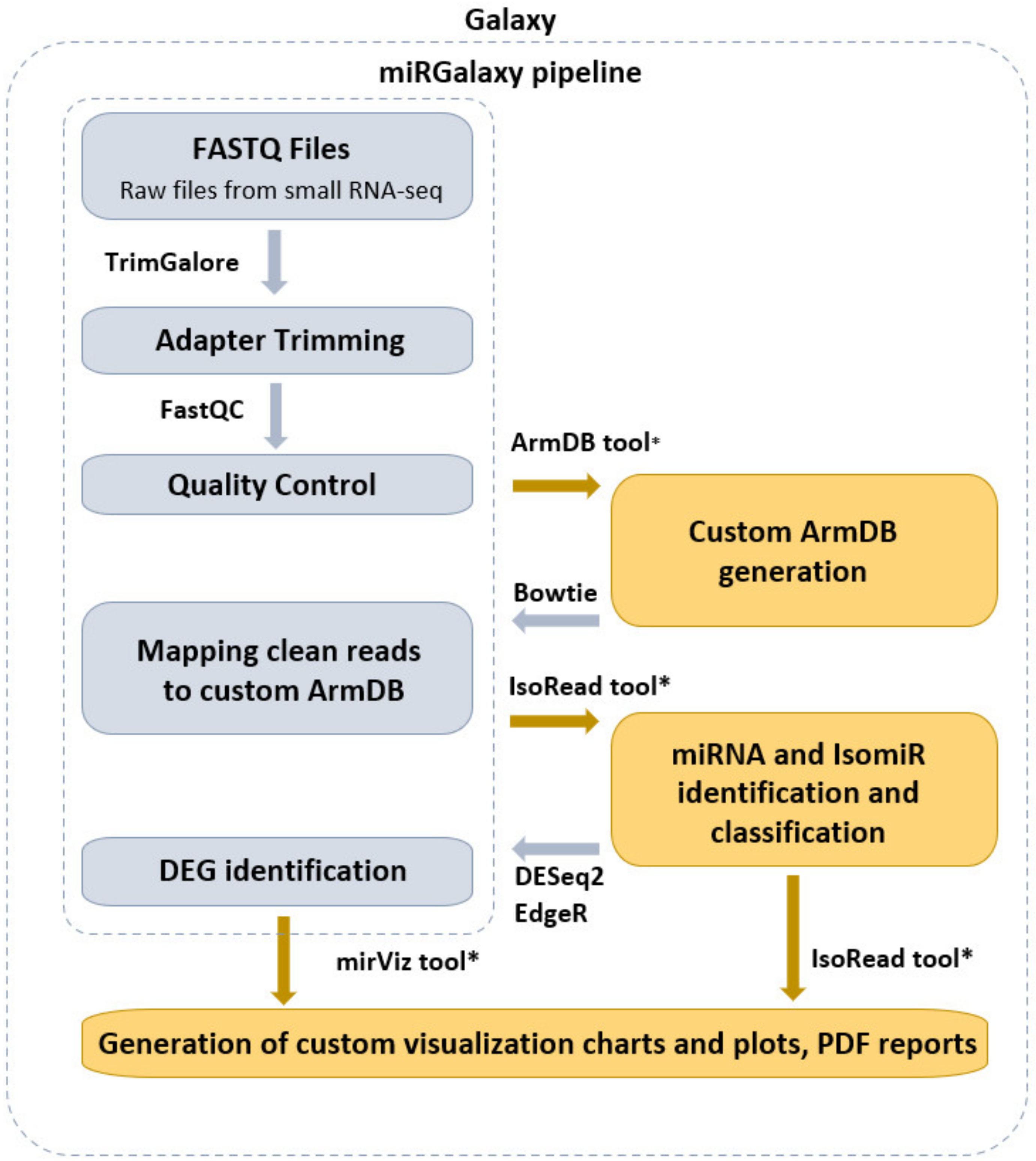
2.1. Pre-Processing Steps
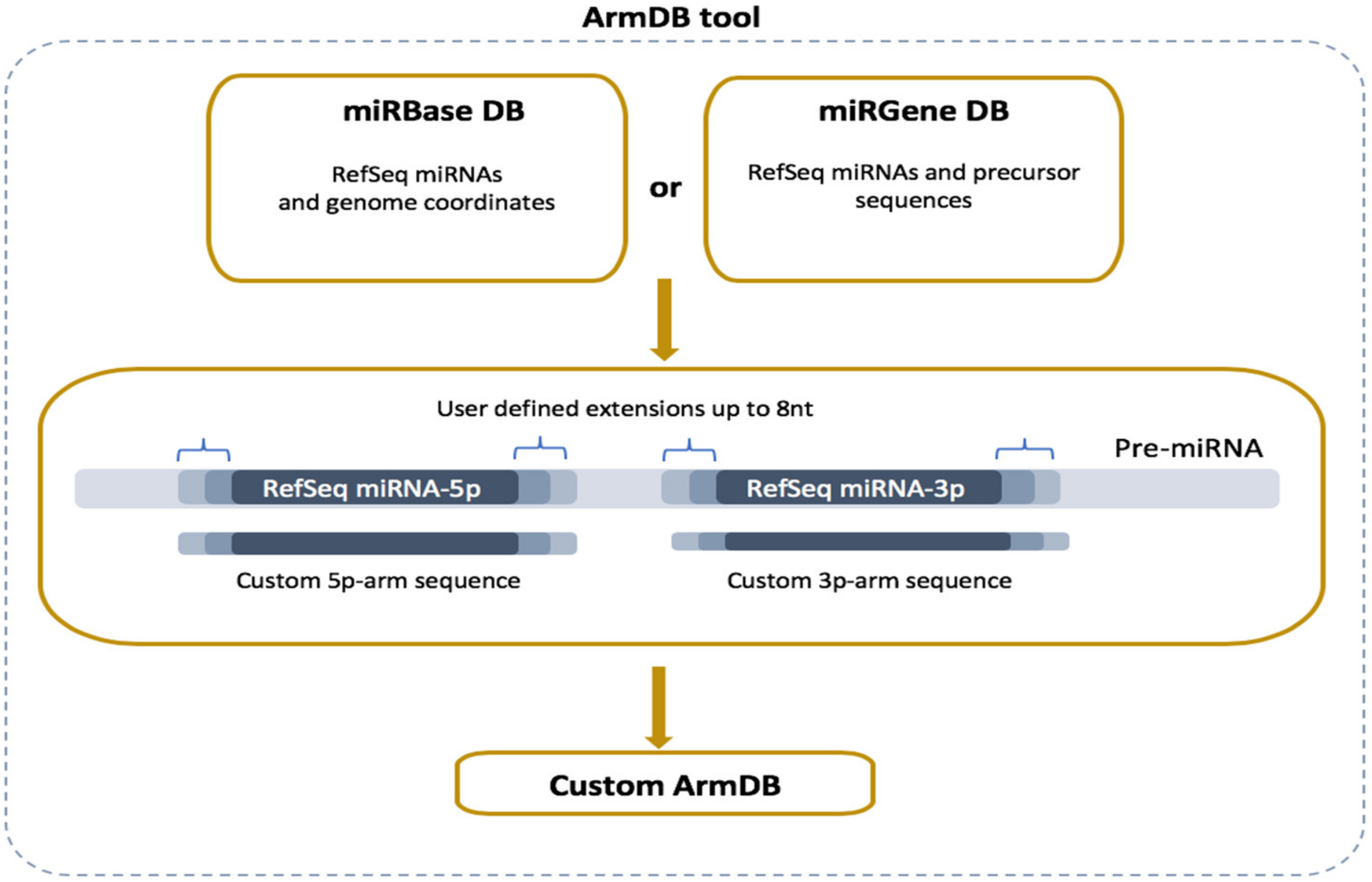
2.2. ArmDB Tool
2.3. Read Mapping
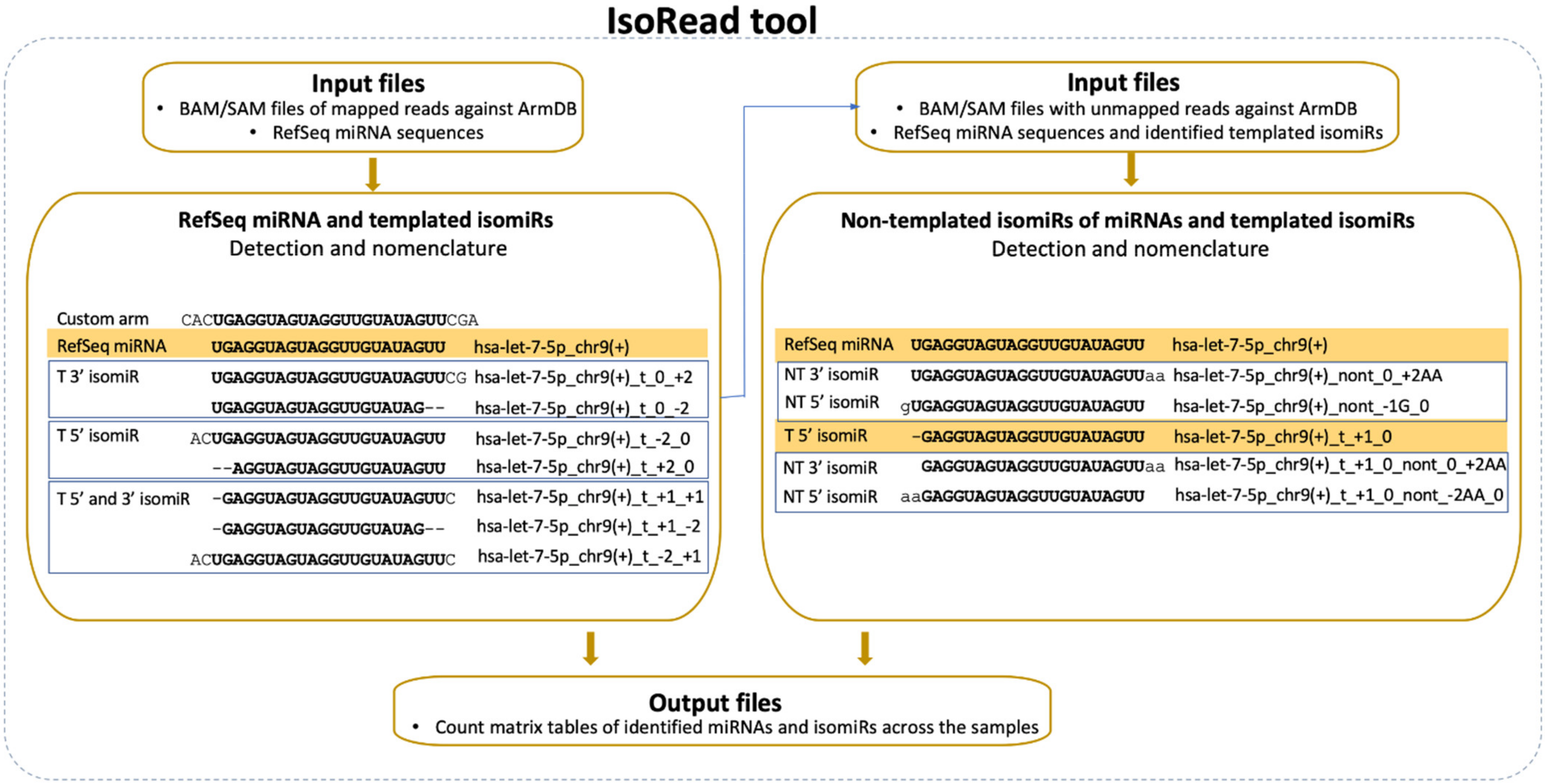
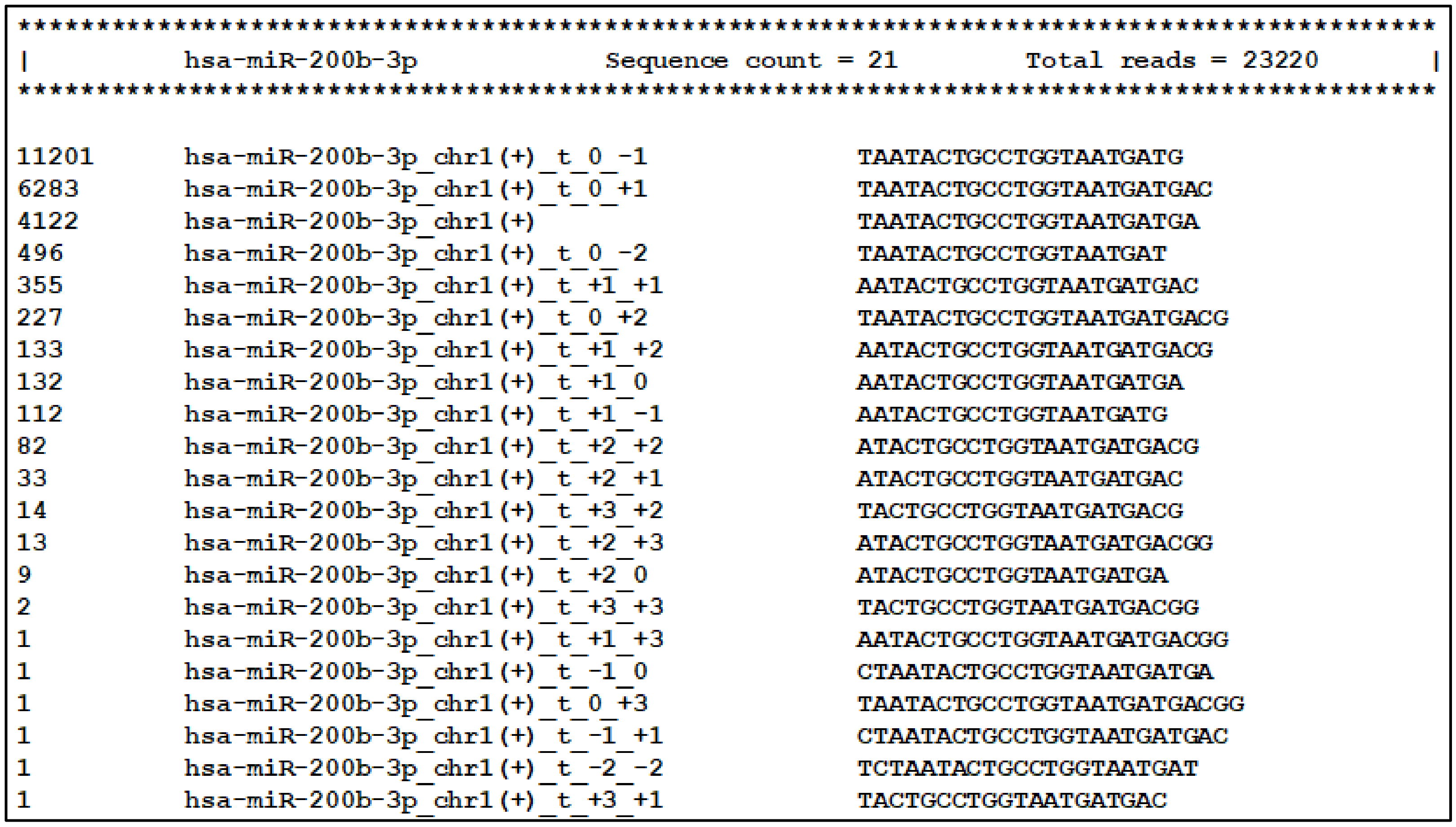
2.4. IsoRead Tool
2.5. DE Step
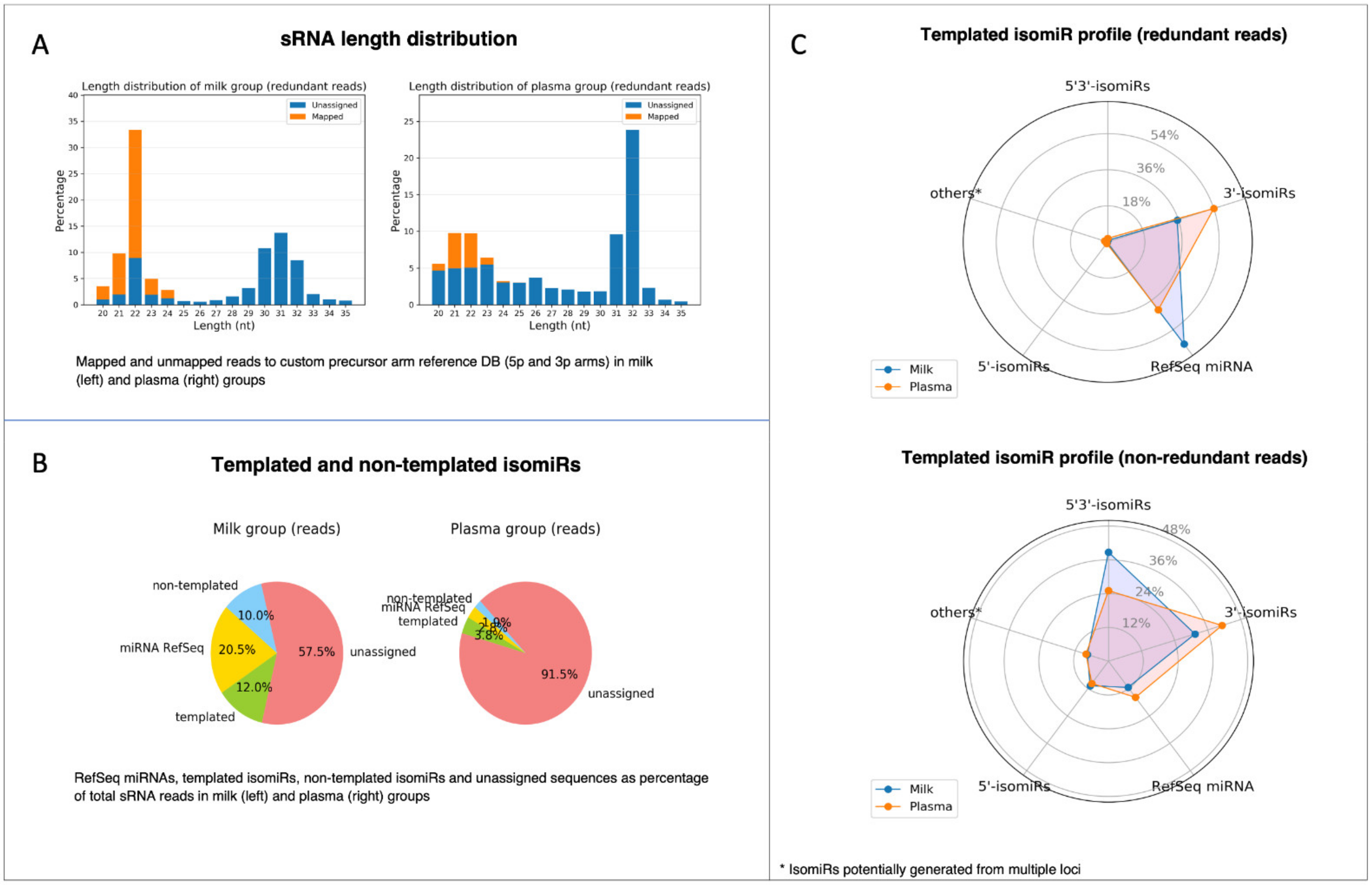
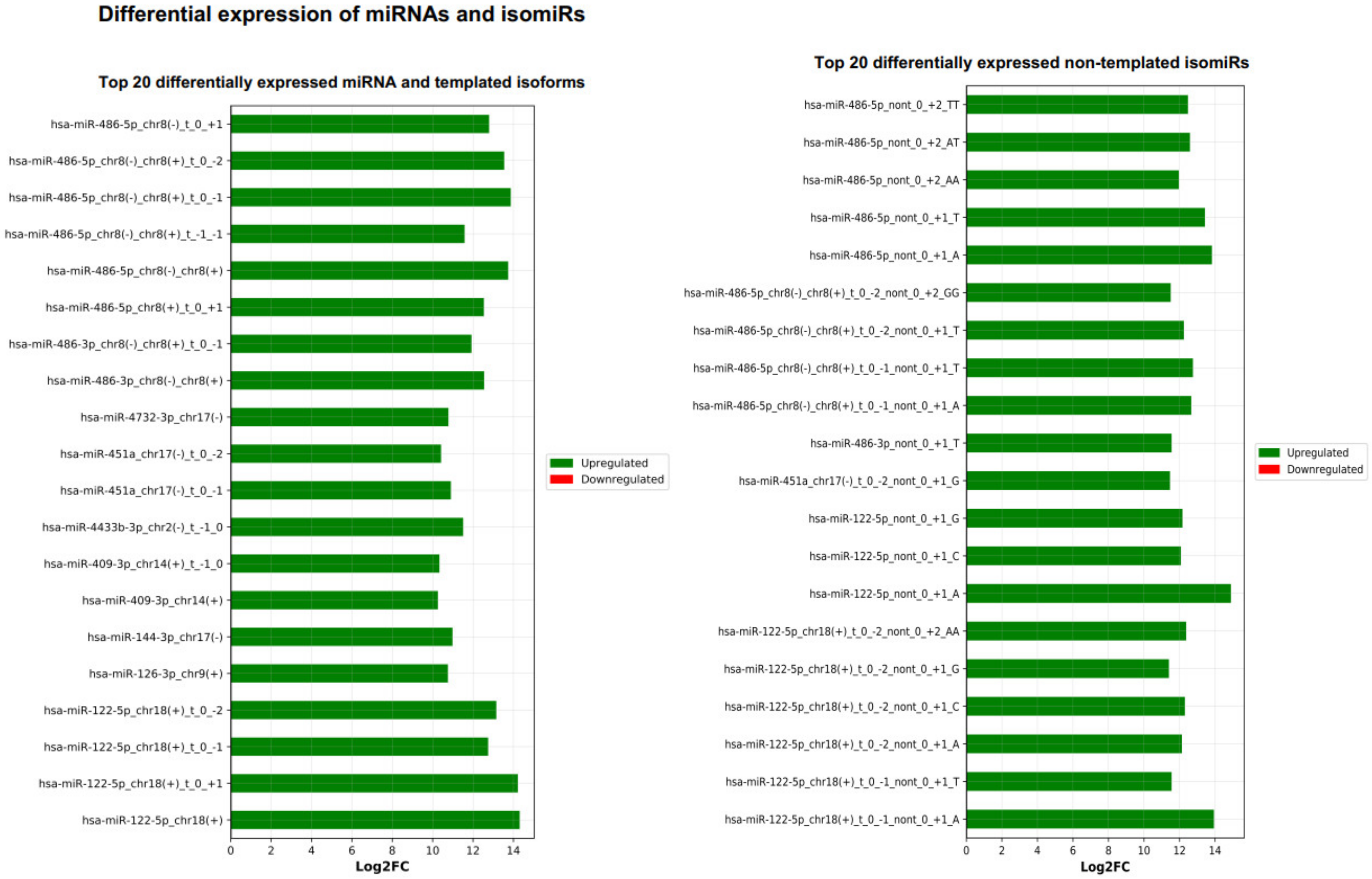
2.6. miRViz Tool
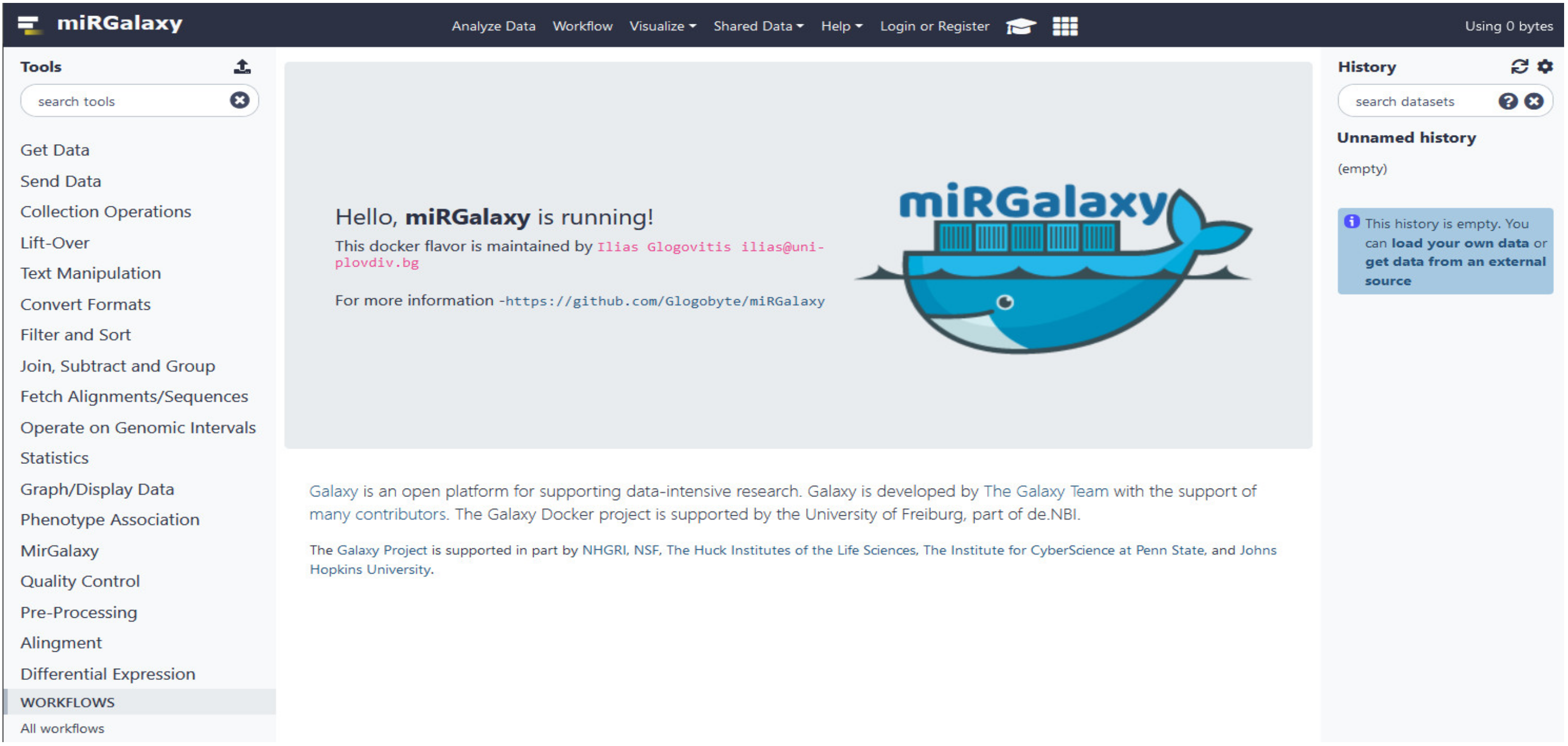
2.7. Availability of the Framework, Installation, and Running miRGalaxy
3. Results and Discussion
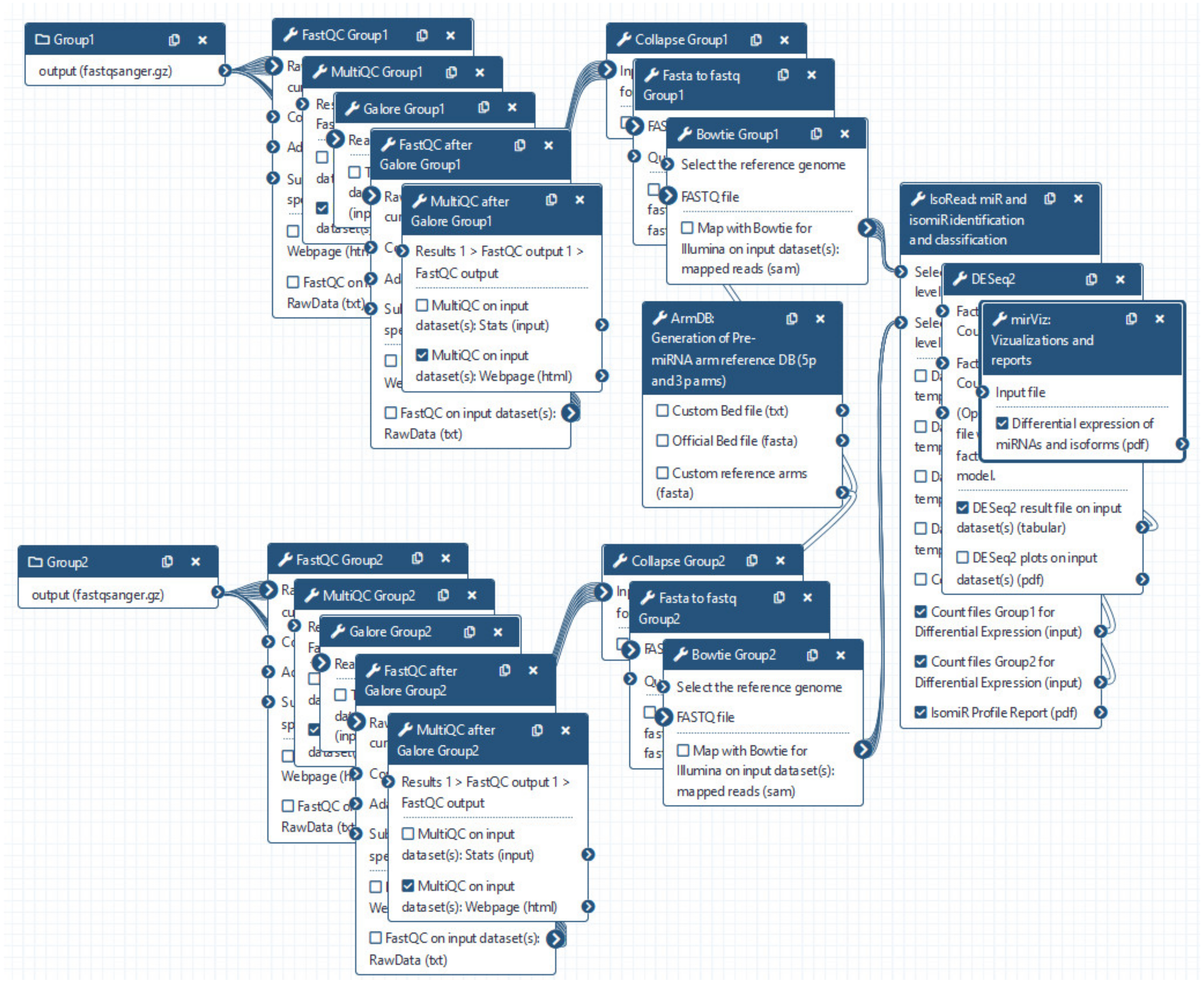
3.1. miRGalaxy Outlines
3.2. miRGalaxy Implementation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morin, R.D.; O’Connor, M.D.; Griffith, M.; Kuchenbauer, F.; Delaney, A.; Prabhu, A.L.; Zhao, Y.; McDonald, H.; Zeng, T.; Hirst, M.; et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008, 18, 610–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burroughs, A.M.; Ando, Y.; de Hoon, M.L.; Tomaru, Y.; Suzuki, H.; Hayashizaki, Y.; Daub, C.O. Deep-sequencing of human Argonaute-associated small RNAs provides insight into miRNA sorting and reveals Argonaute association with RNA fragments of diverse origin. RNA Biol. 2011, 8, 158–177. [Google Scholar] [CrossRef] [PubMed]
- Neilsen, C.T.; Goodall, G.J.; Bracken, C.P. IsomiRs–The overlooked repertoire in the dynamic microRNAome. Trends Genet. 2012, 28, 544–549. [Google Scholar] [CrossRef]
- Tan, G.C.; Chan, E.; Molnar, A.; Sarkar, R.; Alexieva, D.; Isa, I.M.; Robinson, S.; Zhang, S.; Ellis, P.; Langford, C.F.; et al. 5′ isomiR variation is of functional and evolutionary importance. Nucleic Acids Res. 2014, 42, 9424–9435. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Desvignes, T.; Batzel, P.; Berezikov, E.; Eilbeck, K.; Eppig, J.T.; McAndrews, M.S.; Singer, A.; Postlethwait, J.H. MiRNA Nomenclature: A View Incorporating Genetic Origins, Biosynthetic Pathways, and Sequence Variants. Trends Genet. 2015, 31, 613–626. [Google Scholar] [CrossRef] [Green Version]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [Green Version]
- Borges, F.; Martienssen, R.A. The expanding world of small RNAs in plants. Nat. Rev. Mol. Cell Biol. 2015, 16, 727–741. [Google Scholar] [CrossRef] [Green Version]
- Parafioriti, A.; Cifola, I.; Gissi, C.; Pinatel, E.; Vilardo, L.; Armiraglio, E.; Di Bernardo, A.; Daolio, P.A.; Felsani, A.; D’Agnano, I.; et al. Expression profiling of microRNAs and isomiRs in conventional central chondrosarcoma. Cell Death Discov. 2020, 6, 46. [Google Scholar] [CrossRef]
- Li, S.C.; Liao, Y.L.; Ho, M.R.; Tsai, K.W.; Lai, C.H.; Lin, W.C. MiRNA arm selection and isomiR distribution in gastric cancer. BMC Genom. 2012, 13, S13. [Google Scholar] [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; Van Den Beek, M.; Bouvier, D.; Ech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Glogovitis, I.; Yahubyan, G.; Würdinger, T.; Koppers-Lalic, D.; Baev, V. Isomirs–hidden soldiers in the mirna regulatory army, and how to find them? Biomolecules 2021, 11, 41. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 11 November 2021).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F. Trim Galore: A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 11 November 2021).
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Holik, A.Z.; Su, S.; Jansz, N.; Chen, K.; Leong, H.S.; Blewitt, M.E.; Asselin-Labat, M.L.; Smyth, G.K.; Ritchie, M.E. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res. 2015, 43, e97. [Google Scholar] [CrossRef]
- Reingart, M. PyFPDF. Available online: https://github.com/reingart/pyfpdf (accessed on 11 November 2021).
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference (SciPy), Austin, TX, USA, 28 June–3 July 2010; Volume 1, pp. 56–61. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Tareen, A.; Kinney, J.B. Logomaker: Beautiful sequence logos in Python. Bioinformatics 2020, 36, 2272–2274. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, 140–144. [Google Scholar] [CrossRef]
- Fromm, B.; Domanska, D.; Høye, E.; Ovchinnikov, V.; Kang, W.; Aparicio-Puerta, E.; Johansen, M.; Flatmark, K.; Mathelier, A.; Hovig, E.; et al. MirGeneDB 2.0: The metazoan microRNA complement. Nucleic Acids Res. 2020, 48, D132–D141. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Merkel, D. Docker: Lightweight linux containers for consistent development and deployment. Linux J. 2014, 2014, 2. [Google Scholar]
- Blankenberg, D.; Von Kuster, G.; Bouvier, E.; Baker, D.; Afgan, E.; Stoler, N.; Taylor, J.; Nekrutenko, A.; Clements, D.; Coraor, N.; et al. Dissemination of scientific software with Galaxy ToolShed. Genome Biol. 2014, 15, 2–4. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Hu, S.; Zhang, L.; Xin, J.; Sun, C.; Wang, L.; Ding, K.; Wang, B. Tumor circulome in the liquid biopsies for cancer diagnosis and prognosis. Theranostics 2020, 10, 4544–4556. [Google Scholar] [CrossRef]
- Mathai, R.; Vidya, R.; Reddy, B.; Thomas, L.; Udupa, K.; Kolesar, J.; Rao, M. Potential Utility of Liquid Biopsy as a Diagnostic and Prognostic Tool for the Assessment of Solid Tumors: Implications in the Precision Oncology. J. Clin. Med. 2019, 8, 373. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Lázaro, D.; Hernández, J.L.G.; García, A.C.; Martínez, A.C.; Mielgo-Ayuso, J.; Cruz-Hernández, J.J. Liquid biopsy as novel tool in precision medicine: Origins, properties, identification and clinical perspective of cancer’s biomarkers. Diagnostics 2020, 10, 215. [Google Scholar] [CrossRef] [Green Version]
- Rubio, M.; Bustamante, M.; Hernandez-Ferrer, C.; Fernandez-Orth, D.; Pantano, L.; Sarria, Y.; Piqué-Borras, M.; Vellve, K.; Agramunt, S.; Carreras, R.; et al. Circulating miRNAs, isomiRs and small RNA clusters in human plasma and breast milk. PLoS ONE 2018, 13, e0193527. [Google Scholar] [CrossRef]
- Mantini, G.; Meijer, L.L.; Glogovitis, I.; In’T Veld, S.G.J.G.; Paleckyte, R.; Capula, M.; Le Large, T.Y.S.; Morelli, L.; Pham, T.V.; Piersma, S.R.; et al. Omics analysis of educated platelets in cancer and benign disease of the pancreas. Cancers 2021, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Yang, Y.J.; Wang, L.Y.; Qiao, S.B.; Lu, X.F.; Wu, Y.J.; Xu, B.; Li, H.F.; Gu, D.F. Elevated plasma miRNA-122, -140-3p, -720, -2861, and -3149 during early period of acute coronary syndrome are derived from peripheral blood mononuclear cells. PLoS ONE 2017, 12, e0184256. [Google Scholar] [CrossRef] [Green Version]
- Gilje, P.; Frydland, M.; Bro-Jeppesen, J.; Dankiewicz, J.; Friberg, H.; Rundgren, M.; Devaux, Y.; Stammet, P.; Al-Mashat, M.; Jögi, J.; et al. The association between plasma miR-122-5p release pattern at admission and all-cause mortality or shock after out-of-hospital cardiac arrest. Biomarkers 2019, 24, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.L.; Zhao, H.; Yang, S.G.; Chen, E.M.; Chen, W.Q.; Li, L.J. Plasma miRNA-122-5p and miRNA-151a-3p identified as potential biomarkers for liver injury among CHB patients with PNALT. Hepatol. Int. 2018, 12, 277–287. [Google Scholar] [CrossRef]
- Alsaweed, M.; Lai, C.T.; Hartmann, P.E.; Geddes, D.T.; Kakulas, F. Human milk miRNAs primarily originate from the mammary gland resulting in unique miRNA profiles of fractionated milk. Sci. Rep. 2016, 6, 20680. [Google Scholar] [CrossRef] [PubMed]
- Benmoussa, A.; Provost, P. Milk MicroRNAs in Health and Disease. Compr. Rev. Food Sci. Food Saf. 2019, 18, 703–722. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. miRs and isomiRs | |||
|---|---|---|---|
| Plasma | Milk | ||
| Name | Raw Counts | Name | Raw Counts |
| RefSeq miRs | |||
| miR-486-5p_chr8(−)_chr8(+) | 341,413 | miR-148a-3p_chr7(−) | 14,357,338 |
| miR-122-5p_chr18(+) | 190,578 | miR-30a-5p_chr6(−) | 1,753,796 |
| miR-320a-3p_chr8(−) | 66,951 | miR-146b-5p_chr10(+) | 915,663 |
| miR-92a-3p_chr13(+)_chrX(−) | 51,339 | miR-26a-5p_chr12(−)_chr3(+) | 667,270 |
| miR-423-5p_chr17(+) | 49,904 | let-7f-5p_chr9(+)_chrX(−) | 616,265 |
| Template isomiRs | |||
| miR-122-5p_chr18(+)_t_0_−1 | 201,561 | miR-200a-3p_chr1(+)_t_0_−1 | 1,628,344 |
| miR-486-5p_chr8(−)_chr8(+) _t_0_−1 | 177,874 | miR-148a-3p_chr7(−)_t_0_−1 | 1,170,712 |
| miR-423-5p_chr17(+)_t_0_−2 | 171,672 | miR-146b-5p_chr10(+)_t_0_−1 | 620,583 |
| miR-451a_chr17(−)_t_0_−1 | 163,779 | miR-146b-5p_chr10(+)_t_0_+1 | 562,077 |
| miR-486-5p_chr8(−)_chr8(+)_t_0_−2 | 75,436 | miR-21-5p_chr17(+)_t_0_−1 | 558,349 |
| Non-template isomiRs | |||
| miR-486-5p_nont_0_+1_A | 146,122 | miR-148a-3p_chr7(−)_t_0_−1_ nont_0_+1_C | 3,242,405 |
| miR-486-5p_nont_0_+1_T | 93,666 | miR-148a-3p_chr7(−)_t_0_−1_ nont_0_+1_A | 708,482 |
| miR-122-5p_nont_0_+1_A | 67,547 | miR-148a-3p_chr7(−)_t_0_−1_ nont_0_+1_G | 540,724 |
| miR-320a-3p_chr8(−)_t_0_−1_ nont_0_+1_T | 22,925 | miR-200a-3p_chr1(+)_t_0_−2_ nont_0_+1_A | 162,583 |
| miR-122-5p_chr18(+)_t_0_−2_ nont_0_+1_C | 19,211 | miR-26a-5p_chr12(−)_chr3(+)_t_0_−1_nont_0_+1_C | 153,539 |
| B. miRs and isomiRs | |||
| Cancer (PDAC) | Control (Asymptomatic Individuals) | ||
| Name | Raw Counts | Name | Raw Counts |
| RefSeq miRs | |||
| hsa-miR-26a-5p_chr12(−)_chr3(+) | 6,005,560 | hsa-miR-26a-5p_chr12(−)_chr3(+) | 5,825,421 |
| hsa-let-7f-5p_chr9(+)_chrX(−) | 3,307,664 | hsa-miR-191-5p_chr3(−) | 3,375,427 |
| hsa-miR-191-5p_chr3(−) | 3,150,135 | hsa-let-7a-5p_chr11(−)_chr9(+)_chr22(+) | 3,227,687 |
| hsa-let-7a-5p_chr11(−)_chr9(+)_chr22(+) | 3,041,788 | hsa-let-7f-5p_chr9(+)_chrX(−) | 2,844,363 |
| hsa-miR-22-3p_chr17(−) | 2,616,893 | hsa-miR-22-3p_chr17(−) | 2,530,803 |
| Template isomiRs | |||
| hsa-miR-30d-5p_chr8(−)_t_0_+2 | 1,103,932 | hsa-miR-30d-5p_chr8(−)_t_0_+2 | 1,167,321 |
| hsa-miR-486-5p_chr8(−)_chr8(+)_t_0_−1 | 1,033,505 | hsa-miR-486-5p_chr8(−)_chr8(+)_t_0_−1 | 1,114,672 |
| hsa-miR-181a-5p_chr1(−)_chr9(+)_t_0_−1 | 866,979 | hsa-miR-142-5p_chr17(−)_t_-2_−1 | 971,608 |
| hsa-miR-21-5p_chr17(+)_t_0_+2 | 810,654 | hsa-miR-181a-5p_chr1(−)_chr9(+)_t_0_−1 | 936,358 |
| hsa-miR-142-5p_chr17(−)_t_-2_−1 | 808,717 | hsa-miR-21-5p_chr17(+)_t_0_+2 | 718,611 |
| Non-template isomiRs | |||
| hsa-miR-92a-3p_nont_0_+2_AT | 1,940,172 | hsa-miR-92a-3p_nont_0_+2_AT | 2,105,693 |
| hsa-miR-486-5p_nont_0_+1_T | 1,304,573 | hsa-miR-486-5p_nont_0_+1_T | 1,573,373 |
| hsa-miR-92a-3p_nont_0_+2_AA | 864,072 | hsa-miR-92a-3p_nont_0_+2_AA | 903,167 |
| hsa-miR-486-5p_nont_0_+1_A | 582,067 | hsa-miR-486-5p_nont_0_+1_A | 619,989 |
| hsa-miR-92a-3p_nont_0_+2_TT | 459,988 | hsa-miR-92a-3p_nont_0_+2_TT | 474,848 |
| miR and isomiR Names | Base Mean | Log2FC | p-adj |
|---|---|---|---|
| Rubio and co-workers [34] | |||
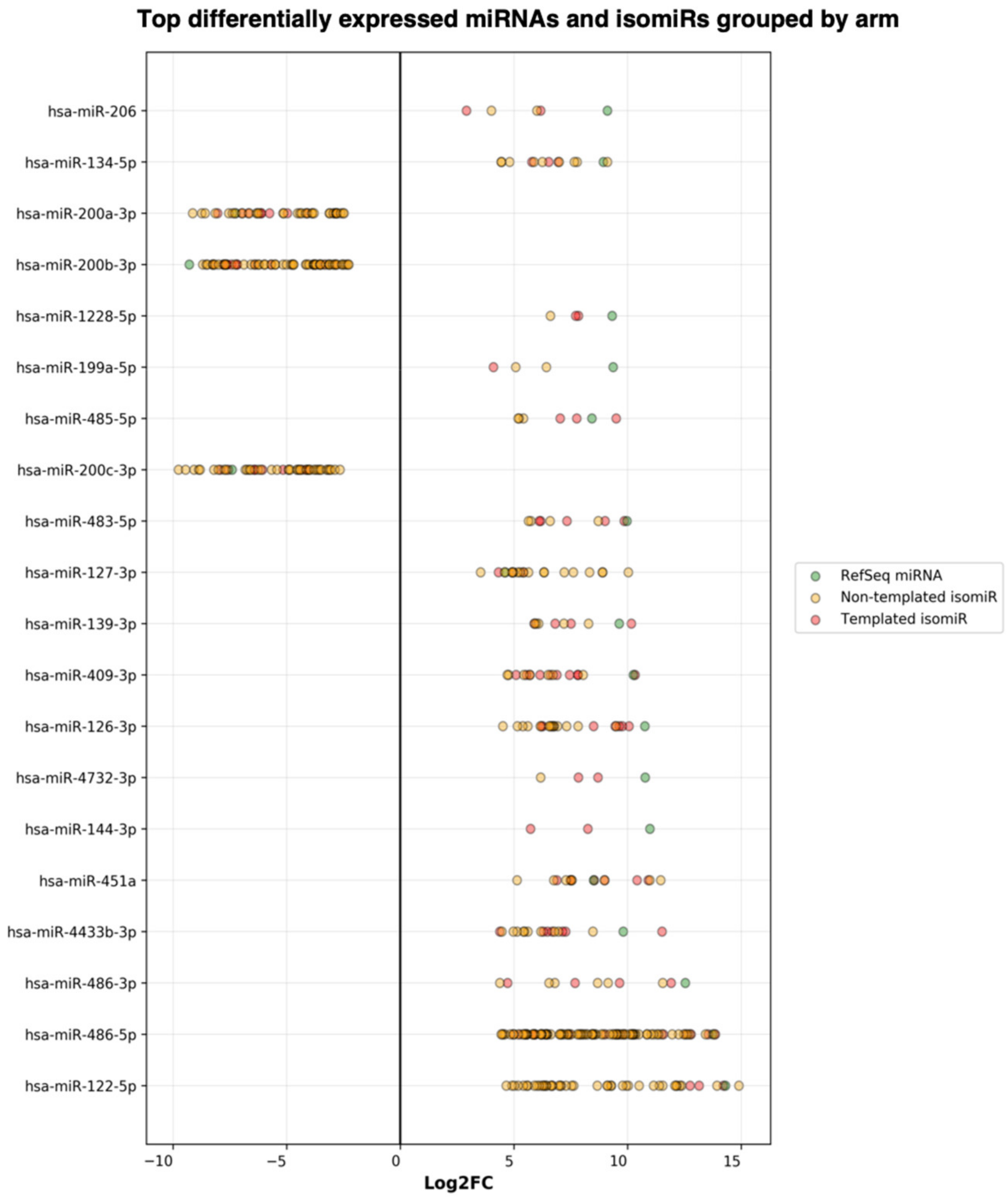
| miR-200b-3p | 22349.79 | −7.74 | 1.67 × 10−17 |
| Output from miRGalaxy | |||
| miR-200b-3p_chr1(+) | 2838.44 | −9.29 | 1.15 × 10−43 |
| miR-200b-3p_chr1(+)_t_+1_-1 | 80.73 | −7.19 | 2.68 × 10−13 |
| miR-200b-3p_chr1(+)_t_+1_+1 | 358.91 | −8.26 | 2.18 × 10−26 |
| miR-200b-3p_chr1(+)_t_+1_+1_nont_0_+1_A | 693.30 | −8.49 | 1.42 × 10−25 |
| miR-200b-3p_chr1(+)_t_+1_+1_nont_0_+1_C | 276.25 | −8.16 | 4.46 × 10−22 |
| miR-200b-3p_chr1(+)_t_+1_+1_nont_0_+1_T | 1267.51 | −7.70 | 4.16 × 10−28 |
| miR-200b-3p_chr1(+)_t_+1_+2 | 101.54 | −7.63 | 8.54 × 10−18 |
| miR-200b-3p_chr1(+)_t_+1_0 | 92.47 | −7.43 | 3.36 × 10−15 |
| miR-200b-3p_chr1(+)_t_+1_0_nont_0_+1_T | 56.09 | −6.57 | 1.75 × 10−09 |
| miR-200b-3p_chr1(+)_t_+2_+1_nont_0_+1_A | 43.01 | −6.41 | 2.49 × 10−12 |
| miR-200b-3p_chr1(+)_t_+2_+1_nont_0_+1_T | 106.81 | −7.71 | 1.48 × 10−18 |
| miR-200b-3p_chr1(+)_t_+2_+2 | 41.27 | −6.40 | 2.34 × 10−13 |
| miR-200b-3p_chr1(+)_t_0_-1 | 6656.60 | −7.18 | 5.29 × 10−25 |
| miR-200b-3p_chr1(+)_t_0_-1_nont_0_+1_C | 32.03 | −5.50 | 3.22 × 10−09 |
| miR-200b-3p_chr1(+)_t_0_-1_nont_0_+1_G | 118.22 | −7.28 | 1.14 × 10−14 |
| miR-200b-3p_chr1(+)_t_0_-1_nont_0_+1_T | 129.70 | −6.88 | 2.39 × 10−17 |
| miR-200b-3p_chr1(+)_t_0_-1_nont_0_+2_GC | 119.02 | −6.26 | 3.31 × 10−09 |
| miR-200b-3p_chr1(+)_t_0_-2 | 268.42 | −7.65 | 1.51 × 10−18 |
| miR-200b-3p_chr1(+)_t_0_-2_nont_0_+1_A | 568.39 | −8.70 | 8.14 × 10−24 |
| miR-200b-3p_chr1(+)_t_0_-2_nont_0_+1_C | 35.22 | −5.96 | 2.94 × 10−08 |
| miR-200b-3p_chr1(+)_t_0_-2_nont_0_+1_T | 40.11 | −6.22 | 4.41 × 10−10 |
| miR-200b-3p_chr1(+)_t_0_+1 | 5005.92 | −7.80 | 1.58 × 10−32 |
| miR-200b-3p_chr1(+)_t_0_+2 | 150.83 | −7.75 | 2.02 × 10−20 |
| miR-200b-3p_nont_0_+1_A | 260.75 | −7.99 | 4.85 × 10−19 |
| miR-200b-3p_nont_0_+1_G | 33.11 | −6.00 | 3.95 × 10−10 |
| miR-200b-3p_nont_0_+1_T | 861.69 | −7.72 | 2.96 × 10−15 |
| miR-200b-3p_nont_0_+2_CA | 881.83 | −8.24 | 6.25 × 10−19 |
| miR-200b-3p_nont_0_+2_CC | 331.61 | −8.54 | 6.48 × 10−23 |
| miR-200b-3p_nont_0_+2_CT | 1424.88 | −8.24 | 5.22 × 10−26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glogovitis, I.; Yahubyan, G.; Würdinger, T.; Koppers-Lalic, D.; Baev, V. miRGalaxy: Galaxy-Based Framework for Interactive Analysis of microRNA and isomiR Sequencing Data. Cancers 2021, 13, 5663. https://doi.org/10.3390/cancers13225663
Glogovitis I, Yahubyan G, Würdinger T, Koppers-Lalic D, Baev V. miRGalaxy: Galaxy-Based Framework for Interactive Analysis of microRNA and isomiR Sequencing Data. Cancers. 2021; 13(22):5663. https://doi.org/10.3390/cancers13225663
Chicago/Turabian StyleGlogovitis, Ilias, Galina Yahubyan, Thomas Würdinger, Danijela Koppers-Lalic, and Vesselin Baev. 2021. "miRGalaxy: Galaxy-Based Framework for Interactive Analysis of microRNA and isomiR Sequencing Data" Cancers 13, no. 22: 5663. https://doi.org/10.3390/cancers13225663
APA StyleGlogovitis, I., Yahubyan, G., Würdinger, T., Koppers-Lalic, D., & Baev, V. (2021). miRGalaxy: Galaxy-Based Framework for Interactive Analysis of microRNA and isomiR Sequencing Data. Cancers, 13(22), 5663. https://doi.org/10.3390/cancers13225663