
Microtubule-Based Mitochondrial Dynamics as a Valuable Therapeutic Target in Cancer


Abstract
:Simple Summary
Abstract
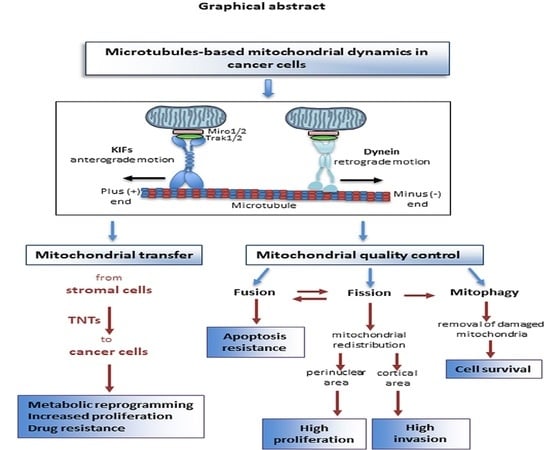
{kind=link}
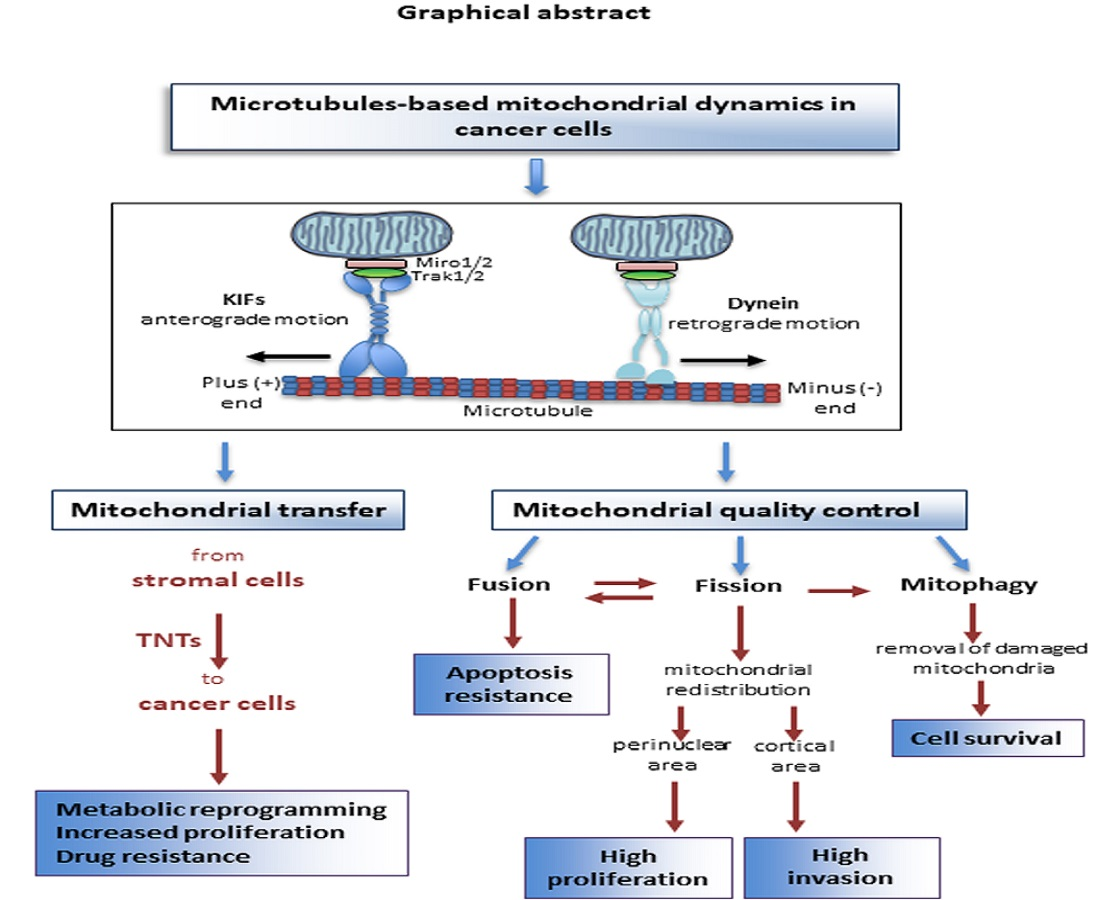
{kind=link}
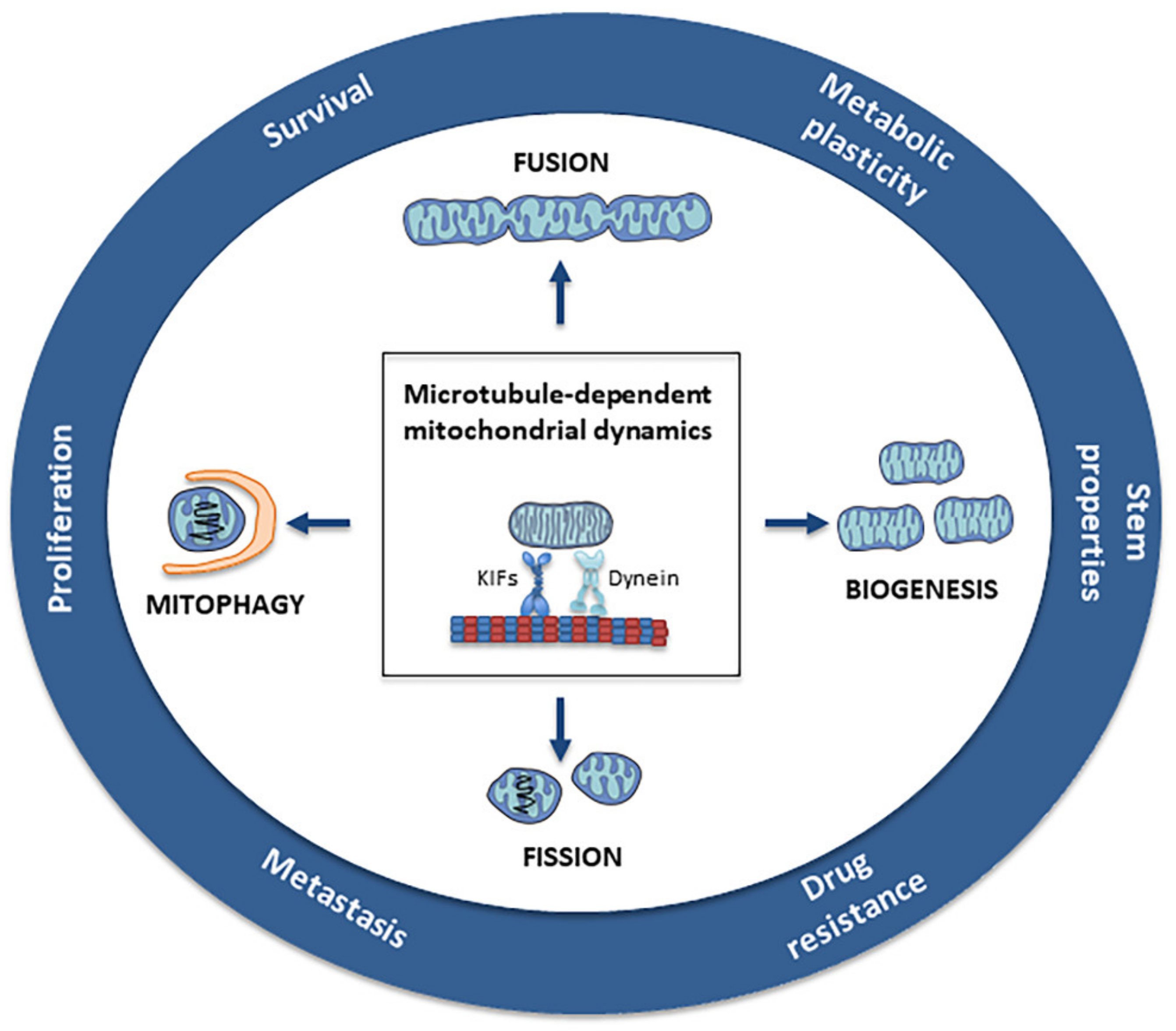
{kind=link}
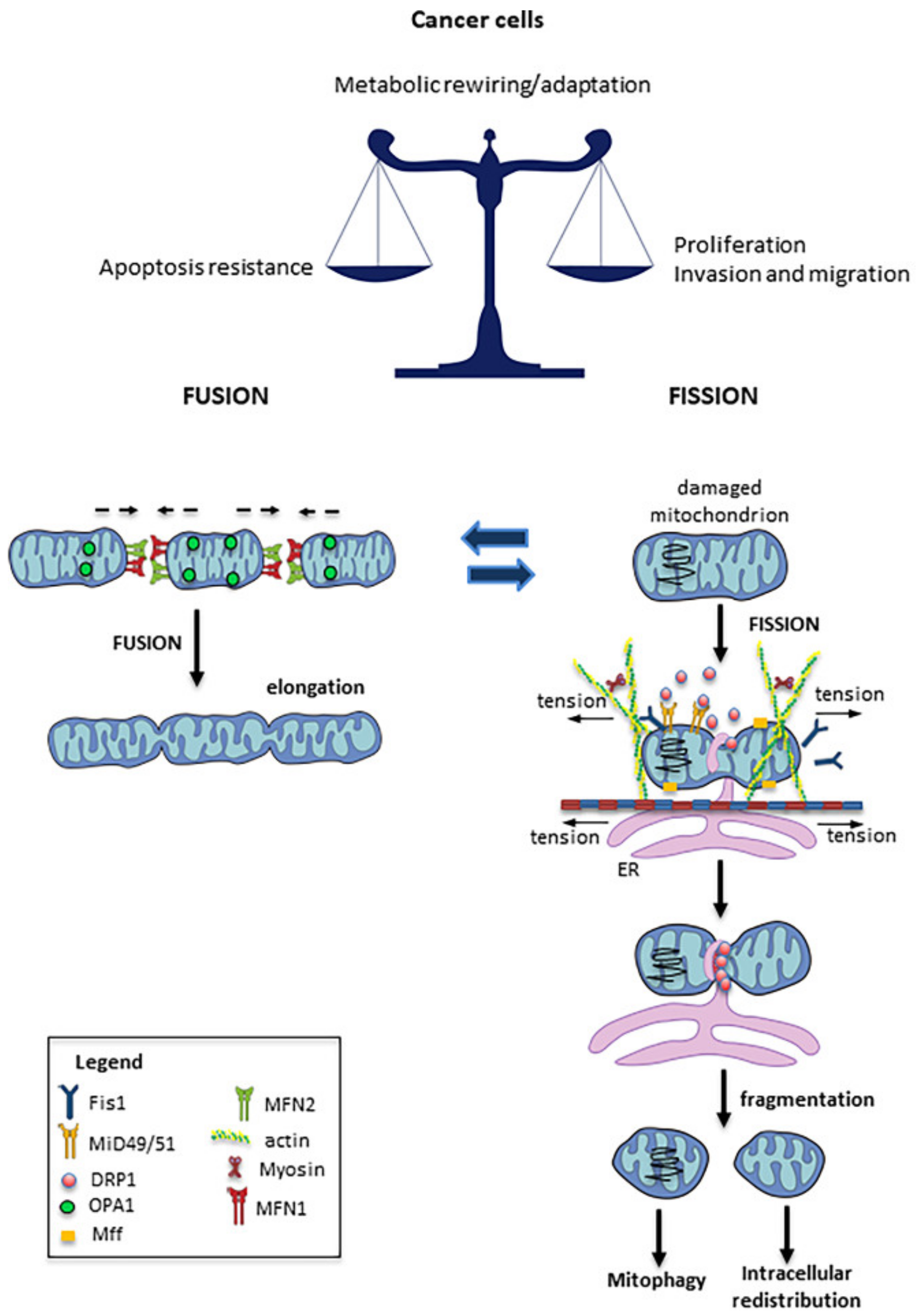
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Mitochondria
3. Mitochondrial Fission and Fusion
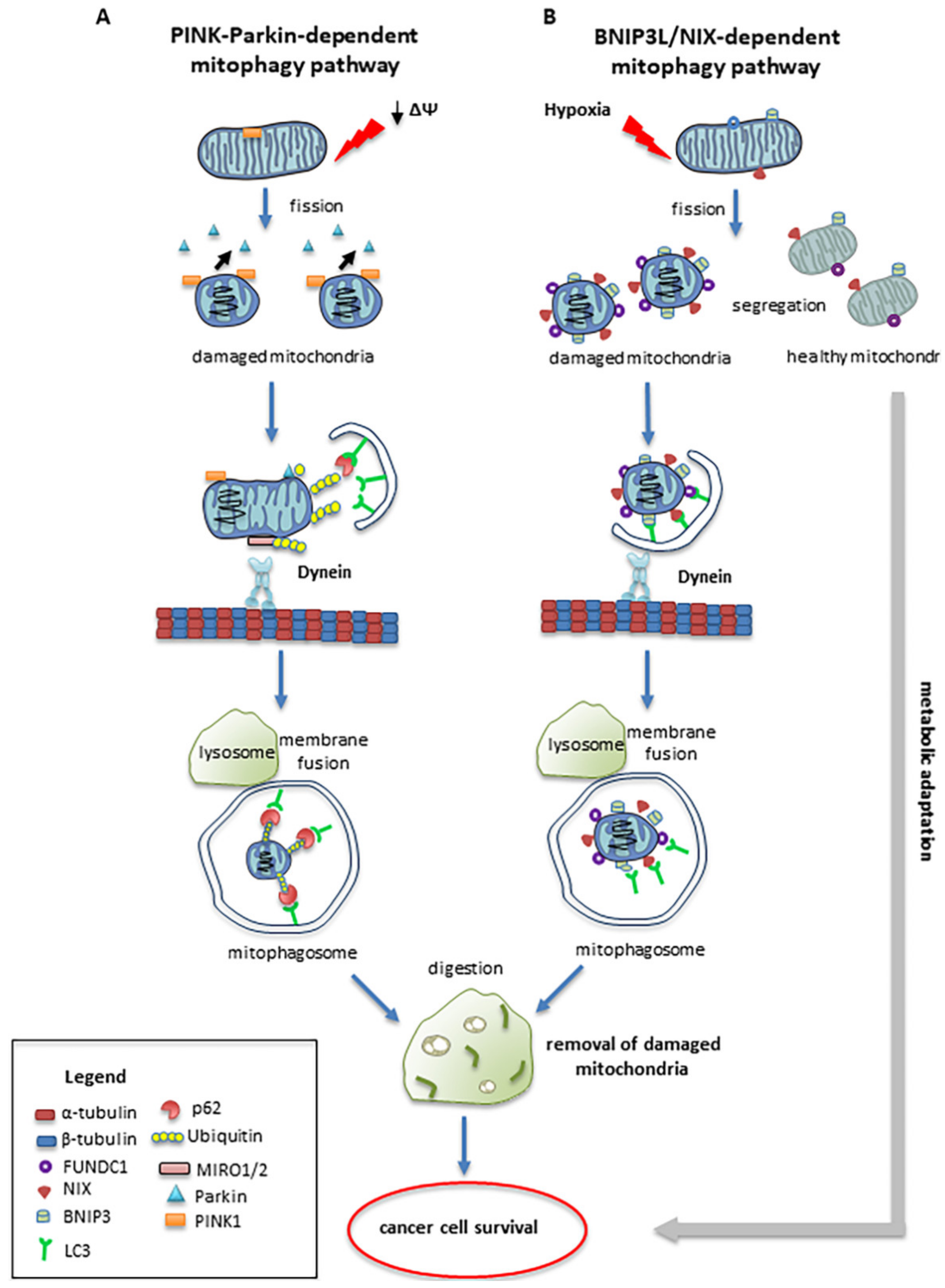
4. Mitophagy
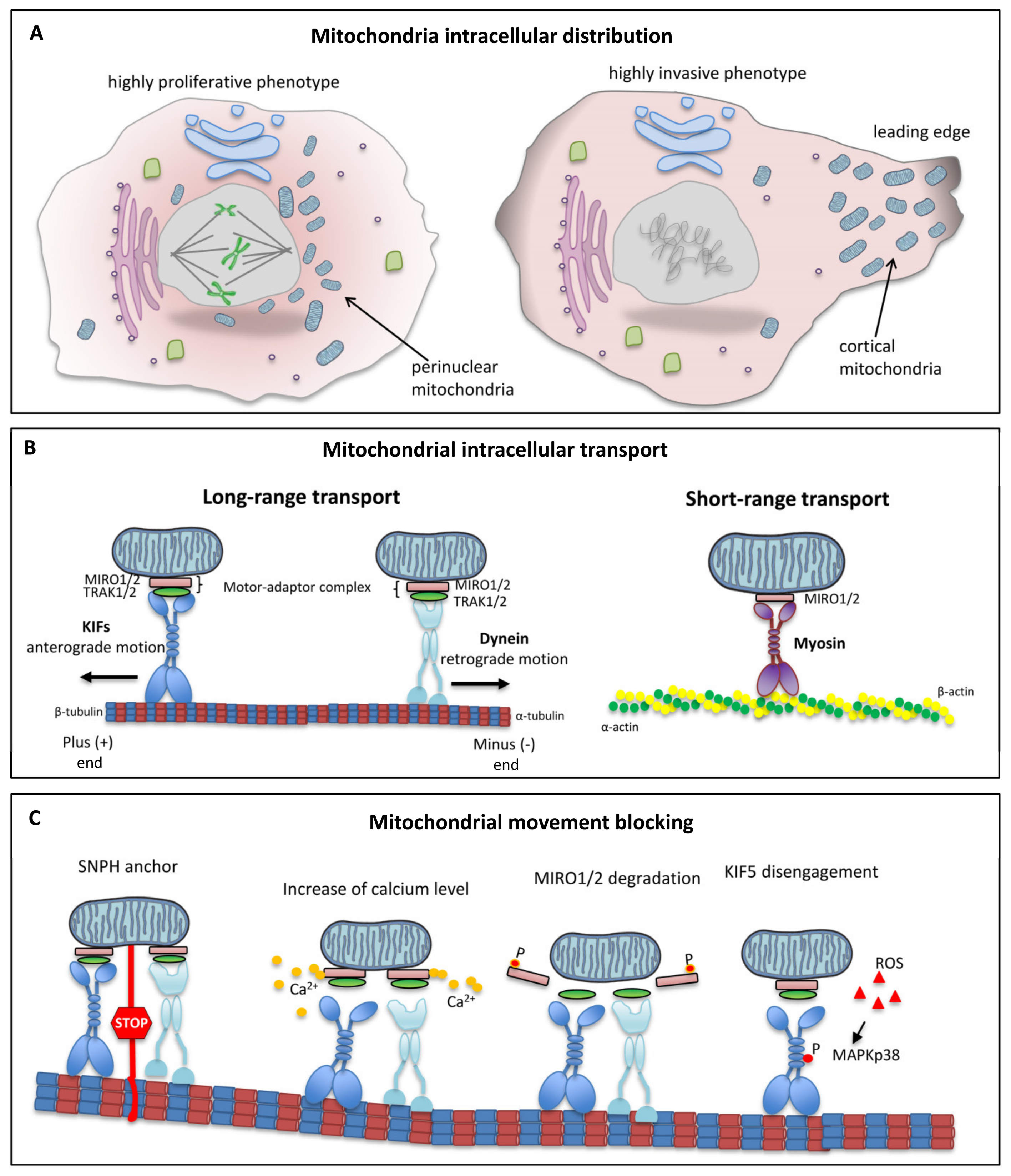
5. Intracellular Mitochondrial Trafficking
6. Role of Microtubules in Mitochondrial Dynamics
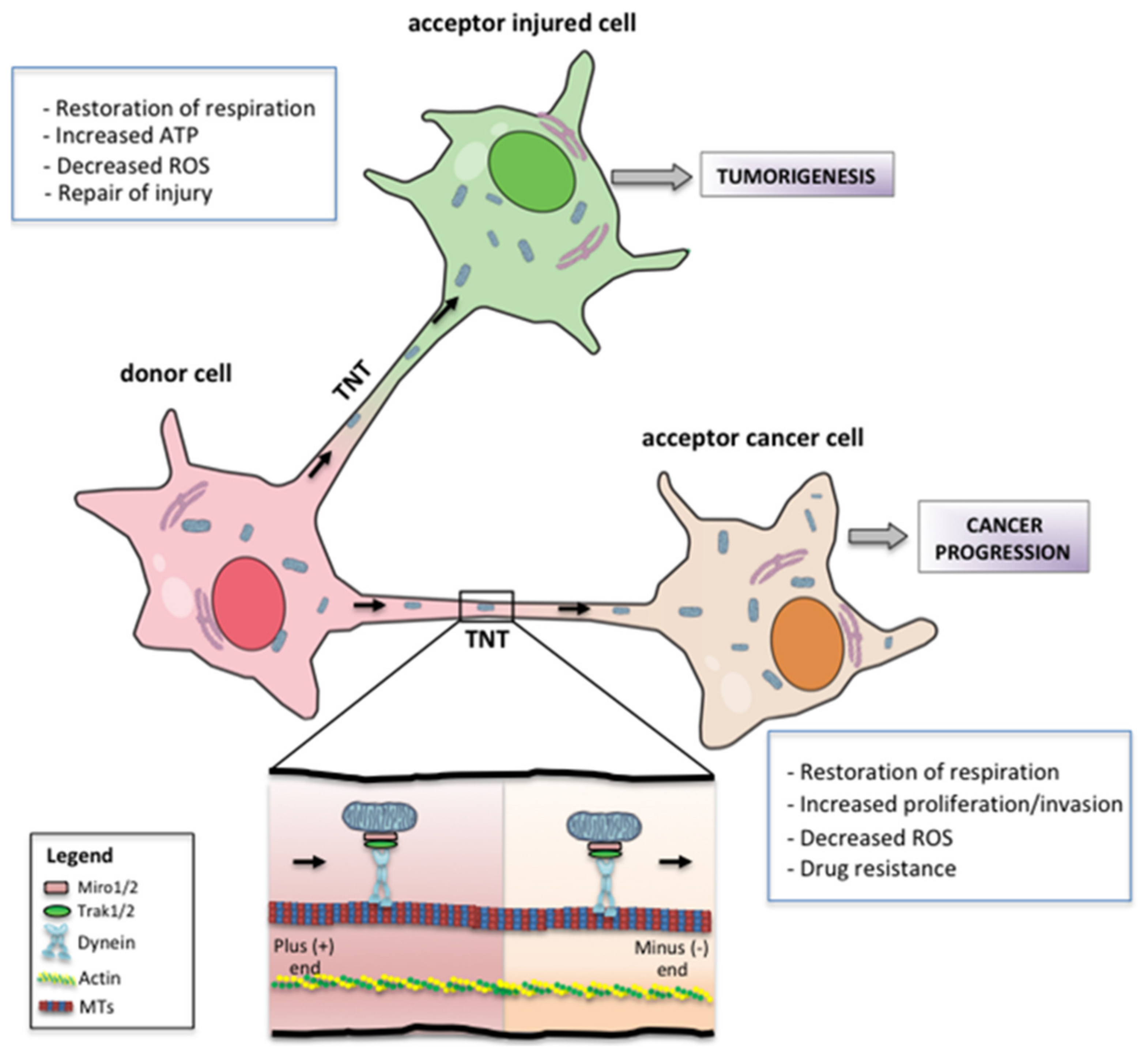
7. Metabolic and Phenotypic Consequences of Mitochondrial Transfer
8. Mitochondrial Dynamics and Cancer Therapy
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nogales, E. Structural insight into microtubule function. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 397–420. [Google Scholar] [CrossRef] [PubMed]
- Heald, R.; Nogales, E. Microtubule dynamics. J. Cell Sci. 2002, 115, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, C.; Lalli, E. Targeting the cytoskeleton against metastatic dissemination. Cancer Metastasis Rev. 2021, 40, 89–140. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, T.; Dehghani, F. The Cytoskeleton-A Complex Interacting Meshwork. Cells 2019, 8, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appaix, F.; Kuznetsov, A.V.; Usson, Y.; Kay, L.; Andrienko, T.; Olivares, J.; Kaambre, T.; Sikk, P.; Margreiter, R.; Saks, V. Possible role of cytoskeleton in intracellular arrangement and regulation of mitochondria. Exp. Physiol. 2003, 88, 175–190. [Google Scholar] [CrossRef] [Green Version]
- Donhauser, Z.J.; Jobs, W.B.; Binka, E.C. Mechanics of microtubules: Effects of protofilament orientation. Biophys. J. 2010, 99, 1668–1675. [Google Scholar] [CrossRef] [Green Version]
- Straight, A.F.; Field, C.M. Microtubules, membranes and cytokinesis. Curr. Biol. 2000, 10, R760–R770. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Weaver, D.; Shirihai, O.; Hajnoczky, G. Mitochondrial ‘kiss-and-run’: Interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 2009, 28, 3074–3089. [Google Scholar] [CrossRef] [Green Version]
- Bulinski, J.C. Microtubule modification: Acetylation speeds anterograde traffic flow. Curr. Biol. 2007, 17, R18–R20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelieres, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef]
- Ligon, L.A.; Steward, O. Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 2000, 427, 351–361. [Google Scholar] [CrossRef]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Schwarz, T.L. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotta, A.P.; Chipuk, J.E. Mitochondrial dynamics as regulators of cancer biology. Cell. Mol. Life Sci. 2017, 74, 1999–2017. [Google Scholar] [CrossRef] [PubMed]
- Saxton, W.M.; Hollenbeck, P.J. The axonal transport of mitochondria. J. Cell Sci. 2012, 125, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Puurand, M.; Tepp, K.; Timohhina, N.; Aid, J.; Shevchuk, I.; Chekulayev, V.; Kaambre, T. Tubulin betaII and betaIII Isoforms as the Regulators of VDAC Channel Permeability in Health and Disease. Cells 2019, 8, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilling, A.D.; Horiuchi, D.; Lively, C.M.; Saxton, W.M. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell 2006, 17, 2057–2068. [Google Scholar] [CrossRef] [Green Version]
- Frederick, R.L.; Shaw, J.M. Moving mitochondria: Establishing distribution of an essential organelle. Traffic 2007, 8, 1668–1675. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Fife, C.M.; McCarroll, J.A.; Kavallaris, M. Movers and shakers: Cell cytoskeleton in cancer metastasis. Br. J. Pharmacol. 2014, 171, 5507–5523. [Google Scholar] [CrossRef] [Green Version]
- Liesa, M.; Palacin, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, S.G.; Zomorodipour, A.; Andersson, J.O.; Sicheritz-Ponten, T.; Alsmark, U.C.; Podowski, R.M.; Naslund, A.K.; Eriksson, A.S.; Winkler, H.H.; Kurland, C.G. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 1998, 396, 133–140. [Google Scholar] [CrossRef]
- Kuhlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altieri, D.C. Mitochondria on the move: Emerging paradigms of organelle trafficking in tumour plasticity and metastasis. Br. J. Cancer 2017, 117, 301–305. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yao, J.; Wang, B.; Qin, L. Microfluidics-Based Single-Cell Protrusion Analysis for Screening Drugs Targeting Subcellular Mitochondrial Trafficking in Cancer Progression. Anal. Chem. 2020, 92, 3095–3102. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senft, D.; Ronai, Z.A. Regulators of mitochondrial dynamics in cancer. Curr. Opin. Cell Biol. 2016, 39, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Gandre-Babbe, S.; van der Bliek, A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [Green Version]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 2013, 288, 27584–27593. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-da-Silva, A.; Valacca, C.; Rios, E.; Populo, H.; Soares, P.; Sobrinho-Simoes, M.; Scorrano, L.; Maximo, V.; Campello, S. Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS ONE 2015, 10, e0122308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.Y.; Zhang, J.F.; Yang, Z.J.; Jiang, L.P.; Wei, Y.F.; Lai, Q.N.; Wang, J.B.; Xin, H.B.; Han, X.J. Involvement of Drp1 in hypoxia-induced migration of human glioblastoma U251 cells. Oncol. Rep. 2014, 32, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Tomkova, V.; Sandoval-Acuna, C.; Torrealba, N.; Truksa, J. Mitochondrial fragmentation, elevated mitochondrial superoxide and respiratory supercomplexes disassembly is connected with the tamoxifen-resistant phenotype of breast cancer cells. Free Radic. Biol. Med. 2019, 143, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, T.F.; Lin, C.W.; Wu, Y.Y.; Chen, Y.J.; Han, C.L.; Chang, Y.L.; Wu, C.T.; Hsiao, T.H.; Hong, T.M.; Yang, P.C. Mitochondrial translocation of EGFR regulates mitochondria dynamics and promotes metastasis in NSCLC. Oncotarget 2015, 6, 37349–37366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012, 26, 2175–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, B.; Fu, G.; Pan, H.; Cheng, X.; Zhou, L.; Lv, J.; Chen, G.; Zheng, S. Anti-tumour efficacy of mitofusin-2 in urinary bladder carcinoma. Med. Oncol. 2011, 28 (Suppl. S1), S373–S380. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cao, H.; Zhan, L.; Yin, C.; Wang, G.; Liang, P.; Li, J.; Wang, Z.; Liu, B.; Huang, Q.; et al. Mitochondrial fission promotes cell migration by Ca(2+)/CaMKII/ERK/FAK pathway in hepatocellular carcinoma. Liver Int. 2018, 38, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Bowes, T.; Gupta, R.S. Novel mitochondrial extensions provide evidence for a link between microtubule-directed movement and mitochondrial fission. Biochem. Biophys. Res. Commun. 2008, 376, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Weaver, D.; Hajnoczky, G. Control of mitochondrial motility and distribution by the calcium signal: A homeostatic circuit. J. Cell Biol. 2004, 167, 661–672. [Google Scholar] [CrossRef]
- Mahecic, D.; Carlini, L.; Kleele, T.; Colom, A.; Goujon, A.; Matile, S.; Roux, A.; Manley, S. Mitochondrial membrane tension governs fission. Cell Rep. 2021, 35, 108947. [Google Scholar] [CrossRef]
- Yang, C.; Svitkina, T.M. Ultrastructure and dynamics of the actin-myosin II cytoskeleton during mitochondrial fission. Nat. Cell Biol. 2019, 21, 603–613. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Ji, W.K.; Stan, R.V.; de Juan Sanz, J.; Ryan, T.A.; Higgs, H.N. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol. 2018, 217, 251–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.; Chacko, L.A.; Joseph, J.P.; Ananthanarayanan, V. Mitochondrial dynamics, positioning and function mediated by cytoskeletal interactions. Cell. Mol. Life Sci. 2021, 78, 3969–3986. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Chacko, L.A.; Chug, M.K.; Jhunjhunwala, S.; Ananthanarayanan, V. Association of mitochondria with microtubules inhibits mitochondrial fission by precluding assembly of the fission protein Dnm1. J. Biol. Chem. 2019, 294, 3385–3396. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zheng, F.; Cheung, M.; Wang, F.; Fu, C. Fission yeast mitochondria are distributed by dynamic microtubules in a motor-independent manner. Sci. Rep. 2015, 5, 11023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Stockum, S.; Nardin, A.; Schrepfer, E.; Ziviani, E. Mitochondrial dynamics and mitophagy in Parkinson’s disease: A fly point of view. Neurobiol. Dis. 2016, 90, 58–67. [Google Scholar] [CrossRef]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway: A mitochondrial quality control system? J. Bioenerg. Biomembr. 2009, 41, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chourasia, A.H.; Boland, M.L.; Macleod, K.F. Mitophagy and cancer. Cancer Metab. 2015, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, H.; Van Eygen, S.; Krysko, D.V.; Vandenabeele, P.; Nys, K.; Rillaerts, K.; Garg, A.D.; Verfaillie, T.; Agostinis, P. BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis. 2014, 5, e1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panuzzo, C.; Jovanovski, A.; Pergolizzi, B.; Pironi, L.L.; Stanga, S.; Fava, C.; Cilloni, D. Mitochondria: A Galaxy in the Hematopoietic and Leukemic Stem Cell Universe. Int. J. Mol. Sci. 2020, 21, 3928. [Google Scholar] [CrossRef] [PubMed]
- Hinge, A.; He, J.; Bartram, J.; Javier, J.; Xu, J.; Fjellman, E.; Sesaki, H.; Li, T.; Yu, J.; Wunderlich, M.; et al. Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 2020, 26, 420–430.e6. [Google Scholar] [CrossRef] [PubMed]
- Katajisto, P.; Dohla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.K.; Mukherjee, S. Mitophagy in Carcinogenesis and Tumor progression—A New paradigm with Emerging Importance. Anticancer Agents Med. Chem. 2021, 21, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Marusawa, H.; Wang, H.Q.; Iwai, A.; Ikeuchi, K.; Imai, Y.; Kataoka, A.; Nukina, N.; Takahashi, R.; Chiba, T. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 2008, 27, 6002–6011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Srinivasainagendra, V.; Sandel, M.W.; Singh, B.; Sundaresan, A.; Mooga, V.P.; Bajpai, P.; Tiwari, H.K.; Singh, K.K. Migration of mitochondrial DNA in the nuclear genome of colorectal adenocarcinoma. Genome Med. 2017, 9, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [Green Version]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, P.K.; Bollrath, J.; Pallangyo, C.K.; Matsutani, T.; Canli, O.; De Oliveira, T.; Diamanti, M.A.; Muller, N.; Gamrekelashvili, J.; Putoczki, T.; et al. Mitophagy in Intestinal Epithelial Cells Triggers Adaptive Immunity during Tumorigenesis. Cell 2018, 174, 88–101.e16. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caino, M.C.; Altieri, D.C. Molecular Pathways: Mitochondrial Reprogramming in Tumor Progression and Therapy. Clin. Cancer Res. 2016, 22, 540–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunniff, B.; McKenzie, A.J.; Heintz, N.H.; Howe, A.K. AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol. Biol. Cell 2016, 27, 2662–2674. [Google Scholar] [CrossRef]
- Schuler, M.H.; Lewandowska, A.; Caprio, G.D.; Skillern, W.; Upadhyayula, S.; Kirchhausen, T.; Shaw, J.M.; Cunniff, B. Miro1-mediated mitochondrial positioning shapes intracellular energy gradients required for cell migration. Mol. Biol. Cell 2017, 28, 2159–2169. [Google Scholar] [CrossRef] [PubMed]
- Van Spronsen, M.; Mikhaylova, M.; Lipka, J.; Schlager, M.A.; van den Heuvel, D.J.; Kuijpers, M.; Wulf, P.S.; Keijzer, N.; Demmers, J.; Kapitein, L.C.; et al. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 2013, 77, 485–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.H.; Agarwal, E.; Bryant, K.G.; Caino, M.C.; Kim, E.T.; Kossenkov, A.V.; Tang, H.Y.; Languino, L.R.; Gabrilovich, D.I.; Cohen, A.R.; et al. Syntaphilin Ubiquitination Regulates Mitochondrial Dynamics and Tumor Cell Movements. Cancer Res. 2018, 78, 4215–4228. [Google Scholar] [CrossRef] [Green Version]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M.; Iyadurai, S.J.; Gassman, A.; Gindhart, J.G., Jr.; Hays, T.S.; Saxton, W.M. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol. Biol. Cell 1999, 10, 3717–3728. [Google Scholar] [CrossRef] [Green Version]
- Campello, S.; Lacalle, R.A.; Bettella, M.; Manes, S.; Scorrano, L.; Viola, A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J. Exp. Med. 2006, 203, 2879–2886. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.M.; Brocardo, M.G.; Henderson, B.R. APC binds the Miro/Milton motor complex to stimulate transport of mitochondria to the plasma membrane. Mol. Biol. Cell 2016, 27, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.Y.; Sheng, Z.H. Regulation of mitochondrial transport in neurons. Exp. Cell Res. 2015, 334, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Macleod, G.T.; Wellington, A.; Hu, F.; Panchumarthi, S.; Schoenfield, M.; Marin, L.; Charlton, M.P.; Atwood, H.L.; Zinsmaier, K.E. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 2005, 47, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrichs, V.; Grycova, L.; Barinka, C.; Nahacka, Z.; Neuzil, J.; Diez, S.; Rohlena, J.; Braun, M.; Lansky, Z. Mitochondria-adaptor TRAK1 promotes kinesin-1 driven transport in crowded environments. Nat. Commun. 2020, 11, 3123. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Domenech, G.; Covill-Cooke, C.; Ivankovic, D.; Halff, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 2018, 37, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Caino, M.C.; Seo, J.H.; Aguinaldo, A.; Wait, E.; Bryant, K.G.; Kossenkov, A.V.; Hayden, J.E.; Vaira, V.; Morotti, A.; Ferrero, S.; et al. A neuronal network of mitochondrial dynamics regulates metastasis. Nat. Commun. 2016, 7, 13730. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Sepp, K.J.; Hollenbeck, P.J. Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. J. Neurosci. 2010, 30, 8984–8992. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Liu, Y.; Sun, D.; Zhou, C.; Liu, A.; Xu, C.; Hao, Y.; Li, D.; Yan, C.; Sun, H. Chronic activation of the G protein-coupled receptor 30 with agonist G-1 attenuates heart failure. PLoS ONE 2012, 7, e48185. [Google Scholar] [CrossRef]
- Schwarz, T.L. Mitochondrial trafficking in neurons. Cold Spring Harb. Perspect. Biol. 2013, 5, a011304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef]
- Rintoul, G.L.; Filiano, A.J.; Brocard, J.B.; Kress, G.J.; Reynolds, I.J. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J. Neurosci. 2003, 23, 7881–7888. [Google Scholar] [CrossRef] [Green Version]
- Macaskill, A.F.; Rinholm, J.E.; Twelvetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; Aspenstrom, P.; Attwell, D.; Kittler, J.T. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 2009, 61, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furnish, M.; Caino, M.C. Altered mitochondrial trafficking as a novel mechanism of cancer metastasis. Cancer Rep. 2020, 3, e1157. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.P.; Bhatia, S.N.; Toner, M.; Irimia, D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys. J. 2013, 104, 2077–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Sheng, Z.H. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J. Cell Biol. 2013, 202, 351–364. [Google Scholar] [CrossRef]
- Caino, M.C.; Seo, J.H.; Wang, Y.; Rivadeneira, D.B.; Gabrilovich, D.I.; Kim, E.T.; Weeraratna, A.T.; Languino, L.R.; Altieri, D.C. Syntaphilin controls a mitochondrial rheostat for proliferation-motility decisions in cancer. J. Clin. Investig. 2017, 127, 3755–3769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.; Bourdette, D.; Banker, G. Oxidative stress inhibits axonal transport: Implications for neurodegenerative diseases. Mol. Neurodegener. 2012, 7, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debattisti, V.; Gerencser, A.A.; Saotome, M.; Das, S.; Hajnoczky, G. ROS Control Mitochondrial Motility through p38 and the Motor Adaptor Miro/Trak. Cell Rep. 2017, 21, 1667–1680. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lim, S.; Hoffman, D.; Aspenstrom, P.; Federoff, H.J.; Rempe, D.A. HUMMR, a hypoxia- and HIF-1alpha-inducible protein, alters mitochondrial distribution and transport. J. Cell Biol. 2009, 185, 1065–1081. [Google Scholar] [CrossRef] [Green Version]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Gerdes, H.H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domhan, S.; Ma, L.; Tai, A.; Anaya, Z.; Beheshti, A.; Zeier, M.; Hlatky, L.; Abdollahi, A. Intercellular communication by exchange of cytoplasmic material via tunneling nano-tube like structures in primary human renal epithelial cells. PLoS ONE 2011, 6, e21283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panasiuk, M.; Rychlowski, M.; Derewonko, N.; Bienkowska-Szewczyk, K. Tunneling Nanotubes as a Novel Route of Cell-to-Cell Spread of Herpesviruses. J. Virol 2018, 92, e00090-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onfelt, B.; Nedvetzki, S.; Benninger, R.K.; Purbhoo, M.A.; Sowinski, S.; Hume, A.N.; Seabra, M.C.; Neil, M.A.; French, P.M.; Davis, D.M. Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J. Immunol. 2006, 177, 8476–8483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, Y.M.; Inaba, M.; Buszczak, M. Specialized Intercellular Communications via Cytonemes and Nanotubes. Annu. Rev. Cell Dev. Biol. 2018, 34, 59–84. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, H.H.; Bukoreshtliev, N.V.; Barroso, J.F. Tunneling nanotubes: A new route for the exchange of components between animal cells. FEBS Lett. 2007, 581, 2194–2201. [Google Scholar] [CrossRef] [Green Version]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Bajzikova, M.; Kovarova, J.; Coelho, A.R.; Boukalova, S.; Oh, S.; Rohlenova, K.; Svec, D.; Hubackova, S.; Endaya, B.; Judasova, K.; et al. Reactivation of Dihydroorotate Dehydrogenase-Driven Pyrimidine Biosynthesis Restores Tumor Growth of Respiration-Deficient Cancer Cells. Cell Metab. 2019, 29, 399–416.e10. [Google Scholar] [CrossRef] [Green Version]
- Desir, S.; Dickson, E.L.; Vogel, R.I.; Thayanithy, V.; Wong, P.; Teoh, D.; Geller, M.A.; Steer, C.J.; Subramanian, S.; Lou, E. Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells. Oncotarget 2016, 7, 43150–43161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matejka, N.; Reindl, J. Perspectives of cellular communication through tunneling nanotubes in cancer cells and the connection to radiation effects. Radiat. Oncol. 2019, 14, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdebenito, S.; Audia, A.; Bhat, K.P.L.; Okafo, G.; Eugenin, E.A. Tunneling Nanotubes Mediate Adaptation of Glioblastoma Cells to Temozolomide and Ionizing Radiation Treatment. iScience 2020, 23, 101450. [Google Scholar] [CrossRef]
- Korenkova, O.; Pepe, A.; Zurzolo, C. Fine intercellular connections in development: TNTs, cytonemes, or intercellular bridges? Cell Stress 2020, 4, 30–43. [Google Scholar] [CrossRef]
- Walters, H.E.; Cox, L.S. Intercellular Transfer of Mitochondria between Senescent Cells through Cytoskeleton-Supported Intercellular Bridges Requires mTOR and CDC42 Signalling. Oxid. Med. Cell. Longev. 2021, 2021, 6697861. [Google Scholar] [CrossRef]
- Kretschmer, A.; Zhang, F.; Somasekharan, S.P.; Tse, C.; Leachman, L.; Gleave, A.; Li, B.; Asmaro, I.; Huang, T.; Kotula, L.; et al. Stress-induced tunneling nanotubes support treatment adaptation in prostate cancer. Sci. Rep. 2019, 9, 7826. [Google Scholar] [CrossRef] [PubMed]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling Nanotubes and Tumor Microtubes in Cancer. Cancers 2020, 12, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, A.M.; Nakhle, J.; Griessinger, E.; Vignais, M.L. Intercellular mitochondria trafficking highlighting the dual role of mesenchymal stem cells as both sensors and rescuers of tissue injury. Cell Cycle 2018, 17, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Mohammadalipour, A.; Dumbali, S.P.; Wenzel, P.L. Mitochondrial Transfer and Regulators of Mesenchymal Stromal Cell Function and Therapeutic Efficacy. Front. Cell Dev. Biol. 2020, 8, 603292. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife 2017, 6, e22187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Zheng, X.; Li, F.; Yu, Y.; Chen, Z.; Liu, Z.; Wang, Z.; Xu, H.; Yang, W. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget 2017, 8, 15539–15552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caicedo, A.; Fritz, V.; Brondello, J.M.; Ayala, M.; Dennemont, I.; Abdellaoui, N.; de Fraipont, F.; Moisan, A.; Prouteau, C.A.; Boukhaddaoui, H.; et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 2015, 5, 9073. [Google Scholar] [CrossRef] [Green Version]
- Inoue-Yamauchi, A.; Oda, H. Depletion of mitochondrial fission factor DRP1 causes increased apoptosis in human colon cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 81–85. [Google Scholar] [CrossRef]
- Xie, Q.; Wu, Q.; Horbinski, C.M.; Flavahan, W.A.; Yang, K.; Zhou, W.; Dombrowski, S.M.; Huang, Z.; Fang, X.; Shi, Y.; et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat. Neurosci. 2015, 18, 501–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Wieder, S.Y.; Serasinghe, M.N.; Sung, J.C.; Choi, D.C.; Birge, M.B.; Yao, J.L.; Bernstein, E.; Celebi, J.T.; Chipuk, J.E. Activation of the Mitochondrial Fragmentation Protein DRP1 Correlates with BRAF(V600E) Melanoma. J. Investig. Dermatol. 2015, 135, 2544–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, L.; Cao, H.; Wang, G.; Lyu, Y.; Sun, X.; An, J.; Wu, Z.; Huang, Q.; Liu, B.; Xing, J. Drp1-mediated mitochondrial fission promotes cell proliferation through crosstalk of p53 and NF-kappaB pathways in hepatocellular carcinoma. Oncotarget 2016, 7, 65001–65011. [Google Scholar] [CrossRef] [Green Version]
- Lennon, F.E.; Cianci, G.C.; Kanteti, R.; Riehm, J.J.; Arif, Q.; Poroyko, V.A.; Lupovitch, E.; Vigneswaran, W.; Husain, A.; Chen, P.; et al. Unique fractal evaluation and therapeutic implications of mitochondrial morphology in malignant mesothelioma. Sci. Rep. 2016, 6, 24578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Wang, J.; Bonamy, G.M.; Meeusen, S.; Brusch, R.G.; Turk, C.; Yang, P.; Schultz, P.G. A small molecule promotes mitochondrial fusion in mammalian cells. Angew. Chem. Int. Ed. 2012, 51, 9302–9305. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Qvit, N.; Su, Y.C.; Mochly-Rosen, D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosdah, A.A.; Holien, J.K.; Delbridge, L.M.; Dusting, G.J.; Lim, S.Y. Mitochondrial fission—A drug target for cytoprotection or cytodestruction? Pharmacol. Res. Perspect. 2016, 4, e00235. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hansen, K.; Edwards, R.; Van Houten, B.; Qian, W. Mitochondrial division inhibitor 1 (mdivi-1) enhances death receptor-mediated apoptosis in human ovarian cancer cells. Biochem. Biophys. Res. Commun. 2015, 456, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar] [CrossRef]
- Soria, J.C.; Smit, E.; Khayat, D.; Besse, B.; Yang, X.; Hsu, C.P.; Reese, D.; Wiezorek, J.; Blackhall, F. Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in combination with paclitaxel, carboplatin, and bevacizumab in patients with advanced non-squamous non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 1527–1533. [Google Scholar] [CrossRef]
- Qian, W.; Wang, J.; Roginskaya, V.; McDermott, L.A.; Edwards, R.P.; Stolz, D.B.; Llambi, F.; Green, D.R.; Van Houten, B. Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget 2014, 5, 4180–4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, J.C.B.; Campos, J.C.; Qvit, N.; Qi, X.; Bozi, L.H.M.; Bechara, L.R.G.; Lima, V.M.; Queliconi, B.B.; Disatnik, M.H.; Dourado, P.M.M.; et al. A selective inhibitor of mitofusin 1-betaIIPKC association improves heart failure outcome in rats. Nat. Commun. 2019, 10, 329. [Google Scholar] [CrossRef]
- Sheng, Z.H. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 2014, 204, 1087–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, J.; Nabi, I.R. Actin cytoskeleton regulation of epithelial mesenchymal transition in metastatic cancer cells. PLoS ONE 2015, 10, e0119954. [Google Scholar]
- Taulet, N.; Delorme-Walker, V.D.; DerMardirossian, C. Reactive oxygen species regulate protrusion efficiency by controlling actin dynamics. PLoS ONE 2012, 7, e41342. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Caino, M.C.; Chae, Y.C.; Vaira, V.; Ferrero, S.; Nosotti, M.; Martin, N.M.; Weeraratna, A.; O’Connell, M.; Jernigan, D.; Fatatis, A.; et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Investig. 2013, 123, 2907–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porporato, P.E.; Payen, V.L.; Perez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vona, R.; Mileo, A.M.; Matarrese, P. Microtubule-Based Mitochondrial Dynamics as a Valuable Therapeutic Target in Cancer. Cancers 2021, 13, 5812. https://doi.org/10.3390/cancers13225812
Vona R, Mileo AM, Matarrese P. Microtubule-Based Mitochondrial Dynamics as a Valuable Therapeutic Target in Cancer. Cancers. 2021; 13(22):5812. https://doi.org/10.3390/cancers13225812
Chicago/Turabian StyleVona, Rosa, Anna Maria Mileo, and Paola Matarrese. 2021. "Microtubule-Based Mitochondrial Dynamics as a Valuable Therapeutic Target in Cancer" Cancers 13, no. 22: 5812. https://doi.org/10.3390/cancers13225812
APA StyleVona, R., Mileo, A. M., & Matarrese, P. (2021). Microtubule-Based Mitochondrial Dynamics as a Valuable Therapeutic Target in Cancer. Cancers, 13(22), 5812. https://doi.org/10.3390/cancers13225812