
Review: Challenges of In Vitro CAF Modelling in Liver Cancers




Abstract
:Simple Summary
Abstract
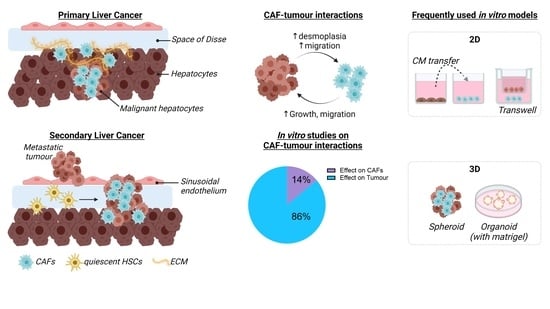
1. Introduction
2. Epidemiology of Liver Cancer
2.1. Primary Liver Cancer
2.2. Secondary Liver Cancer
3. Role of Microenvironment in Liver Tumour Development
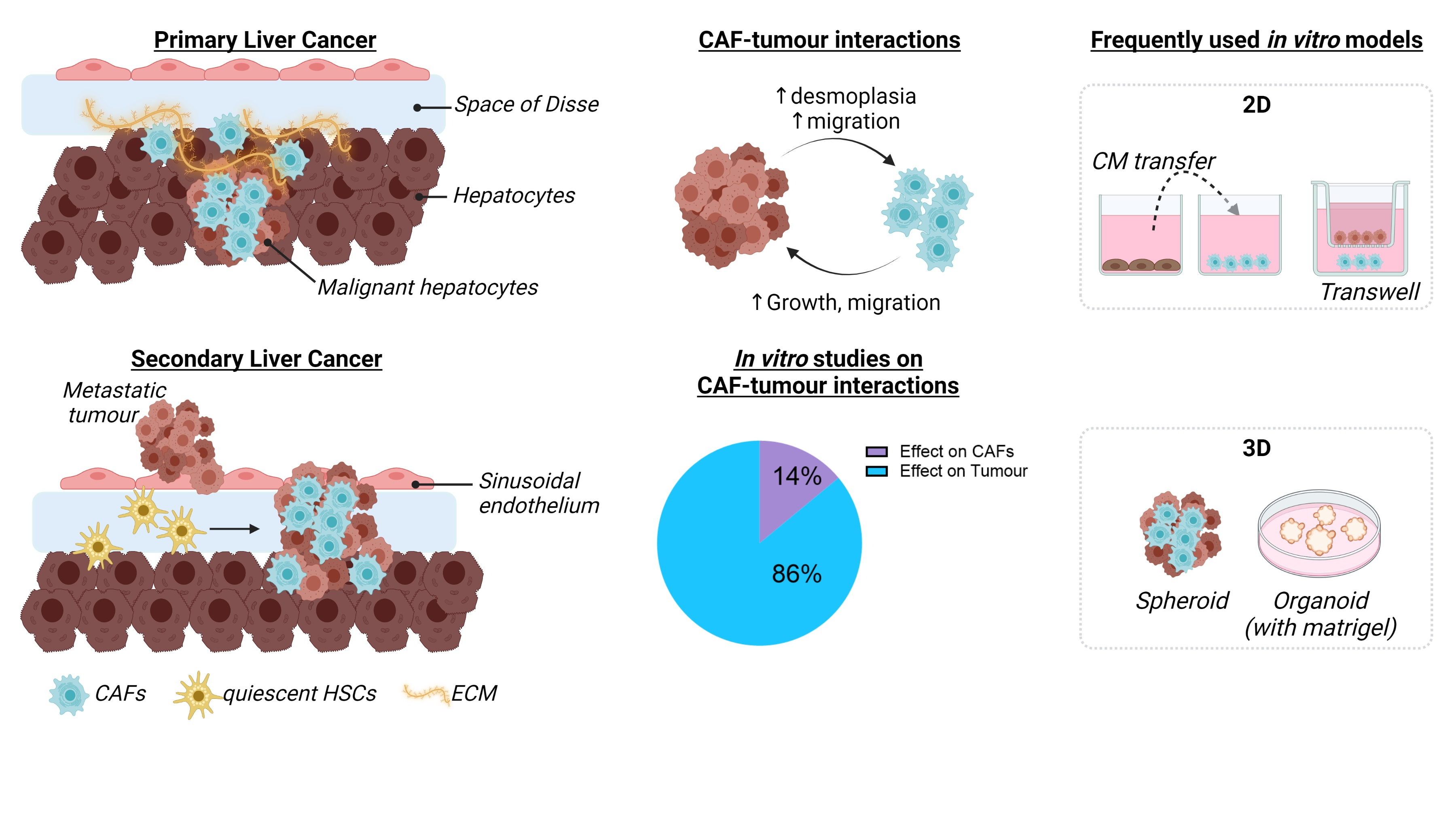
4. Definition and Use of CAFs for In Vitro Liver Cancer Studies
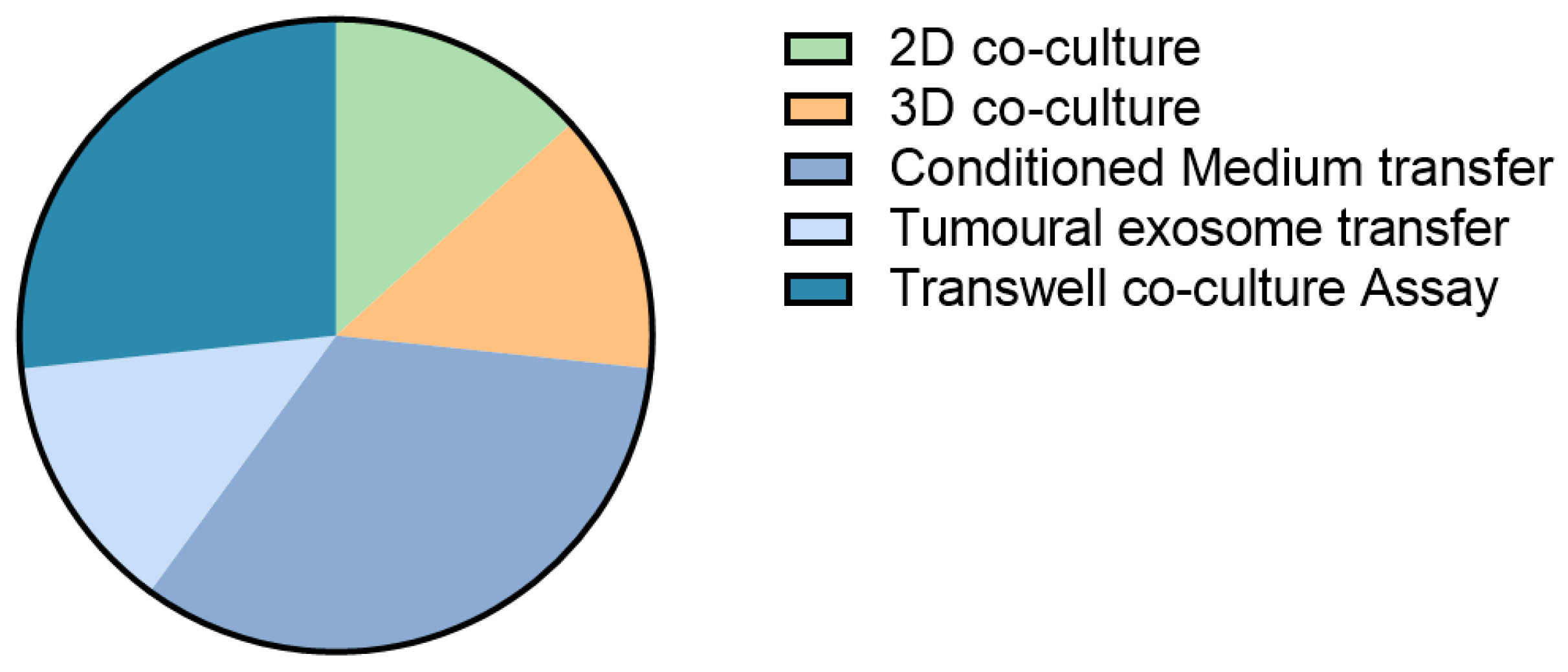
5. Models for CAF-Tumour Interaction in Liver Cancer
6. In Vitro Models Studying the Role of HSCs in Liver Metastases
7. Conclusions and Future Steps in CAF-Tumour Interaction Studies
Funding
Acknowledgments
Conflicts of Interest
References
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef] [PubMed]
- Valderrama-Treviño, A.I.; Barrera-Mera, B.; Ceballos-Villalva, J.C.; Montalvo-Javé, E.E. Hepatic Metastasis from Colorectal Cancer. Euroasian J. Hepatogastroenterol. 2017, 7, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hoffmann, A.D.; Liu, H.; Liu, X. Organotropism: New insights into molecular mechanisms of breast cancer metastasis. NPJ Precis Oncol. 2018, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Kurishima, K.; Nakazawa, K.; Kagohashi, K.; Ishikawa, H.; Satoh, H.; Hizawa, N. Specific organ metastases and survival in metastatic non-small-cell lung cancer. Mol. Clin. Oncol. 2015, 3, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Rofstad, E.K. Integrins as therapeutic targets in the organ-specific metastasis of human malignant melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 92. [Google Scholar] [CrossRef]
- Zeng, X.; Ward, S.E.; Zhou, J.; Cheng, A.S.L. Liver Immune Microenvironment and Metastasis from Colorectal Cancer-Pathogenesis and Therapeutic Perspectives. Cancers 2021, 13, 2418. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Affo, S.; Nair, A.; Brundu, F.; Ravichandra, A.; Bhattacharjee, S.; Matsuda, M.; Chin, L.; Filliol, A.; Wen, W.; Song, X.; et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell. 2021, 39, 883. [Google Scholar] [CrossRef]
- Affo, S.; Yu, L.X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Hamberger, F.; Ravichandra, A.; Miller, M.; Nair, A.; Affo, S.; Filliol, A.; Chin, L.; Savage, T.M.; Yin, D.; et al. Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhou, L.; Zhou, J.; Li, Q.; Ji, Q. Underlying mechanisms and drug intervention strategies for the tumour microenvironment. J. Exp. Clin. Cancer Res. 2021, 40, 97. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ozakyol, A. Global Epidemiology of Hepatocellular Carcinoma (HCC Epidemiology). J. Gastrointest. Cancer 2017, 48, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [Green Version]
- Kanda, T.; Goto, T.; Hirotsu, Y.; Moriyama, M.; Omata, M. Molecular Mechanisms Driving Progression of Liver Cirrhosis towards Hepatocellular Carcinoma in Chronic Hepatitis B and C Infections: A Review. Int. J. Mol. Sci. 2019, 20, 1358. [Google Scholar] [CrossRef] [Green Version]
- Vo Quang, E.; Shimakawa, Y.; Nahon, P. Epidemiological projections of viral-induced hepatocellular carcinoma in the perspective of WHO global hepatitis elimination. Liver Int. 2021, 41, 915–927. [Google Scholar] [CrossRef]
- Ferreira, R.G.; Cardoso, M.V.; de Souza Furtado, K.M.; Espíndola, K.M.M.; Amorim, R.P.; Monteiro, M.C. Epigenetic alterations caused by aflatoxin b1: A public health risk in the induction of hepatocellular carcinoma. Transl. Res. 2019, 204, 51–71. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, F. Global burden of aflatoxin-induced hepatocellular carcinoma: A risk assessment. Environ. Health Perspect. 2010, 118, 818–824. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.A.; Lee, H.C.; Choe, J.; Kim, M.J.; Lee, M.J.; Chang, H.S.; Bae, I.Y.; Kim, H.K.; An, J.; Shim, J.H.; et al. Association between non-alcoholic fatty liver disease and cancer incidence rate. J. Hepatol. 2017, 68, 140–146. [Google Scholar] [CrossRef]
- Raza, A.; Sood, G.K. Hepatocellular carcinoma review: Current treatment, and evidence-based medicine. World J. Gastroenterol. 2014, 20, 4115–4127. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Zavyalova, M.V.; Denisov, E.V.; Tashireva, L.A.; Savelieva, O.E.; Kaigorodova, E.V.; Krakhmal, N.V.; Perelmuter, V.M. Intravasation as a Key Step in Cancer Metastasis. Biochemistry 2019, 84, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Vilahur, N.; Bianchini, F.; Guha, N.; Straif, K. The IARC Perspective on Colorectal Cancer Screening. N. Engl. J. Med. 2018, 378, 1734–1740. [Google Scholar] [CrossRef]
- Gunter, M.J.; Alhomoud, S.; Arnold, M.; Brenner, H.; Burn, J.; Casey, G.; Chan, A.T.; Cross, A.J.; Giovannucci, E.; Hoover, R.; et al. Meeting report from the joint IARC-NCI international cancer seminar series: A focus on colorectal cancer. Ann. Oncol. 2019, 30, 510–519. [Google Scholar] [CrossRef]
- von Mehren, M.; Joensuu, H. Gastrointestinal Stromal Tumors. J. Clin. Oncol. 2018, 36, 136–143. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Hollie, N.; Asakrah, S. MALT lymphoma of the colon: A clinicopathological review. J. Clin. Pathol. 2020, 73, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.G.; Geiger, W.J.; Davis, N.B. Carcinoid tumors. Am. Fam. Physician 2006, 74, 429–434. [Google Scholar] [PubMed]
- Kow, A.W.C. Hepatic metastasis from colorectal cancer. J. Gastrointest. Oncol. 2019, 10, 1274–1298. [Google Scholar] [CrossRef]
- Zarour, L.R.; Anand, S.; Billingsley, K.G.; Bisson, W.H.; Cercek, A.; Clarke, M.F.; Coussens, L.M.; Gast, C.E.; Geltzeiler, C.B.; Hansen, L.; et al. Colorectal Cancer Liver Metastasis: Evolving Paradigms and Future Directions. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, F.C.; Chok, K.S. Colorectal liver metastases: An update on multidisciplinary approach. World J. Hepatol. 2019, 11, 150–172. [Google Scholar] [CrossRef] [PubMed]
- Hackl, C.; Neumann, P.; Gerken, M.; Loss, M.; Klinkhammer-Schalke, M.; Schlitt, H.J. Treatment of colorectal liver metastases in Germany: A ten-year population-based analysis of 5772 cases of primary colorectal adenocarcinoma. BMC Cancer 2014, 14, 810. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahmasebi Birgani, M.; Carloni, V. Tumor Microenvironment, a Paradigm in Hepatocellular Carcinoma Progression and Therapy. Int. J. Mol. Sci. 2017, 18, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodt, P. Role of the Microenvironment in Liver Metastasis: From Pre- to Prometastatic Niches. Clin. Cancer Res. 2016, 22, 5971–5982. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.S.; Tang, X.T.; Lin, M.; Yuan, J.; Peng, Y.J.; Yin, X.; Shang, G.; Ge, G.; Ren, Z.; Zhou, B.O. Perivenous Stellate Cells Are the Main Source of Myofibroblasts and Cancer-Associated Fibroblasts Formed After Chronic Liver Injuries. Hepatology 2021, 74, 1578–1594. [Google Scholar] [CrossRef]
- Baglieri, J.; Brenner, D.A.; Kisseleva, T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int. J. Mol. Sci 2019, 20, 1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Jia, Y.L.; Shi, L.; Zhou, J.N.; Fu, C.J.; Chen, L.; Yuan, H.F.; Wang, Y.F.; Yan, X.L.; Xu, Y.C.; Zeng, Q.; et al. Epimorphin promotes human hepatocellular carcinoma invasion and metastasis through activation of focal adhesion kinase/extracellular signal-regulated kinase/matrix metalloproteinase-9 axis. Hepatology 2011, 54, 1808–1818. [Google Scholar] [CrossRef]
- Jia, C.; Wang, G.; Wang, T.; Fu, B.; Zhang, Y.; Huang, L.; Deng, Y.; Chen, G.; Wu, X.; Chen, J.; et al. Cancer-associated Fibroblasts induce epithelial-mesenchymal transition via the Transglutaminase 2-dependent IL-6/IL6R/STAT3 axis in Hepatocellular Carcinoma. Int. J. Biol. Sci. 2020, 16, 2542–2558. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Kuroda, K.; Sugitani, M.; Takayama, T.; Hasegawa, K.; Esumi, M. Transglutaminase 2 is upregulated in primary hepatocellular carcinoma with early recurrence as determined by proteomic profiles. Int. J. Oncol. 2017, 50, 1749–1759. [Google Scholar] [CrossRef]
- Song, T.; Dou, C.; Jia, Y.; Tu, K.; Zheng, X. TIMP-1 activated carcinoma-associated fibroblasts inhibit tumor apoptosis by activating SDF1/CXCR4 signaling in hepatocellular carcinoma. Oncotarget 2015, 6, 12061–12079. [Google Scholar] [CrossRef] [Green Version]
- Owusu, B.Y.; Galemmo, R.; Janetka, J.; Klampfer, L. Hepatocyte Growth Factor, a Key Tumor-Promoting Factor in the Tumor Microenvironment. Cancers 2017, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Sakai, R. Direct Interaction between Carcinoma Cells and Cancer Associated Fibroblasts for the Regulation of Cancer Invasion. Cancers 2015, 7, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Qiu, M.; Wan, L.; Wang, G.; Huang, T.; Chen, Z.; Jiang, S.; Li, X.; Xie, L.; Cai, L. TGF-β1 Promotes Hepatocellular Carcinoma Invasion and Metastasis via ERK Pathway-Mediated FGFR4 Expression. Cell Physiol. Biochem. 2018, 45, 1690–1699. [Google Scholar] [CrossRef]
- Reichl, P.; Dengler, M.; van Zijl, F.; Huber, H.; Führlinger, G.; Reichel, C.; Sieghart, W.; Peck-Radosavljevic, M.; Grubinger, M.; Mikulits, W. Axl activates autocrine transforming growth factor-β signaling in hepatocellular carcinoma. Hepatology 2015, 61, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Yeldag, G.; Rice, A.; Del Río Hernández, A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers 2018, 10, 471. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Wu, J.; Shi, H.; Wang, Z.; Zhang, G.; Cao, Y.; Jiang, C.; Ding, Y. Hepatic stellate cell coculture enables sorafenib resistance in Huh7 cells through HGF/c-Met/Akt and Jak2/Stat3 pathways. Biomed. Res. Int. 2014, 2014, 764981. [Google Scholar] [CrossRef]
- Peng, H.; Xue, R.; Ju, Z.; Qiu, J.; Wang, J.; Yan, W.; Gan, X.; Tian, Y.; Shen, H.; Wang, X.; et al. Cancer-associated fibroblasts enhance the chemoresistance of CD73(+) hepatocellular carcinoma cancer cells via HGF-Met-ERK1/2 pathway. Ann. Transl. Med. 2020, 8, 856. [Google Scholar] [CrossRef]
- Gao, L.; Morine, Y.; Yamada, S.; Saito, Y.; Ikemoto, T.; Tokuda, K.; Miyazaki, K.; Okikawa, S.; Takasu, C.; Shimada, M. The BAFF/NFκB axis is crucial to interactions between sorafenib-resistant HCC cells and cancer-associated fibroblasts. Cancer Sci. 2021, 112, 3545–3554. [Google Scholar] [CrossRef]
- Liu, J.; Li, P.; Wang, L.; Li, M.; Ge, Z.; Noordam, L.; Lieshout, R.; Verstegen, M.M.A.; Ma, B.; Su, J.; et al. Cancer-Associated Fibroblasts Provide a Stromal Niche for Liver Cancer Organoids That Confers Trophic Effects and Therapy Resistance. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 407–431. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, L.; Yin, Z.; Su, W.; Ren, G.; Zhou, C.; You, J.; Fan, J.; Wang, X. Activated hepatic stellate cells promote hepatocellular carcinoma development in immunocompetent mice. Int. J. Cancer 2011, 129, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhao, W.; Xu, J.; Li, J.; Hong, Z.; Yin, Z.; Wang, X. Activated hepatic stellate cells promote liver cancer by induction of myeloid-derived suppressor cells through cyclooxygenase-2. Oncotarget 2016, 7, 8866–8878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: More mechanisms for inhibiting antitumor immunity. Cancer Immunol. Immunother. 2010, 59, 1593–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaps, L.; Schuppan, D. Targeting Cancer Associated Fibroblasts in Liver Fibrosis and Liver Cancer Using Nanocarriers. Cells 2020, 9, 2027. [Google Scholar] [CrossRef]
- Coulouarn, C.; Corlu, A.; Glaise, D.; Guénon, I.; Thorgeirsson, S.S.; Clément, B. Hepatocyte-stellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer Res. 2012, 72, 2533–2542. [Google Scholar] [CrossRef] [Green Version]
- Taura, K.; De Minicis, S.; Seki, E.; Hatano, E.; Iwaisako, K.; Osterreicher, C.H.; Kodama, Y.; Miura, K.; Ikai, I.; Uemoto, S.; et al. Hepatic stellate cells secrete angiopoietin 1 that induces angiogenesis in liver fibrosis. Gastroenterology 2008, 135, 1729–1738. [Google Scholar] [CrossRef]
- Sanz-Cameno, P.; Martín-Vílchez, S.; Lara-Pezzi, E.; Borque, M.J.; Salmerón, J.; Muñoz de Rueda, P.; Solís, J.A.; López-Cabrera, M.; Moreno-Otero, R. Hepatitis B virus promotes angiopoietin-2 expression in liver tissue: Role of HBV x protein. Am. J. Pathol. 2006, 169, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Tovoli, F.; Negrini, G.; Benevento, F.; Faggiano, C.; Goio, E.; Granito, A. Systemic treatments for hepatocellular carcinoma: Challenges and future perspectives. Hepat. Oncol. 2018, 5, Hep01. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colangelo, T.; Polcaro, G.; Muccillo, L.; D’Agostino, G.; Rosato, V.; Ziccardi, P.; Lupo, A.; Mazzoccoli, G.; Sabatino, L.; Colantuoni, V. Friend or foe? The tumour microenvironment dilemma in colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Kubo, N.; Araki, K.; Kuwano, H.; Shirabe, K. Cancer-associated fibroblasts in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 6841–6850. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weimer, J.; Meurer, S.K.; Kron, A.; Seipel, B.; Vater, I.; Arnold, N.; Siebert, R.; Xu, L.; Friedman, S.L.; et al. Genetic characteristics of the human hepatic stellate cell line LX-2. PLoS ONE 2013, 8, e75692. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Caruso, S.; Calatayud, A.L.; Pilet, J.; La Bella, T.; Rekik, S.; Imbeaud, S.; Letouzé, E.; Meunier, L.; Bayard, Q.; Rohr-Udilova, N.; et al. Analysis of Liver Cancer Cell Lines Identifies Agents With Likely Efficacy Against Hepatocellular Carcinoma and Markers of Response. Gastroenterology 2019, 157, 760–776. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, H.; Wan, L.; Wang, Z.; Wang, H.; Ge, C.; Liu, Y.; Hao, Y.; Zhang, D.; Shi, G.; et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ren, H.; Dai, B.; Li, J.; Shang, L.; Huang, J.; Shi, X. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J. Exp. Clin. Cancer Res. 2018, 37, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, N.; Chen, Z.; Lu, Y.; Li, Y.; Hu, K.; Xu, R. Role of activated hepatic stellate cells in proliferation and metastasis of hepatocellular carcinoma. Hepatol. Res. 2015, 45, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Bandapalli, O.R.; Macher-Goeppinger, S.; Schirmacher, P.; Brand, K. Paracrine signalling in colorectal liver metastases involving tumor cell-derived PDGF-C and hepatic stellate cell-derived PAK-2. Clin. Exp. Metastasis 2012, 29, 409–417. [Google Scholar] [CrossRef]
- Dominijanni, A.; Devarasetty, M.; Soker, S. Manipulating the Tumor Microenvironment in Tumor Organoids Induces Phenotypic Changes and Chemoresistance. iScience 2020, 23, 101851. [Google Scholar] [CrossRef] [PubMed]
- Myojin, Y.; Hikita, H.; Sugiyama, M.; Sasaki, Y.; Fukumoto, K.; Sakane, S.; Makino, Y.; Takemura, N.; Yamada, R.; Shigekawa, M.; et al. Hepatic Stellate Cells in Hepatocellular Carcinoma Promote Tumor Growth Via Growth Differentiation Factor 15 Production. Gastroenterology 2021, 160, 1741–1754.e1716. [Google Scholar] [CrossRef]
- Wang, C.; Shang, C.; Gai, X.; Song, T.; Han, S.; Liu, Q.; Zheng, X. Sulfatase 2-Induced Cancer-Associated Fibroblasts Promote Hepatocellular Carcinoma Progression via Inhibition of Apoptosis and Induction of Epithelial-to-Mesenchymal Transition. Front. Cell Dev. Biol. 2021, 9, 631931. [Google Scholar] [CrossRef]
- Benedicto, A.; Herrero, A.; Romayor, I.; Marquez, J.; Smedsrød, B.; Olaso, E.; Arteta, B. Liver sinusoidal endothelial cell ICAM-1 mediated tumor/endothelial crosstalk drives the development of liver metastasis by initiating inflammatory and angiogenic responses. Sci. Rep. 2019, 9, 13111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero, A.; Benedicto, A.; Romayor, I.; Olaso, E.; Arteta, B. Inhibition of COX-2 Impairs Colon Cancer Liver Metastasis through Reduced Stromal Cell Reaction. Biomol. Ther. 2021, 29, 342–351. [Google Scholar] [CrossRef]
- Ronen, J.; Hayat, S.; Akalin, A. Evaluation of colorectal cancer subtypes and cell lines using deep learning. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.X.; Gong, W.Z.; Zhou, K.; Xiao, Z.G.; Hou, F.T.; Huang, T.; Zhang, L.; Dong, H.Y.; Zhang, W.L.; Liu, Y.; et al. CXCR4/TGF-β1 mediated hepatic stellate cells differentiation into carcinoma-associated fibroblasts and promoted liver metastasis of colon cancer. Cancer Biol. Ther. 2020, 21, 258–268. [Google Scholar] [CrossRef]
- Mueller, L.; von Seggern, L.; Schumacher, J.; Goumas, F.; Wilms, C.; Braun, F.; Broering, D.C. TNF-alpha similarly induces IL-6 and MCP-1 in fibroblasts from colorectal liver metastases and normal liver fibroblasts. Biochem. Biophys. Res. Commun. 2010, 397, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Hu, Z.; Lu, L.; Lu, H.; Xu, X. Three-dimensional cell culture: A powerful tool in tumor research and drug discovery. Oncol. Lett. 2017, 14, 6999–7010. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, L.P.; Gaspar, V.M.; Mano, J.F. Design of spherically structured 3D in vitro tumor models -Advances and prospects. Acta Biomater. 2018, 75, 11–34. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.T.; Wang, J.Y.; Lin, Y.F.; Wo, A.M.; Chen, B.P.C.; Lee, H. Three-dimensional spheroid culture targeting versatile tissue bioassays using a PDMS-based hanging drop array. Sci. Rep. 2017, 7, 4363. [Google Scholar] [CrossRef] [PubMed]
- Haisler, W.L.; Timm, D.M.; Gage, J.A.; Tseng, H.; Killian, T.C.; Souza, G.R. Three-dimensional cell culturing by magnetic levitation. Nat. Protoc. 2013, 8, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- Hurrell, T.; Ellero, A.A.; Masso, Z.F.; Cromarty, A.D. Characterization and reproducibility of HepG2 hanging drop spheroids toxicology in vitro. Toxicol. In Vitro 2018, 50, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Kamatar, A.; Gunay, G.; Acar, H. Natural and Synthetic Biomaterials for Engineering Multicellular Tumor Spheroids. Polymers 2020, 12, 2506. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, E.M.; Yamamoto, M.; Park, H.; Shin, H. Engineering Multi-Cellular Spheroids for Tissue Engineering and Regenerative Medicine. Adv. Healthc. Mater. 2020, 23, e2000608. [Google Scholar] [CrossRef]
- Mehta, G.; Hsiao, A.Y.; Ingram, M.; Luker, G.D.; Takayama, S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J. Control. Release 2012, 164, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.; Hoorens, A.; Reynaert, H.; Halder, G.; van Grunsven, L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Seki, E.; Uchinami, H.; Kluwe, J.; Zhang, Y.; Brenner, D.A.; Schwabe, R.F. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology 2007, 132, 1937–1946. [Google Scholar] [CrossRef] [PubMed]
- Nii, T.; Makino, K.; Tabata, Y. Three-Dimensional Culture System of Cancer Cells Combined with Biomaterials for Drug Screening. Cancers 2020, 12, 2754. [Google Scholar] [CrossRef] [PubMed]
- Burdett, E.; Kasper, F.K.; Mikos, A.G.; Ludwig, J.A. Engineering tumors: A tissue engineering perspective in cancer biology. Tissue Eng. Part B Rev. 2010, 16, 351–359. [Google Scholar] [CrossRef]
- Han, S.J.; Kwon, S.; Kim, K.S. Challenges of applying multicellular tumor spheroids in preclinical phase. Cancer Cell Int. 2021, 21, 152. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Fong, E.L.S.; Toh, T.B.; Lin, Q.X.X.; Liu, Z.; Hooi, L.; Mohd Abdul Rashid, M.B.; Benoukraf, T.; Chow, E.K.; Huynh, T.H.; Yu, H. Generation of matched patient-derived xenograft in vitro-in vivo models using 3D macroporous hydrogels for the study of liver cancer. Biomaterials 2018, 159, 229–240. [Google Scholar] [CrossRef]
- Li, L.; Knutsdottir, H.; Hui, K.; Weiss, M.J.; He, J.; Philosophe, B.; Cameron, A.M.; Wolfgang, C.L.; Pawlik, T.M.; Ghiaur, G.; et al. Human primary liver cancer organoids reveal intratumor and interpatient drug response heterogeneity. JCI Insight. 2019, 4. [Google Scholar] [CrossRef]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Q.; Wang, B.; Li, K.; Sun, H.; Hai, T.; Zhang, Y.; Kang, H. Upregulating MMP-1 in carcinoma-associated fibroblasts reduces the efficacy of Taxotere on breast cancer synergized by Collagen IV. Oncol. Lett. 2018, 16, 3537–3544. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Huang, L.; Hu, K. Natural products remodel cancer-associated fibroblasts in desmoplastic tumors. Acta Pharm. Sin. B 2020, 10, 2140–2155. [Google Scholar] [CrossRef] [PubMed]
- Benedicto, A.; Romayor, I.; Arteta, B. CXCR4 receptor blockage reduces the contribution of tumor and stromal cells to the metastatic growth in the liver. Oncol. Rep. 2018, 39, 2022–2030. [Google Scholar] [CrossRef]
- Dou, C.; Liu, Z.; Tu, K.; Zhang, H.; Chen, C.; Yaqoob, U.; Wang, Y.; Wen, J.; van Deursen, J.; Sicard, D.; et al. P300 Acetyltransferase Mediates Stiffness-Induced Activation of Hepatic Stellate Cells Into Tumor-Promoting Myofibroblasts. Gastroenterology 2018, 154, 2209–2221.e2214. [Google Scholar] [CrossRef] [Green Version]
- Vidal-Vanaclocha, F. The liver prometastatic reaction of cancer patients: Implications for microenvironment-dependent colon cancer gene regulation. Cancer Microenviron. 2011, 4, 163–180. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Variation | Cell Name | Cellosaurus [67] | Species | Origin | References |
|---|---|---|---|---|---|
| CAFs | Primary CAF | NA | Mouse | DEN model | [56] |
| Primary CAF | NA | Human | CCA, HCC | [56,72] | |
| LX-2 | CVCL_5792 | Human | Hepatic stellate cells | [55,61,73,74,75,76,77,78] | |
| Primary HSCs | NA | Mouse | Liver digest | [79,80] | |
| 3T3-NIH | CVCL_KS54 | Mouse | Fibroblast | [80] | |
| Liver cancer cells [71] | HepaRG | CVCL_9720 | Human | HCC | [61] |
| Huh-7 | CVCL_0336 | Human | HCC | [55,77] | |
| HepG2 | CVCL_0027 | Human | Hepatoblastoma | [74,77] | |
| LM3 | CVCL_D269 | Mouse | Malignant neoplasms of the mouse mammary gland | [73] | |
| MHCC97-H | CVCL_4972 | Human | HCC | [73] | |
| HEpB3 | CVCL_0326 | Paediatric Human | HCC | [77,78] | |
| HuCCT1 | CVCL_0324 | Human | cholangiocarcinoma | [72] | |
| FRH0201 | Not mentioned | Human | cholangiocarcinoma | [72] | |
| RBE | CVCL_4896 | Human | cholangiocarcinoma | [72] | |
| QBC939 | CVCL_6942 | Human | cholangiocarcinoma | [72] | |
| CRC cell lines [81] | HCT-116 | CVCL_0291 | Human | Colon carcinoma | [76,82] |
| LS174T | CVCL_1384 | Human | Colon adenocarcinoma | [75] | |
| HT-29 | CVCL_0320 | Human | Colon carcinoma | [82,83] | |
| CT-26 | CVCL_7254 | Mouse | Colon carcinoma | [79,80] |
| Manuscript | Culture Model | Culture Conditions | CAF Source | Tumour Source | Main Result | Ref. |
|---|---|---|---|---|---|---|
| Zhou Y, 2018 | Tumour cell exosomes transfer to 2D | CM was collected from HCC cells in 10 CM plates with DMEM 10% FBS after 48 h. Exosomes were isolated through untracentrifugation. | LX-2 | LM3, MHCC97-H | Tumour cells facilitate the transition of HSCs into CAFs (increased α-SMA, FAP, FSP1, VEGF-α, MMP2, MMP9, bFGF and TGF-β) via miR-21 and AKT. | [73] |
| Zhang M, 2020 | Tumour cell exosomes transfer to 2D | Exosomes were derived from the supernatant of ICC cells collected from 48h serum-free cultures by ultracentrifugation. | Passaged primary CD146+ cells | HuCCT1, FRH0201, RBE, QBC939 | Tumour-cell-derived exosomal miR-9-5p induces IL-6 expression in vascular CAFs, which enhances ICC malignancy. | [72] |
| Tumour sphere-CAF transwell | CAFs were cultured for 24 h in the upper insert of a transwell in α-MEM 10% FBS, moved to fresh wells and supplemented with tumour sphere culture medium. A total of 2000 tumour cells were seeded in each chamber of a 6-well plate in DMEM 0% FBS. | HuCCT1 | Tumour-cell-derived exosomal miR-9-5p induces IL-6 expression in vascular CAFs, which increases tumour sphere formation. | |||
| Coulouarn C, 2012 | Tumour-CAF transwell | LX-2 and HepaRG cells were co-cultured in serum- and DMSO-free William’s E medium in 6-well plates with 1 µm pore size transwell inserts. | LX-2 | HepaRG | Tumour cells induce the enrichment of pro-fibrogenic and pro-inflammatory cytokines, acute phase proteins, and growth factors in CAFs. | [61] |
| Lin N, 2015 | Tumour-CAF transwell | HepG2 CM was added to the lower chamber and LX2 suspension (cultured in serum free DMEM) was added to the lower chamber. LX2 were incubated with the tumour cell CM for 24 h. | LX-2 | HepG2 | PDGF-bb release by HepG2 induces LX-2 migration | [74] |
| Gao L, 2021 | Tumour CM transfer | (Sorafenib resistant) Huh7 cells were cultured in DMEM 10% FBS for 48 h after which CM was collected which was added to LX2 cells cultured in DMEM at a 1:1 ratio for at least 48 h. | LX-2 | Huh7, Huh7-SR | Sorafenib-resistant tumour cells facilitate the transition of HSCs into CAFs (increased α-SMA and FAP expression) through the induction of BAFF/NFκB signalling in CAFs. | [55] |
| Wang C, 2021 | Direct co-culture in 2D | Sulfatase 2 overexpressing Hep3B cells were co-cultured with LX2 cells in DMEM 10% FBS for 72 h. | LX-2 | Hep3B | Sulfatase 2-overexpressing tumour cells promote HSC to CAF differentation (increased ACTA2, FAP, and POSTN) via TGF-β/SMAD3 signalling. | [78] |
| Myojin Y, 2021 | Direct co-culture in 2D | LX2 cells were co-cultured with the same number of hepatoma cells for 48 h. | LX-2 | HepG2, Hep3B, Huh7 | Co-culturing HSCs with tumour cells induces GDF15 expression in HSCs. | [77] |
| Liu J, 2020 | Direct co-culture in 3D | Tumour organoids were dissociated and co-cultured with CAFs (grown in 2D flasks) by sorting the cells in well plates containing mouse organoid basic medium (0% serum) and 1% Matrigel using FACS. | α-SMA+ FAP+ primary cells from DEN mice, HCC, CCA patients | Primary cells from DEN mice, HCC, CCA patients | Tumour medium transfer increases gremlin-1 expression in CAFs with a suggested role for BMP signalling. | [56] |
| Manuscript | Culture Model | Culture Conditions | CAF Source | Tumour Source | Main Result | Ref. |
|---|---|---|---|---|---|---|
| Bandapalli OR, 2012 | Tumour CM transfer | Supernatans from wild type or PDGF-C silenced LS174T cells (cultured in RPMI 10% FCS) was transferred to LX-2 cells (cultured in DMEM 1% FCS). | LX-2 | LS174T | Tumour-derived PDGF-C promotes LX-2 activation through PAK-2 signaling. | [75] |
| Mueller L, 2010 | Tumour CM transfer | CAFs were seeded in the upper chamber and HT-29 cells in the lower chamber of a Boyden chamber in DMEM 10% FBS. | Primary human CAFs | HT-29 | CAFs express IL-6 and MCP-1 induced by tumour TNF-alpha. | [83] |
| Herrero A, 2021 | Tumour CM transfer | 3T3 cells were cultured in DMEM/F-12 10% FBS and HSCs and CT-26 in RPMI-1640 with 0% and 10% FBS, respectively. CM was collected from these cells after 24 h of culture in RPMI-1640 without FBS. | Primary mouse HSCs and 3T3 | CT-26 | Tumour cells promote the migratory capacity of HSCs through ICAM-1/COX-2. | [80] |
| Benedicto A, 2018 | Tumour CM transfer | HSCs were cultured in serum-free DMEM and treated with CM of CT26 cells, which were cultured in RPMI-1640 1% FCS. | Primary mouse HSCs | CT-26 | Tumour cells induce CXCR4 expression in HSCs which reduces the cytotoxic capacity of T cells. | [79] |
| Tan Hao-Xiang, 2020 | Tumour-CAF transwell | HCT-116 or HT-29 cells were seeded onto a transwell membrane and LX-2 cells were grown in the lower chambers. Cells were incubated in RPMI-1640 2% FBS. | LX-2 | HCT-116, HT-29 | Tumours cells induce SDF-1 expression in HSCs and tumour cell-derived CXCR4 and TGF-β mediate the differentiation of HSCs into CAFs. | [82] |
| Dominijanni A, 2020 | Direct co-culture in 3D | LX-2 and HCT-116 cells were cultured in DMEM 10% FBS followed by a co-culture in organoids. | LX-2 | HCT-116 | Activated HSCs (by TGF-β presence) modulate the stiffness of ECM and reduce the chemotherapy response. | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrero, A.; Knetemann, E.; Mannaerts, I. Review: Challenges of In Vitro CAF Modelling in Liver Cancers. Cancers 2021, 13, 5914. https://doi.org/10.3390/cancers13235914
Herrero A, Knetemann E, Mannaerts I. Review: Challenges of In Vitro CAF Modelling in Liver Cancers. Cancers. 2021; 13(23):5914. https://doi.org/10.3390/cancers13235914
Chicago/Turabian StyleHerrero, Alba, Elisabeth Knetemann, and Inge Mannaerts. 2021. "Review: Challenges of In Vitro CAF Modelling in Liver Cancers" Cancers 13, no. 23: 5914. https://doi.org/10.3390/cancers13235914
APA StyleHerrero, A., Knetemann, E., & Mannaerts, I. (2021). Review: Challenges of In Vitro CAF Modelling in Liver Cancers. Cancers, 13(23), 5914. https://doi.org/10.3390/cancers13235914