
The Use of Oncolytic Viruses in the Treatment of Multiple Myeloma


Abstract
:Simple Summary
Abstract
1. Introduction
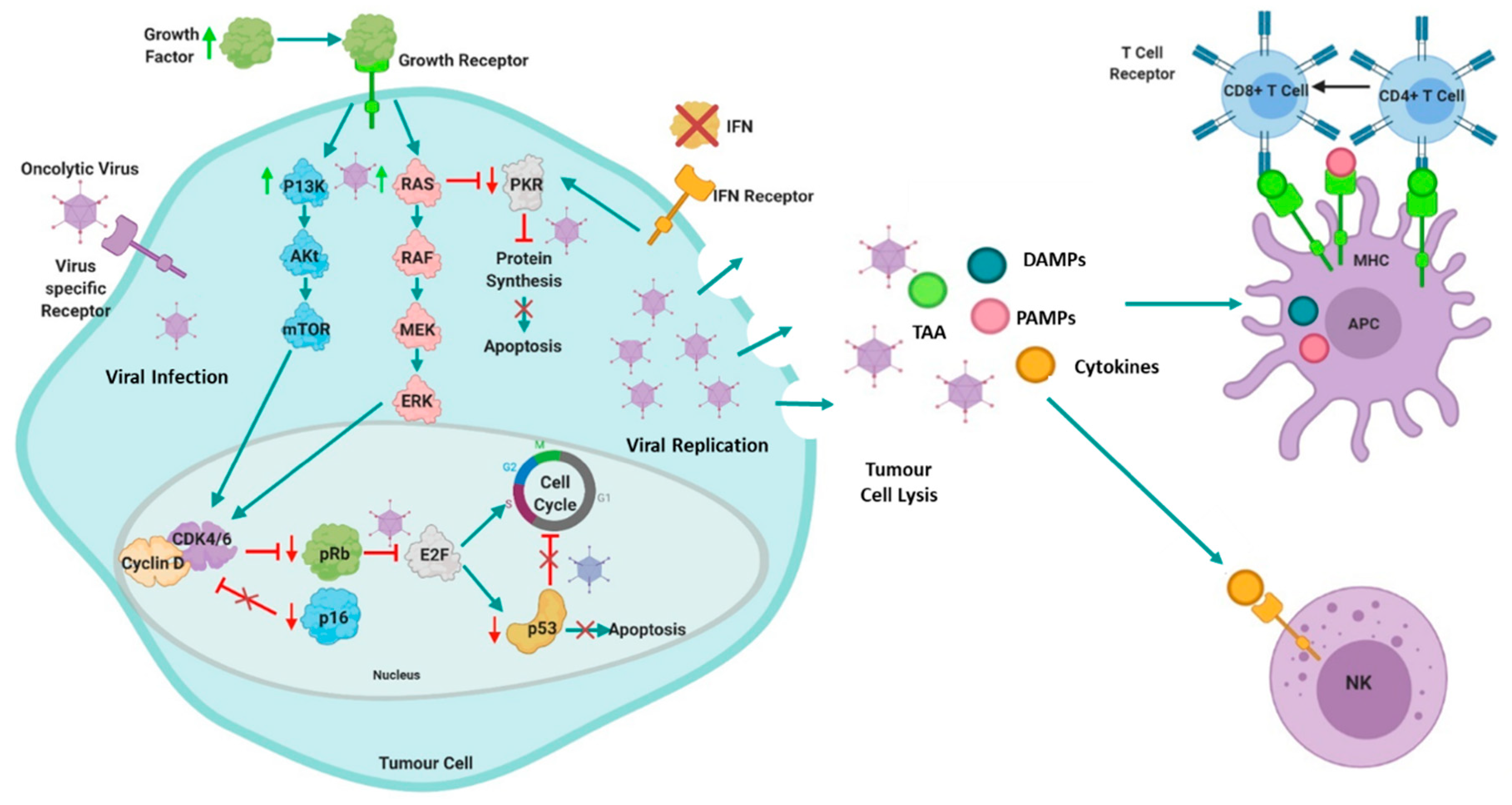
2. Oncolytic Virotherapy in Cancer
3. The Use of Oncolytic Viruses in the Treatment of Multiple Myeloma
3.1. The Use of DNA Oncolytic Viruses in the Treatment of Myeloma
3.1.1. Preclinical Studies with Adenovirus in the Treatment of Myeloma
3.1.2. Preclinical Studies with Vaccinia Virus in the Treatment of Myeloma
3.1.3. Clinical Use of Vaccinia Virus in the Treatment of Myeloma
3.1.4. Preclinical Studies with Myxoma Virus in the Treatment of Myeloma
3.1.5. Preclinical Studies with Herpes Simplex Virus in the Treatment of Myeloma
3.2. The Use of RNA Oncolytic Viruses in the Treatment of Myeloma
3.2.1. Preclinical Studies with Reovirus in the Treatment of Myeloma
3.2.2. Clinical Use of Reovirus in the Treatment of Myeloma
3.2.3. Preclinical Studies with Coxsackie Virus in the Treatment of Myeloma
3.2.4. Preclinical Studies with Measles Virus in the Treatment of Myeloma
3.2.5. Clinical Use of Measles Virus in the Treatment of Myeloma
3.2.6. Preclinical Studies with Bovine Viral Diarrhea Virus in the Treatment of Myeloma
3.2.7. Preclinical Studies with Vesicular Stomatitis Virus
3.2.8. Clinical Use of VSV-IFNβ-NIS in the Treatment of Myeloma
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 123I | Iodine-123 |
| Ad | Adenovirus |
| Ad5 | Adenovirus serotype 5 |
| Ad35 | Adenovirus serotype 5 |
| ASCT | Autologous stem cell transplant |
| BCMA | B cell mature antigen |
| BITES | Bi-specific T cell engagers |
| BMSC | Bone marrow stromal cells |
| BVDV | Bovine viral diarrhoea virus |
| CAR | Coxsackievirus and adenovirus receptor |
| DC | Dendritic cell |
| FLC | Free light chain |
| GFP | Green fluorescent protein |
| GM-CSF | Granulocyte macrophage-colony stimulating factor |
| HDACi | Histone deacetylase inhibitors |
| HSV | Herpes simplex virus |
| IFNβ | Interferon beta |
| IMiDs | Immunomodulatory drugs |
| mAb | Monoclonal antibody |
| MPC | Malignant plasma cell |
| MV | Measles virus |
| MV-Edm | Measles virus Edmonton strain |
| MYXV | Myxoma virus |
| NIS | Sodium/iodine symporter |
| NK | Natural killer |
| LOAd | Lokon oncolytic adenovirus |
| OV | Oncolytic virus |
| PBMC | Peripheral blood mononuclear cell |
| PHA | Phytohemagglutinin |
| PI | Proteasome inhibitor |
| Reo | Reovirus |
| scFV | Single chain variable fragment |
| SIRT-1 | Sirtuin-1 |
| TAA | Tumour-associated antigens |
| TAMS | Tumour-associated macrophages |
| TK | Thymidine kinase |
| VGF | Vaccinia growth factor |
| VSV | Vesicular stomatitis virus |
| VV | Vaccinia virus |
| VVDD | Double-deleted VV |
| V-BECN-1 | TK-deleted VV expressing beclin-1 |
| WT | Wildtype |
References
- Myeloma Incidence Statistics. Cancer Research UK. 2016. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/myeloma (accessed on 21 September 2020).
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863. [Google Scholar] [CrossRef]
- Persona, E.P.; Mesa, M.G.; Sánchez, P.J.G.; Rodríguez, A.P.G. Lenalidomide treatment for patients with multiple myeloma: Diagnosis and management of most frequent adverse events. Adv. Ther. 2011, 28 (Suppl. 1), 11–16. [Google Scholar] [CrossRef]
- Laubach, J.P.; Mitsiades, C.S.; Hideshima, T.; Schlossman, R.; Chauhan, D.; Munshi, N.; Ghobrial, I.; Carreau, N.; Anderson, K.C.; Richardson, P.G. Bortezomib in the management of multiple myeloma. Cancer Manag. Res. 2009, 1, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.K.; Bailey, H.; Stenehjem, D. Panobinostat for the treatment of multiple myeloma: The evidence to date. J. Blood Med. 2015, 6, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dima, D.; Dower, J.; Comenzo, R.L.; Varga, C. Evaluating Daratumumab in the Treatment of Multiple Myeloma: Safety, Efficacy and Place in Therapy. Cancer Manag. Res. 2020, 12, 7891–7903. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Isatuximab: A Review of Its Use in Multiple Myeloma. Target. Oncol. 2021, 16, 675–686. [Google Scholar] [CrossRef]
- Starr, P. Elotuzumab, First-in-Class Monoclonal Antibody Immunotherapy, Improves Outcomes in Patients with Multiple Myeloma. Am. Health Drug Benefits 2015, 8, 17. [Google Scholar]
- Munshi, N.C.; Anderson, J.L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Su, C.T.; Ye, J.C. Emerging therapies for relapsed/refractory multiple Myeloma. J. Hematol. Oncol. 2021, 14, 115. [Google Scholar] [CrossRef]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Bluming, A.Z.; Ziegler, J.L. Regression of Burkitt’s lymphoma in association with measles infection. Lancet 1971, 2, 105–106. [Google Scholar] [CrossRef]
- Taqi, A.M.; Abdurrahman, M.B.; Yakubu, A.M.; Fleming, A.F. Regression of Hodgkin’s disease after measles. Lancet 1981, 1, 1112. [Google Scholar] [CrossRef]
- Kawa, A.; Arakawa, S. The effect of attenuated vaccinia virus AS strain on multiple myeloma; a case report. Jpn. J. Exp. Med. 1987, 57, 79–81. [Google Scholar]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Oliva, S.; Gambella, M.; Boccadoro, M.; Bringhen, S. Systemic virotherapy for multiple myeloma. Expert Opin. Biol. Ther. 2017, 17, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.G.; Feng, X.; DiFrancesco, L.M.; Fonseca, K.; Forsyth, P.A.; Paterson, A.H.; Coffey, M.C.; Thompson, B. REO-001: A phase I trial of percutaneous intralesional administration of reovirus type 3 dearing (Reolysin®) in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 696–706. [Google Scholar] [CrossRef]
- Gollamudi, R.; Ghalib, M.H.; Desai, K.K.; Chaudhary, I.; Wong, B.; Einstein, M.; Coffey, M.; Gill, G.M.; Mettinger, K.; Mariadason, J.; et al. Intravenous administration of Reolysin®, a live replication competent RNA virus is safe in patients with advanced solid tumors. Investig. New Drugs 2010, 28, 641–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, E.A.; Sampson, V.; Stabley, D.; Walter, A.; Sol-Church, K.; Cripe, T.; Hingorani, P.; Ahern, C.H.; Weigel, B.J.; Zwiebel, J.; et al. A phase I trial and viral clearance study of reovirus (Reolysin) in children with relapsed or refractory extra-cranial solid tumors: A Children’s Oncology Group Phase I Consortium report. Pediatr. Blood Cancer 2015, 62, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immun-ity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Bartee, E. Potential of oncolytic viruses in the treatment of multiple myeloma. Oncolytic Virother. 2018, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sborov, D.W.; Nuovo, G.J.; Stiff, A.; Mace, T.; Lesinski, G.B.; Benson, D.M.; Efebera, Y.A.; Rosko, A.E.; Pichiorri, F.; Grever, M.R.; et al. A Phase I Trial of Single-Agent Reolysin in Patients with Relapsed Multiple Myeloma. Clin. Cancer Res. 2014, 20, 5946–5955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teoh, G.; Chen, L.; Urashima, M.; Tai, Y.T.; Celi, L.A.; Chen, D.; Chauhan, D.; Ogata, A.; Finberg, R.W.; Webb, I.J.; et al. Adenovirus vector-based purging of multiple myeloma cells. Blood 1998, 92, 4591–4601. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.S.; Gomes, E.M.; Butcher, L.D.; Hernandez-Alcoceba, R.; Chang, D.; Kansopon, J.; Newman, J.; Stone, M.J.; Tong, A.W. Growth Inhibition of Human Multiple Myeloma Cells by an Oncolytic Adenovirus Carrying the CD40 Ligand Transgene. Clin. Cancer Res. 2009, 15, 4847–4856. [Google Scholar] [CrossRef] [Green Version]
- Senac, J.S.; Doronin, K.; Russell, S.J.; Jelinek, D.F.; Greipp, P.R.; Barry, M.A. Infection and killing of multiple myeloma by adenoviruses. Hum. Gene Ther. 2010, 21, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Drouin, M.; Cayer, M.-P.; Jung, D. Adenovirus 5 and chimeric adenovirus 5/F35 employ distinct B-lymphocyte intracellular trafficking routes that are independent of their cognate cell surface receptor. Virology 2010, 401, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Lyle, C.; McCormick, F. Integrin alphavbeta5 is a primary receptor for adenovirus in CAR-negative cells. Virol. J. 2010, 7, 148. [Google Scholar] [CrossRef] [Green Version]
- Lavery, D.; Fu, S.M.; Lufkin, T.; Chen-Kiang, S. Productive infection of cultured human lymphoid cells by adenovirus. J. Virol. 1987, 61, 1466–1472. [Google Scholar] [CrossRef] [Green Version]
- Lavery, D.J.; Chen-Kiang, S. Adenovirus E1A and E1B genes are regulated posttranscriptionally in human lymphoid cells. J. Virol. 1990, 64, 5349–5359. [Google Scholar] [CrossRef] [Green Version]
- Wenthe, J.; Naseri, S.; Hellström, A.C.; Wiklund, H.J.; Eriksson, E.; Loskog, A. Immunostimulatory oncolytic virotherapy for mul-tiple myeloma targeting 4-1BB and/or CD40. Cancer Gene Ther. 2020, 27, 948–959. [Google Scholar] [CrossRef]
- Zhang, L.; Hedjran, F.; Larson, C.; Perez, G.L.; Reid, T. A novel immunocompetent murine model for replicating oncolytic ade-noviral therapy. Cancer Gene Ther. 2015, 22, 17–22. [Google Scholar] [CrossRef]
- Ginsberg, H.S.; Moldawer, L.L.; Sehgal, P.B.; Redington, M.; Kilian, P.L.; Chanock, R.M.; Prince, G.A. A mouse model for investigating the molecular pathogenesis of adenovirus pneumonia. Proc. Natl. Acad. Sci. USA 1991, 88, 1651–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, S.J.; Gordon, F.C.; Gregory, D.W.; McPhie, J.L.; Postlethwaite, R.; White, R.; Willcox, H.N.A. Infection of mouse liver by human adeno-virus type 5. J. Gen. Virol. 1978, 40, 45–61. [Google Scholar] [CrossRef]
- Deng, H.; Tang, N.; Stief, A.E.; Mehta, N.; Baig, E.; Head, R.; Sleep, G.; Yang, X.-Z.; McKerlie, C.; Trudel, S.; et al. Oncolytic virotherapy for multiple myeloma using a tumour-specific double-deleted vaccinia virus. Leukemia 2008, 22, 2261–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, W.; Wang, S.; Yang, C.; Huang, X.; Chen, Z.; He, W.; Shen, J.; Liu, X.; Qian, W. Combined expression of miR-34a and Smac mediated by oncolytic vaccinia virus synergistically promote anti-tumor effects in Multiple Myeloma. Sci. Rep. 2016, 6, 32174. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Fan, W.; Yang, C.; Lei, W.; Pan, H.; Tong, X.; Wu, Y.; Wang, S. Beclin1-armed oncolytic Vaccinia virus enhances the therapeutic efficacy of R-CHOP against lymphoma in vitro and in vivo. Oncol. Rep. 2021, 45, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Bartee, M.Y.; Dunlap, K.M.; Bartee, E. Myxoma Virus Induces Ligand Independent Extrinsic Apoptosis in Human Myeloma Cells. Clin. Lymphoma Myeloma Leuk. 2016, 16, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartee, M.Y.; Dunlap, K.M.; Bartee, E. Myxoma virus attenuates expression of activating transcription factor 4 (ATF4) which has implications for the treatment of proteasome inhibitor–resistant multiple myeloma. Oncolytic Virother. 2015, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lilly, C.L.; Villa, N.Y.; de Matos, A.L.; Ali, H.M.; Dhillon, J.-K.S.; Hofland, T.; Rahman, M.M.; Chan, W.; Bogen, B.; Cogle, C.; et al. Ex Vivo Oncolytic Virotherapy with Myxoma Virus Arms Multiple Allogeneic Bone Marrow Transplant Leukocytes to Enhance Graft versus Tumor. Mol. Ther.-Oncolytics 2017, 4, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Villa, N.Y.; Rahman, M.M.; Mamola, J.; D’Isabella, J.; Goras, E.; Kilbourne, J.; Lowe, K.; Daggett-Vondras, J.; Torres, L.; Christie, J.; et al. Autologous Transplantation Using Donor Leu-kocytes Loaded. Mol. Ther.-Oncolytics 2020, 18, 171–188. [Google Scholar] [CrossRef]
- Ghose, J.; Dona, A.; Murtadha, M.; Gunes, E.G.; Caserta, E.; Yoo, J.Y.; Russell, L.; Jaime-Ramirez, A.C.; Barwick, B.G.; Gupta, V.A.; et al. Oncolytic herpes simplex virus infects myeloma cells in vitro and in vivo. Mol. Ther.-Oncolytics 2021, 20, 519–531. [Google Scholar] [CrossRef]
- Oku, M.; Ishino, R.; Uchida, S.; Imataki, O.; Sugimoto, N.; Todo, T.; Kadowaki, N. Oncolytic herpes simplex virus type 1 (HSV-1) in combi-nation with lenalidomide for plasma cell neoplasms. Br. J. Haematol. 2021, 192, 343–353. [Google Scholar] [CrossRef]
- Thirukkumaran, C.M.; Shi, Z.Q.; Luider, J.; Kopciuk, K.; Gao, H.; Bahlis, N.; Neri, P.; Pho, M.; Stewart, D.; Mansoor, A.; et al. Reovirus modulates autophagy during oncolysis of multiple myeloma. Autophagy 2013, 9, 413–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirukkumaran, C.M.; Shi, Z.Q.; Luider, J.; Kopciuk, K.; Bahlis, N.; Neri, P.; Pho, M.; Stewart, D.; Mansoor, A.; Morris, D.G. Reovirus as a successful ex vivo purging modality for multiple myeloma. Bone Marrow Transplant. 2013, 49, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.R.; Espitia, C.M.; Mahalingam, D.; Oyajobi, B.O.; Coffey, M.; Giles, F.; Carew, J.S.; Nawrocki, S.T. Reovirus therapy stimulates endoplasmic reticular stress, NOXA induction, and augments bortezomib-mediated apoptosis in multiple myeloma. Oncogene 2012, 31, 3023–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiff, A.; Caserta, E.; Sborov, D.W.; Nuovo, G.J.; Mo, X.; Schlotter, S.Y.; Canella, A.; Smith, E.; Badway, J.; Old, M.; et al. Histone Deacetylase Inhibitors Enhance the Therapeutic Potential of Reovirus in Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.R.; Espitia, C.M.; Zhao, W.; Wu, K.; Visconte, V.; Anwer, F.; Calton, C.M.; Carew, J.S.; Nawrocki, S.T. Oncolytic reovirus sensitizes multiple myeloma cells to anti-PD-L1 therapy. Leukemia 2018, 32, 230–233. [Google Scholar] [CrossRef]
- Thirukkumaran, C.M.; Shi, Z.Q.; Nuovo, G.J.; Luider, J.; Kopciuk, K.A.; Dong, Y.; Mostafa, A.A.; Thakur, S.; Gratton, K.; Yang, A.; et al. Oncolytic immunotherapy and bortezomib synergy improves survival of refractory multiple myeloma in a preclinical model. Blood Adv. 2019, 3, 797–812. [Google Scholar] [CrossRef] [Green Version]
- Müller, L.M.E.; Migneco, G.; Scott, G.B.; Down, J.; King, S.; Askar, B.; Jennings, V.; Oyajobi, B.; Scott, K.; West, E.; et al. Reovirus-induced cell-mediated immunity for the treatment of multiple myeloma within the resistant bone marrow niche. J. Immunother. Cancer 2021, 9, e001803. [Google Scholar] [CrossRef]
- Kelly, K.R.; Espitia, C.M.; Zhao, W.; Wendlandt, E.; Tricot, G.; Zhan, F.; Carew, J.S.; Nawrocki, S.T. Junctional adhesion molecule-A is overexpressed in advanced multiple myeloma and determines response to oncolytic reovirus. Oncotarget 2015, 6, 41275–41289. [Google Scholar] [CrossRef] [PubMed]
- Au, G.G.; Lincz, L.F.; Enno, A.; Shafren, D.R. Oncolytic Coxsackievirus A21 as a novel therapy for multiple myeloma. Br. J. Haematol. 2007, 137, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W. Measles Virus for Cancer Therapy. Curr. Top. Microbiol. Immunol. 2009, 330, 213–241. [Google Scholar] [CrossRef] [Green Version]
- Peng, K.W.; Ahmann, G.J.; Pham, L.; Greipp, P.R.; Cattaneo, R.; Russell, S.J. Systemic therapy of myeloma xenografts by an attenu-ated measles virus. Blood 2001, 98, 2002–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dingli, D.; Peng, K.-W.; Harvey, M.E.; Greipp, P.R.; O’Connor, M.K.; Cattaneo, R.; Morris, J.C.; Russell, S.J. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood 2004, 103, 1641–1646. [Google Scholar] [CrossRef] [Green Version]
- Hummel, H.D.; Kuntz, G.; Russell, S.J.; Nakamura, T.; Greiner, A.; Einsele, H.; Topp, M.S. Genetically engineered attenuated measles vi-rus specifically infects and kills primary multiple myeloma cells. J. Gen. Virol. 2009, 90, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Tong, C.; LaPlant, B.; Lacy, M.Q.; Laumann, K.; Dingli, D.; Zhou, Y.; Federspiel, M.J.; Gertz, M.A.; Hayman, S.; et al. Phase I trial of systemic administration of Edmon-ston strain of measles virus genetically engineered to express the sodium iodide symporter in patients with recurrent or refractory multiple myeloma. Leukemia 2017, 31, 2791–2798. [Google Scholar] [CrossRef]
- Lindberg, A.; Houe, H. Characteristics in the epidemiology of bovine viral diarrhea virus (BVDV) of relevance to control. Prev. Veter.-Med. 2005, 72, 55–73. [Google Scholar] [CrossRef]
- Marchica, V.; Franceschi, V.; Vescovini, R.; Storti, P.; Vicario, E.; Toscani, D.; Zorzoli, A.; Airoldi, I.; Palma, B.D.; Campanini, N.; et al. Bovine pestivirus is a new alternative virus for multiple myeloma oncolytic virotherapy. J. Hematol. Oncol. 2020, 13, 89. [Google Scholar] [CrossRef]
- Naik, S.; Nace, R.; Federspiel, M.J.; Barber, G.N.; Peng, K.W.; Russell, S.J. Curative one-shot systemic virotherapy in murine myeloma. Leukemia 2012, 26, 1870–1878. [Google Scholar] [CrossRef]
- Goel, A.; Carlson, S.K.; Classic, K.L.; Greiner, S.; Naik, S.; Power, A.T.; Bell, J.C.; Russell, S.J. Radioiodide imaging and radiovirotherapy of multiple myeloma using VSV(Delta51)-NIS, an attenuated vesicular stomatitis virus encoding the sodium iodide symporter gene. Blood 2007, 110, 2342–2350. [Google Scholar] [CrossRef] [Green Version]
- Naik, S.; Nace, R.; Barber, G.N.; Russell, S.J. Potent systemic therapy of multiple myeloma utilizing oncolytic vesicular stomati-tis virus coding for interferon-β. Cancer Gene Ther. 2012, 19, 443–450. [Google Scholar] [CrossRef]
- Evgin, L.; Acuna, S.A.; Tanese de Souza, C.; Marguerie, M.; Lemay, C.G.; Ilkow, C.S.; Findlay, C.S.; Falls, T.; Parato, K.A.; Hanwell, D.; et al. Complement inhibition prevents onco-lytic vaccinia virus neutralization in immune humans and cynomolgus macaques. Mol. Ther. 2015, 23, 1066–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magge, D.; Guo, Z.S.; O’Malley, M.E.; Francis, L.; Ravindranathan, R.; Bartlett, D.L. Inhibitors of C5 complement enhance vaccinia virus oncolysis. Cancer Gene Ther. 2013, 20, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.-W.; Dogan, A.; Vrana, J.; Liu, C.; Ong, H.T.; Kumar, S.; Dispenzieri, A.; Dietz, A.; Russell, S.J. Tumor-associated macrophages infiltrate plasmacytomas and can serve as cell carriers for oncolytic measles virotherapy of disseminated myeloma. Am. J. Hematol. 2009, 84, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and Lightning: Immunotherapy and Oncolytic Viruses Collide. Mol. Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Meyers, D.E.; Thakur, S.; Thirukkumaran, C.M.; Morris, D.G. Oncolytic virotherapy as an immunotherapeutic strategy for multi-ple myeloma. Blood Cancer J. 2017, 7, 640. [Google Scholar] [CrossRef] [Green Version]
- Romano, A.; Conticello, C.; Cavalli, M.; Vetro, C.; La Fauci, A.; Parrinello, N.L.; Di Raimondo, F. Immunological Dysregulation in Multiple Myeloma Microenvironment. BioMed Res. Int. 2014, 2014, 198539. [Google Scholar] [CrossRef] [Green Version]
- Ohno, S.; Ono, N.; Seki, F.; Takeda, M.; Kura, S.; Tsuzuki, T.; Yanagi, Y. Measles virus infection of SLAM (CD150) knockin mice repro-duces tropism and immunosuppression in human infection. J. Virol. 2007, 81, 1650–1659. [Google Scholar] [CrossRef] [Green Version]
- Comins, C.; Spicer, J.; Protheroe, A.; Roulstone, V.; Twigger, K.; White, C.M.; Vile, R.; Melcher, A.; Coffey, M.C.; Mettinger, K.L.; et al. REO-10: A Phase I Study of Intravenous Reovirus and Docetaxel in Patients with Advanced Cancer. Clin. Cancer Res. 2010, 16, 5564–5572. [Google Scholar] [CrossRef] [Green Version]
- Khuri, F.R.; Nemunaitis, J.; Ganly, I.; Arseneau, J.; Tannock, I.F.; Romel, L.; Gore, M.; Ironside, J.; MacDougall, R.H.; Heise, C.; et al. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat. Med. 2000, 6, 879–885. [Google Scholar] [CrossRef]
- Freytag, S.O.; Stricker, H.; Pegg, J.; Paielli, D.; Pradhan, D.G.; Peabody, J.; DePeralta-Venturina, M.; Xia, X.; Brown, S.; Lu, M.; et al. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate-to-high-risk prostate cancer. Cancer Res. 2003, 63, 7497–7506. [Google Scholar] [PubMed]
- Xia, Z.-J.; Chang, J.-H.; Zhang, L.; Jiang, W.-Q.; Guan, Z.-Z.; Liu, J.-W.; Zhang, Y.; Hu, X.-H.; Wu, G.-H.; Wang, H.-Q.; et al. [Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus]. Chin. J. Cancer 2004, 23, 1666–1670. [Google Scholar]
- Galanis, E.; Okuno, S.H.; Nascimento, A.G.; Lewis, B.D.; Lee, R.A.; Oliveira, A.M.; Sloan, J.A.; Atherton, P.; Edmonson, J.H.; Erlichman, C.; et al. Phase I–II trial of ONYX-015 in combination with MAP chemotherapy in patients with advanced sarcomas. Gene Ther. 2005, 12, 437–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karapanagiotou, E.M.; Roulstone, V.; Twigger, K.; Ball, M.; Tanay, M.; Nutting, C.; Newbold, K.; Gore, M.E.; Larkin, J.; Syrigos, K.N.; et al. Phase I/II trial of carboplatin and paclitax-el chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin. Cancer Res. 2012, 18, 2080–2089. [Google Scholar] [CrossRef] [Green Version]
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimäki, A.; et al. Phase I study with ONCOS-102 for the treatment of solid tumors—An evaluation of clinical response and exploratory analyses of immune markers. J. Immunother. Cancer 2016, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Oncolytic Virus | Genome | Enveloped | Replication Site | Receptors for MM | Mechanisms of Specificity | Mechanisms of Killing | Genetic Manipulation | Combination Therapy |
|---|---|---|---|---|---|---|---|---|
| DNA Viruses | ||||||||
| Adenovirus | dsDNA | No | Nuc/Cyto | UnK/CAR | UnK | Lytic Viral Replication | Easy | N/A |
| Vaccinia | dsDNA | Yes | Cyto | UnK | Engineered | Lytic viral replication | Easy | N/A |
| Myxoma | dsDNA | Yes | Cyto | UnK | Myeloma specific binding (UK receptor) | Induction of apoptosis | Easy | N/A |
| HSV | dsDNA | Yes | Nuc/Cyto | HVEM | UnK | Induction of apoptosis | Easy | Len |
| RNA Viruses | ||||||||
| Reo | dsRNA | No | Cyto | JAM-A | OE of JAM-A | Lytic viral replication, apoptosis, autophagyUPR | Difficult | BTZ, Len, Pom, anti-PD-L1 |
| Coxsackie | ss(+)RNA | No | Cyto | ICAM-1, DAF | Unk | Unk | Easy | N/A |
| MV | ss(−)RNA | Yes | Cyto | CD46 | OE of CD46 | Lytic viral replication | Easy | CP |
| BVDV | ss(+)RNA | Yes | Cyto | CD46 | OE of CD46 | Induction of apoptosis | Unk | BTZ |
| VSV | ss(−)RNA | Yes | Cyto | LDLRs | Defects in interferon response | Lytic viral replication Inhibition of DNA synthesis | Easy | BTZ, CP |
| NDV | ss(−)RNA | Yes | Cyto | Sialic acids | UnK | Lytic viral replication, induction of apoptosis | Unk | N/A |
| Rotavirus | dsRNA | No | Cyto | PDI, HSP, integrin β3 | UnK | Lytic viral replication | UnK | N/A |
| Virotherapy | Clinical Trial Phase | Combination Agents | Clinicaltrials.Gov Identifier |
|---|---|---|---|
| Reovirus (Reolysin) | I | None | NCT01533194 (Completed) |
| I | Lenalidomide or Pomalidomide | NCT03015922 (Active) | |
| I | Bortezomib & Dexamethasone | NCT02514382 (Unknown) | |
| I | Carfilzomib & Dexamethasone | NCT02101944 (Recruiting) | |
| Measles virus (MV-NIS) | I/II | ± Cyclophosphamide | NCT00450814 (Completed) |
| II | Cyclophosphamide | NCT02192775 (Completed) | |
| I | ± Cyclophosphamide | NCT00450814 (Completed) | |
| VSV (VSV-IFNB-NIS) | I | ± Cyclophosphamide | NCT03017820 (Recruiting) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stewart, G.; Chantry, A.; Lawson, M. The Use of Oncolytic Viruses in the Treatment of Multiple Myeloma. Cancers 2021, 13, 5687. https://doi.org/10.3390/cancers13225687
Stewart G, Chantry A, Lawson M. The Use of Oncolytic Viruses in the Treatment of Multiple Myeloma. Cancers. 2021; 13(22):5687. https://doi.org/10.3390/cancers13225687
Chicago/Turabian StyleStewart, Georgia, Andrew Chantry, and Michelle Lawson. 2021. "The Use of Oncolytic Viruses in the Treatment of Multiple Myeloma" Cancers 13, no. 22: 5687. https://doi.org/10.3390/cancers13225687
APA StyleStewart, G., Chantry, A., & Lawson, M. (2021). The Use of Oncolytic Viruses in the Treatment of Multiple Myeloma. Cancers, 13(22), 5687. https://doi.org/10.3390/cancers13225687