
A Functional Network Model of the Metastasis Suppressor PEBP1/RKIP and Its Regulators in Breast Cancer Cells



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Protein-Protein Interactions, Literature Search, and Biological Expression Language
2.2. Knockdown of Metastasis Suppressors and Transcription Factors in MCF7
2.3. Pharmacological Perturbations of Breast Cancer Cell Lines
2.4. Network Perturbation Amplitude (NPA)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Object | Ref. | Interaction |
|---|---|---|---|
| Interactions between metastasis suppressors | |||
| CD44 | CASP8 | [36] | p(CD44) decreases act(p(CASP8)) |
| CD82 | CD44 | [37] | p(CD82) decreases p(CD44) |
| CD82 | BRMS1 | [38] | p(CD82) increases r(BRMS1) |
| CD82 | CDH1 | [39] | p(CD82) increases r(CDH1) |
| CDH1 | CASP8 | [40] | p(CDH1) increases act(p(CASP8)) |
| GSN | CDH2 | [41] | p(GSN) increases r(CDH2) |
| MAP2K3 | MAPK14 | [42] | act(comp(p(MAP2K6), p(MAP2K3)), ma(kin)) directlyIncreases p(MAPK14) |
| MAP2K4 | MAP2K7 | [43] | p(MAP2K4) decreases act(p(MAP2K7), ma(kin)) |
| MAP2K4 | MAPK14 | [42] | p(MAP2K4) directlyIncreases act(p(:MAPK14), ma(kin)) |
| MAP2K4 | CASP8 | [44] | p(MAPK14) increases p(CASP8) |
| MAP2K6 | MAPK14 | [42] | p(MAP2K6) directlyIncreases p(MAPK14) |
| MAP2K7 | CDH2 | [45] | p(MAP2K7) increases r(CDH2) |
| MAPK14 | CDH1 | [46] | p(MAPK14) increases p(CDH1) |
| MAPK14 | CASP8 | [47] | act(p(MAPK14), ma(kin)) directlyDecreases p(CASP8) |
| MAPK14 | SAP1A | [48] | act(p(MAPK14), ma(kin)) directlyIncreases p(ets-Domain Protein Elk-4) |
| MAPK14 | RUNX2 | [49] | p(MAPK14) increases r(RUNX2) |
| NME1 | BRMS1 | [50] | p(NME1) decreases r(BRMS1) |
| NME1 | AKAP12 | [50] | p(NME1) increases r(AKAP12) |
| NME1 | PEBP1 | [51] | r(NME1) increases r(PEBP1) |
| PEBP1 | MAP2K6 | [52] | p(PEBP1) increases act(p(MAP2K6)) |
| PEBP1 | MAP2K3 | [52] | p(PEBP1) increases act(p(MAP2K3)) |
| PEBP1 | MAPK14 | [52] | p(PEBP1) increases act(p(MAPK14)) |
| PEBP1 | CDH1 | [53] | p(PEBP1) increases r(CDH1) |
| RUNX2 | CDH2 | [54] | p(RUNX2) decreases r(CDH2) |
| RUNX2 | CDH1 | [54] | p(RUNX2) increases r(CDH1) |
| TGFB1 | CDH1 | [55] | p(TGFB1) decreases r(CDH1) |
| TGFB1 | CDH2 | [56] | p(TGFB1) decreases r(CDH2) |
| Interactions between metastasis suppressors and regulators | |||
| CDH1 | YBX1 | [57] | p(CDH1) decreases p(YBX1) |
| CDH1 | FOXM1 | [58] | p(CDH1) decreases p(FOXM1) |
| ESR1 | CDH2 | [59] | p(ESR1) decreases r(CDH2) |
| ESR1 | CDH1 | [60] | p(ESR1) directlyIncreases r(CDH1) |
| ESR1 | BRMS1 | [61] | p(HGNC:ESR1) directlyIncreases r(HGNC:BRMS1) |
| ESR1 | MTA3 | [62] | p(ESR1) increases r(MTA3) |
| ESR1 | RUNX2 | [63] | p(ESR1) increases r(RUNX2) |
| FOS | CDH1 | [55] | p(FOS) decreases r(CDH1) |
| FOS | RUNX2 | [64] | p(FOS) decreases r(RUNX2) |
| FOS | TGFB1 | [55] | p(FOS) directlyDecreases r(TGFB1) |
| FOS | CD44 | [65] | p(FOS) increases r(CD44) |
| GATA3 | CD44 | [66] | r(GATA3) directlyIncreases r(CD44) |
| GATA3 | CD44 | [66] | r(GATA3) directlyIncreases r(CD44) |
| GATA3 | CD44 | [66] | p(GATA3) increases r(CD44) |
| GATA3 | CD44 | [66] | p(GATA3) increases r(CD44) |
| HIF1A | CD44 | [67] | p(HIF1A) increases r(CD44) |
| MAP2K4 | FOS | [68] | p(MAP2K4) increases p(FOS) |
| MAPK14 | FOS | [48] | act(p(MAPK14), ma(kin)) increases p(FOS) |
| MAPK14 | GATA3 | [69] | p(MAPK14) increases act(p(GATA3)) |
| MTA3 | CDH2 | [62] | p(MTA3) decreases r(CDH2) |
| MTA3 | CDH1 | [62] | p(MTA3) increases r(CDH1) |
| RELA | BRMS1 | [70] | p(RELA) directlyDecreases r(BRMS1) |
| RUNX2 | HIF1A | [71] | p(RUNX2) increases act(p(HIF1A)) |
| RUNX2 | SPDEF | [72] | p(RUNX2) directlyDecreases r(SPDEF) |
| SAP1A | FOS | [48] | p(ets-Domain Protein Elk-4) directlyIncreases r(FOS) |
| SATB1 | NME1 | [73] | p(SATB1) directlyDecreases r(NME1) |
| SATB1 | BRMS1 | [73] | p(SATB1) directlyDecreases r(BRMS1) |
| SATB1 | CD82 | [73] | p(SATB1) directlyDecreases r(CD82) |
| SATB1 | CDH1 | [73] | p(SATB1) directlyDecreases r(CDH1) |
| SNAI1 | CDH1 | [74] | p(SNAI1) directlyDecreases r(CDH1) |
| SNAI1 | PEBP1 | [75] | p(SNAI1) directlyDecreases r(PEBP1) |
| SNAI1 | CASP8 | [76] | p(SNAI1) decreases act(p(CASP8)) |
| TFAP2C | CD44 | [77] | p(TFAP2C) directlyDecreases r(CD44) |
| YBX1 | CD44 | [78] | p(YBX1) directlyIncreases r(CD44) |
| Interactions between transcription factors | |||
| ESR1 | HIF1A | [79] | p(ESR1) directlyIncreases r(HIF1A) |
| ESR1 | GATA3 | [20] | p(GATA3) increases p(ESR1) |
| FOXM1 | GATA3 | [80] | p(FOXM1) directlyDecreases r(GATA3) |
| MTA3 | SNAI1 | [81] | p(MTA3) directlyDecreases r(SNAI1) |
| NR5A2 | ESR1 | [82] | p(NR5A2) directlyIncreases r(ESR1) |
| RARA | FOS | [83] | p(RARA) decreases act(p(FOS)) |
| RELA | SNAI1 | [84] | p(RELA) directlyIncreases r(SNAI1) |
| TFAP2C | ESR1 | [85] | p(TFAP2C) directlyIncreases r(ESR1) |
| TNFSF10 | CASP8 | [86] | p(TNFSF10) increases p(CASP8) |
2.5. Measures of Agreement
2.6. Reagents and Drugs
2.7. Cell Culture and Drugs Treatment
2.8. RNA Extraction, and RT-qPCR
2.9. Software Environment and Reproducibility
3. Results
3.1. A Workflow for Building a Network of the Metastasis Suppressors and Their Regulators
3.2. Identifying Possible Interactions of Metastasis Suppressors Using Text Mining of the Literature
3.3. Contextualizing Metastasis Suppressor Interactions in Breast Cancer Cells
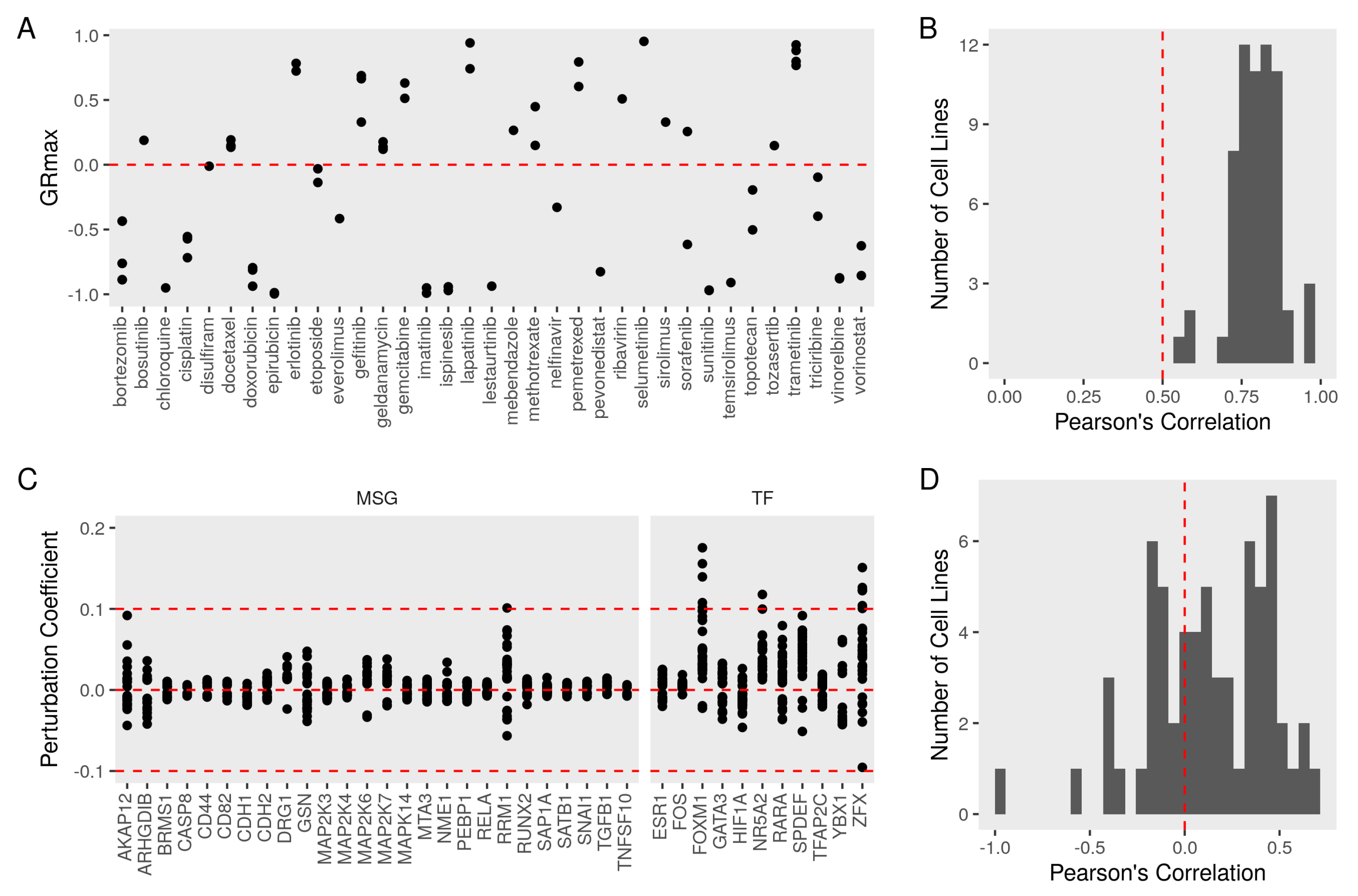
3.4. Evaluating the Metastasis Model Using Drug Perturbation Data
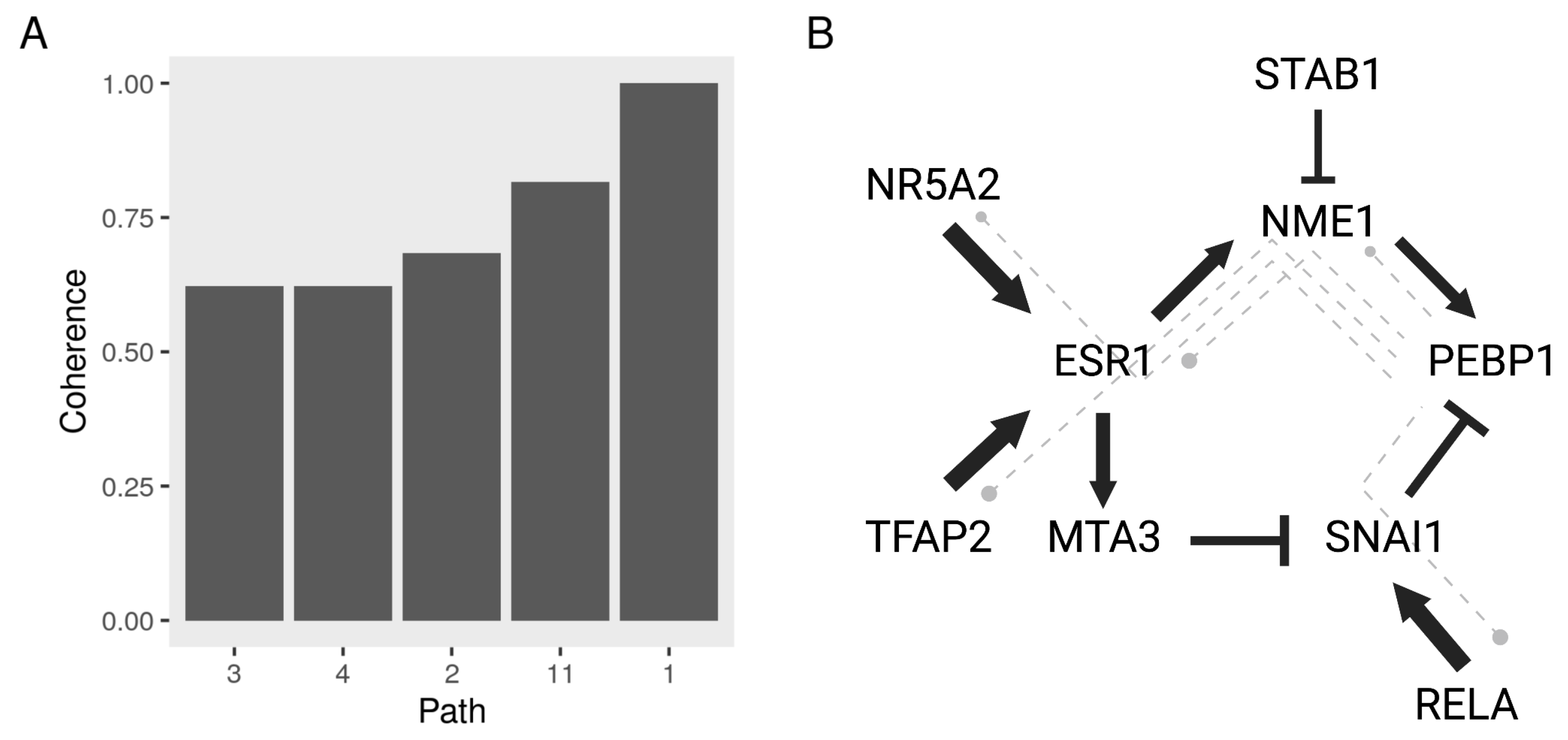
3.5. Constructing a Model of PEBP1 and Its Interaction with Other Metastasis Suppressors
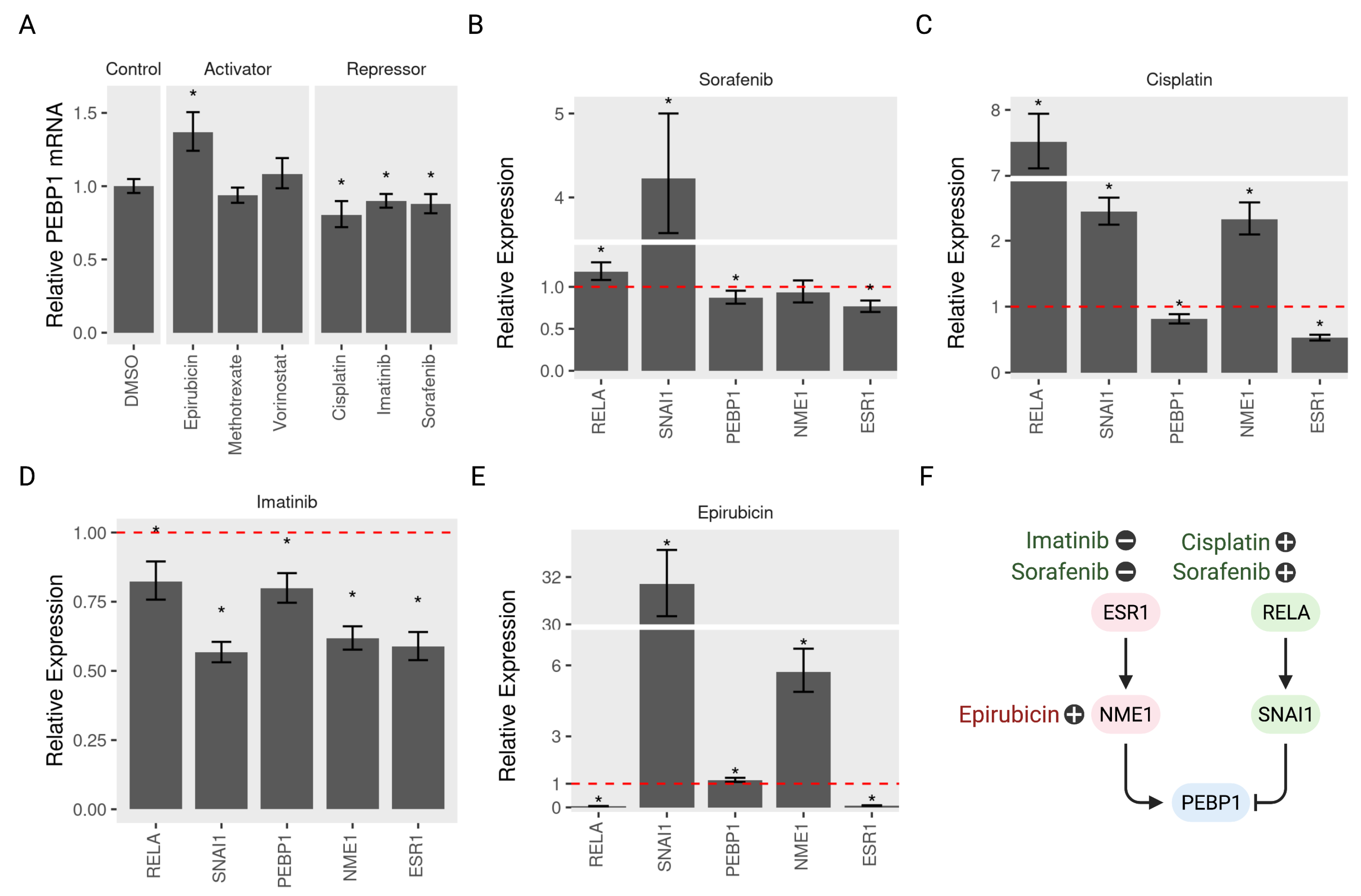
3.6. Validating the Model Predictions
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MSG | Metastasis suppressor genes |
| TF | Transcription factor |
| EMT | Epithelial to mesenchymal transition |
| BEL | Biological expression language |
| GEO | Gene expression omnibus |
| LINCS | Library of network-based integrated cellular signatures |
| NPA | Network perturbation amplitude |
| PPI | Protein-protein interactions |
| KS | Kolmogorov-Smirnov test |
| GRmax | Maximum growth-rate inhibition |
| ECDF | Empirical cumulative distribution function |
References
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- Martin, F.; Thomson, T.M.; Sewer, A.; Drubin, D.A.; Mathis, C.; Weisensee, D.; Pratt, D.; Hoeng, J.; Peitsch, M.C. Assessment of network perturbation amplitudes by applying high-throughput data to causal biological networks. BMC Syst. Biol. 2012, 6, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catlett, N.L.; Bargnesi, A.J.; Ungerer, S.; Seagaran, T.; Ladd, W.; Elliston, K.O.; Pratt, D. Reverse causal reasoning: Applying qualitative causal knowledge to the interpretation of high-throughput data. BMC Bioinform. 2013, 14, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, F.; Sewer, A.; Talikka, M.; Xiang, Y.; Hoeng, J.; Peitsch, M.C. Quantification of biological network perturbations for mechanistic insight and diagnostics using two-layer causal models. BMC Bioinform. 2014, 15, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef]
- Shankavaram, U.T.; Reinhold, W.C.; Nishizuka, S.; Major, S.; Morita, D.; Chary, K.K.; Reimers, M.A.; Scherf, U.; Kahn, A.; Dolginow, D.; et al. Transcript and protein expression profiles of the NCI-60 cancer cell panel: An integromic microarray study. Mol. Cancer Ther. 2007, 6, 820–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala de la Peña, F.; Andrés, R.; Garcia-Sáenz, J.A.; Manso, L.; Margelí, M.; Dalmau, E.; Pernas, S.; Prat, A.; Servitja, S.; Ciruelos, E. SEOM clinical guidelines in early stage breast cancer (2018). Clin. Transl. Oncol. 2019, 21, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Kogure, A.; Yoshioka, Y.; Ochiya, T. Extracellular vesicles in cancer metastasis: Potential as therapeutic targets and materials. Int. J. Mol. Sci. 2020, 21, 4463. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Steeg, P.S. Metastasis suppressors: Functional pathways. Lab Investig. 2018, 98, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, E.T. Metastasis suppressor genes: A role for raf kinase inhibitor protein (RKIP). Anticancer Drugs 2004, 15, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Yeung, M.L.; Beach, S.; Shields, J.M.; Yeung, K.C. RKIP downregulates B-Raf kinase activity in melanoma cancer cells. Oncogene 2005, 24, 3535–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Q.; Wu, H.; Fu, F.; Yi, W.; Pei, L.; Zhou, M. RKIP suppresses the proliferation and metastasis of breast cancer cell lines through up-regulation of miR-185 targeting HMGA2. Arch. Biochem. Biophys. 2016, 610, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Zhang, C.; Liu, G.; Wang, F. RKIP negatively regulates the glucose induced angiogenesis and endothelial-mesenchymal transition in retinal endothelial cells. Exp. Eye Res. 2019, 189, 107851. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Bai, Y.; Wang, Z.; Beach, S.; Mott, S.; Roy, R.; Braastad, C.; Sun, Y.; Mukhopadhyay, A.; Aggarwal, B.B.; et al. RKIP Sensitizes Prostate and Breast Cancer Cells to Drug-induced Apoptosis. J. Biol. Chem. 2004, 279, 17515–17523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, C.T.; Domingo-Fernández, D.; Hofmann-Apitius, M. BEL Commons: An environment for exploration and analysis of networks encoded in Biological Expression Language. Database J. Biol. Databases Curation 2018, 2018, bay126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, C.; Benz, C.C. Genes responsive to both oxidant stress and loss of estrogen receptor function identify a poor prognosis group of estrogen receptor positive primary breast cancers. Breast Cancer Res. BCR 2008, 10, R61. [Google Scholar] [CrossRef] [Green Version]
- Dahlman-Wright, K.; Qiao, Y.; Jonsson, P.; Gustafsson, J.A.; Williams, C.; Zhao, C. Interplay between AP-1 and estrogen receptor α in regulating gene expression and proliferation networks in breast cancer cells. Carcinogenesis 2012, 33, 1684–1691. [Google Scholar] [CrossRef] [Green Version]
- Bergamaschi, A.; Madak-Erdogan, Z.; Kim, Y.J.; Choi, Y.L.; Lu, H.; Katzenellenbogen, B.S. The forkhead transcription factor FOXM1 promotes endocrine resistance and invasiveness in estrogen receptor-positive breast cancer by expansion of stem-like cancer cells. Breast Cancer Res. BCR 2014, 16, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodorou, V.; Stark, R.; Menon, S.; Carroll, J.S. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013, 23, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Elvidge, G.P.; Glenny, L.; Appelhoff, R.J.; Ratcliffe, P.J.; Ragoussis, J.; Gleadle, J.M. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: The role of HIF-1alpha, HIF-2alpha, and other pathways. J. Biol. Chem. 2006, 281, 15215–15226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.F.; Flach, K.D.; Alexi, X.; Fox, S.P.; Ottaviani, S.; Thiruchelvam, P.T.R.; Kyle, F.J.; Thomas, R.S.; Launchbury, R.; Hua, H.; et al. Co-regulated gene expression by oestrogen receptor α and liver receptor homolog-1 is a feature of the oestrogen response in breast cancer cells. Nucleic Acids Res. 2013, 41, 10228–10240. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.D.; Ratnam, M.; Patki, M.; Kisovic, I.; Trumbly, R.; Iman, M.; Ratnam, M. During hormone depletion or tamoxifen treatment of breast cancer cells the estrogen receptor apoprotein supports cell cycling through the retinoic acid receptor α1 apoprotein. Breast Cancer Res. BCR 2011, 13, R18. [Google Scholar] [CrossRef] [Green Version]
- Buchwalter, G.; Hickey, M.M.; Cromer, A.; Selfors, L.M.; Gunawardane, R.N.; Frishman, J.; Jeselsohn, R.; Lim, E.; Chi, D.; Fu, X.; et al. PDEF promotes luminal differentiation and acts as a survival factor for ER-positive breast cancer cells. Cancer Cell 2013, 23, 753–767. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.K.; Lin, Z.H.; Chang, C.W.; Varang, V.; Chng, K.R.; Pan, Y.F.; Yong, E.L.; Sung, W.K.; Sung, W.K.; Cheung, E. AP-2γ regulates oestrogen receptor-mediated long-range chromatin interaction and gene transcription. EMBO J. 2011, 30, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Lasham, A.; Samuel, W.; Cao, H.; Patel, R.; Mehta, R.; Stern, J.L.; Reid, G.; Woolley, A.G.; Miller, L.D.; Black, M.A.; et al. YB-1, the E2F pathway, and regulation of tumor cell growth. J. Natl. Cancer Inst. 2012, 104, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489. [Google Scholar] [CrossRef]
- Marino, N.; Collins, J.W.; Shen, C.; Caplen, N.J.; Merchant, A.S.; Gökmen-Polar, Y.; Goswami, C.P.; Hoshino, T.; Qian, Y.; Sledge, G.W.; et al. Identification and validation of genes with expression patterns inverse to multiple metastasis suppressor genes in breast cancer cell lines. Clin. Exp. Metastasis 2014, 31, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Sean, D.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Song, C.; Liu, Y.; Qian, F.; Gao, Y.; Ning, Z.; Wang, Q.; Jiang, Y.; Li, Y.; Li, M.; et al. KnockTF: A comprehensive human gene expression profile database with knockdown/knockout of transcription factors. Nucleic Acids Res. 2019, 48, D93–D100. [Google Scholar] [CrossRef]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 2016, 13, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koleti, A.; Terryn, R.; Stathias, V.; Chung, C.; Cooper, D.J.; Turner, J.P.; VidoviÄ, D.; Forlin, M.; Kelley, T.T.; D’Urso, A.; et al. Data Portal for the Library of Integrated Network-based Cellular Signatures (LINCS) program: Integrated access to diverse large-scale cellular perturbation response data. Nucleic Acids Res. 2018, 46, D558–D566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kort, E. Slinky: Putting the Fun in LINCS L1000 Data Analysis. Available online: https://bioconductor.org/packages/release/bioc/html/slinky.html (accessed on 29 November 2021).
- Martin, F.; Gubian, S.; Talikka, M.; Hoeng, J.; Peitsch, M.C. NPA: An R package for computing network perturbation amplitudes using gene expression data and two-layer networks. BMC Bioinform. 2019, 20, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baaten, B.J.; Li, C.R.; Bradley, L.M. Multifaceted regulation of T cells by CD44. Commun. Integr. Biol. 2010, 3, 508–512. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Zhang, F.; Richardson, M.M.; Roy, N.H.; Rodgers, W.; Liu, Y.; Zhao, W.; Fu, C.; Ding, Y.; Huang, C.; et al. CD82 restrains pathological angiogenesis by altering lipid raft clustering and CD44 trafficking in endothelial cells. Circulation 2014, 130, 1493–1504. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Cheng, Y.; Martinka, M.; Ong, C.J.; Li, G. Prognostic significance of KAI1/CD82 in human melanoma and its role in cell migration and invasion through the regulation of ING4. Carcinogenesis 2014, 35, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Byun, H.J.; Lee, J.; Jeoung, D.I.; Kim, Y.M.; Lee, H. Tetraspanin CD82 represses Sp1-mediated Snail expression and the resultant E-cadherin expression interrupts nuclear signaling of β-catenin by increasing its membrane localization. Cell. Signal. 2018, 52, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Marsters, S.; Ye, X.; Luis, E.; Gonzalez, L.; Ashkenazi, A. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol. Cell 2014, 54, 987–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.C.; Ha, Y.J.; Tak, K.H.; Roh, S.A.; Kwon, Y.H.; Kim, C.W.; Yoon, Y.S.; Lee, J.L.; Park, Y.; Kim, S.K.; et al. Opposite functions of GSN and OAS2 on colorectal cancer metastasis, mediating perineural and lymphovascular invasion, respectively. PLoS ONE 2018, 13, e0202856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raingeaud, J.; Whitmarsh, A.J.; Barrett, T.; Dérijard, B.; Davis, R.J. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 1996, 16, 1247–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeusgen, W.; Tueffers, L.; Herdegen, T.; Waetzig, V. Map2k4δ-identification and functional characterization of a novel Map2k4 splice variant. Biochim. Biophys. Acta 2014, 1843, 875–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Mchichi, B.; Hadji, A.; Vazquez, A.; Leca, G. p38 MAPK and MSK1 mediate caspase-8 activation in manganese-induced mitochondria-dependent cell death. Cell Death Differ. 2007, 14, 1826–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shintani, Y.; Fukumoto, Y.; Chaika, N.; Svoboda, R.; Wheelock, M.J.; Johnson, K.R. Collagen I-mediated up-regulation of N-cadherin requires cooperative signals from integrins and discoidin domain receptor 1. J. Cell Biol. 2008, 180, 1277–1289. [Google Scholar] [CrossRef] [Green Version]
- Ventura, J.J.; Tenbaum, S.; Perdiguero, E.; Huth, M.; Guerra, C.; Barbacid, M.; Pasparakis, M.; Nebreda, A.R. p38alpha MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat. Genet. 2007, 39, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Kristensson, M.; Melander, F.; Leandersson, K.; Rönnstrand, L.; Wernstedt, C.; Andersson, T. p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J. Exp. Med. 2004, 199, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Janknecht, R.; Hunter, T. Convergence of MAP kinase pathways on the ternary complex factor Sap-1a. EMBO J. 1997, 16, 1620–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchison, M.R. Mice with a conditional deletion of the neurotrophin receptor TrkB are dwarfed, and are similar to mice with a MAPK14 deletion. PLoS ONE 2013, 8, e66206. [Google Scholar] [CrossRef]
- McCorkle, J.R.; Leonard, M.K.; Kraner, S.D.; Blalock, E.M.; Ma, D.; Zimmer, S.G.; Kaetzel, D.M. The metastasis suppressor NME1 regulates expression of genes linked to metastasis and patient outcome in melanoma and breast carcinoma. Cancer Genom. Proteom. 2014, 11, 175–194. [Google Scholar]
- Berger, J.C.; Vander Griend, D.J.; Robinson, V.L.; Hickson, J.A.; Rinker-Schaeffer, C.W. Metastasis suppressor genes: From gene identification to protein function and regulation. Cancer Biol. Ther. 2005, 4, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, R.; Gu, M.; Jiang, W.; Lin, W.; Xu, P.; Liu, Z.; Huang, H.; An, H.; Wang, X. Raf Kinase Inhibitor Protein Preferentially Promotes TLR3-Triggered Signaling and Inflammation. J. Immunol. 2017, 198, 4086–4095. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chen, K.; Xing, G.; Li, L.; Ma, B.; Hu, Z.; Duan, L.; Liu, X. Phospholipid remodeling is critical for stem cell pluripotency by facilitating mesenchymal-to-epithelial transition. Sci. Adv. 2019, 5, eaax7525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Takahashi, K.; Huang, B.; Asahara, M.; Kiso, H.; Togo, Y.; Tsukamoto, H.; Mishima, S.; Nagata, M.; Iida, M.; et al. Loss of Stemness, EMT, and Supernumerary Tooth Formation in Cebpb-/-Runx2+/− Murine Incisors. Sci. Rep. 2018, 8, 5169. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Parish, C.R.; Wong, M.L.; Licinio, J.; Blackburn, A.C. Leptin signals via TGFB1 to promote metastatic potential and stemness in breast cancer. PLoS ONE 2017, 12, e0178454. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Leinwand, L.A.; Anseth, K.S. Roles of transforming growth factor-β1 and OB-cadherin in porcine cardiac valve myofibroblast differentiation. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 4551–4562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Su, L.; Liu, X. Loss of CDH1 up-regulates epidermal growth factor receptor via phosphorylation of YBX1 in non-small cell lung cancer cells. FEBS Lett. 2013, 587, 3995–4000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Li, L.; Xu, S.; Liu, Z.; Zhou, C.; Li, Z.; Liu, Y.; Wu, W.; Huang, Y.; Kuang, M.; et al. A Cdh1-FoxM1-Apc axis controls muscle development and regeneration. Cell Death Dis. 2020, 11, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Kajita, M.; Taysavang, P.; Wade, P.A. Hormonal regulation of metastasis-associated protein 3 transcription in breast cancer cells. Mol. Endocrinol. 2004, 18, 2937–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardamone, M.D.; Bardella, C.; Gutierrez, A.; Di Croce, L.; Rosenfeld, M.G.; Di Renzo, M.F.; De Bortoli, M. ERalpha as ligand-independent activator of CDH-1 regulates determination and maintenance of epithelial morphology in breast cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 7420–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Gollahon, L.S. ERα Mediates Estrogen-Induced Expression of the Breast Cancer Metastasis Suppressor Gene BRMS1. Int. J. Mol. Sci. 2016, 17, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Jaye, D.L.; Kajita, M.; Geigerman, C.; Moreno, C.S.; Wade, P.A. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 2003, 113, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Kammerer, M.; Gutzwiller, S.; Stauffer, D.; Delhon, I.; Seltenmeyer, Y.; Fournier, B. Estrogen Receptor α (ERα) and Estrogen Related Receptor α (ERRα) are both transcriptional regulators of the Runx2-I isoform. Mol. Cell. Endocrinol. 2013, 369, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Hovhannisyan, H.; Zhang, Y.; Hassan, M.Q.; Wu, H.; Glackin, C.; Lian, J.B.; Stein, J.L.; Montecino, M.; Stein, G.S.; van Wijnen, A.J. Genomic occupancy of HLH, AP1 and Runx2 motifs within a nuclease sensitive site of the Runx2 gene. J. Cell. Physiol. 2013, 228, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Ye, D.X.; Zhang, W.B.; Pan, H.Y.; Zhang, Z.Y.; Zhang, L. Overexpression of c-fos promotes cell invasion and migration via CD44 pathway in oral squamous cell carcinoma. J. Oral Pathol. Med. Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2015, 44, 353–360. [Google Scholar] [CrossRef]
- Chen, H.J.; Huang, R.L.; Liew, P.L.; Su, P.H.; Chen, L.Y.; Weng, Y.C.; Chang, C.C.; Wang, Y.C.; Chan, M.W.Y.; Lai, H.C. GATA3 as a master regulator and therapeutic target in ovarian high-grade serous carcinoma stem cells. Int. J. Cancer 2018, 143, 3106–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamachary, B.; Penet, M.F.; Nimmagadda, S.; Mironchik, Y.; Raman, V.; Solaiyappan, M.; Semenza, G.L.; Pomper, M.G.; Bhujwalla, Z.M. Hypoxia regulates CD44 and its variant isoforms through HIF-1α in triple negative breast cancer. PLoS ONE 2012, 7, e44078. [Google Scholar] [CrossRef]
- Xue, Z.; Vis, D.J.; Bruna, A.; Sustic, T.; van Wageningen, S.; Batra, A.S.; Rueda, O.M.; Bosdriesz, E.; Caldas, C.; Wessels, L.F.A.; et al. MAP3K1 and MAP2K4 mutations are associated with sensitivity to MEK inhibitors in multiple cancer models. Cell Res. 2018, 28, 719–729. [Google Scholar] [CrossRef]
- Wan, Y.Y. GATA3: A master of many trades in immune regulation. Trends Immunol. 2014, 35, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mayo, M.W.; Nagji, A.S.; Smith, P.W.; Ramsey, C.S.; Li, D.; Jones, D.R. Phosphorylation of RelA/p65 promotes DNMT-1 recruitment to chromatin and represses transcription of the tumor metastasis suppressor gene BRMS1. Oncogene 2012, 31, 1143–1154. [Google Scholar] [CrossRef]
- Lee, S.H.; Che, X.; Jeong, J.H.; Choi, J.Y.; Lee, Y.J.; Lee, Y.H.; Bae, S.C.; Lee, Y.M. Runx2 protein stabilizes hypoxia-inducible factor-1α through competition with von Hippel-Lindau protein (pVHL) and stimulates angiogenesis in growth plate hypertrophic chondrocytes. J. Biol. Chem. 2012, 287, 14760–14771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, N.; Zhang, J.; Chen, S.Y. Runx2, a novel regulator for goblet cell differentiation and asthma development. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Han, H.J.; Russo, J.; Kohwi, Y.; Kohwi-Shigematsu, T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature 2008, 452, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Very, N.; Lefebvre, T.; El Yazidi-Belkoura, I. Drug resistance related to aberrant glycosylation in colorectal cancer. Oncotarget 2018, 9, 1380–1402. [Google Scholar] [CrossRef] [Green Version]
- Beach, S.; Tang, H.; Park, S.; Dhillon, A.S.; Keller, E.T.; Kolch, W.; Yeung, K.C. Snail is a repressor of RKIP transcription in metastatic prostate cancer cells. Oncogene 2008, 27, 2243–2248. [Google Scholar] [CrossRef] [Green Version]
- Vega, S.; Morales, A.V.; Ocaña, O.H.; Valdés, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Spanheimer, P.M.; Askeland, R.W.; Kulak, M.V.; Wu, T.; Weigel, R.J. High TFAP2C/low CD44 expression is associated with an increased rate of pathologic complete response following neoadjuvant chemotherapy in breast cancer. J. Surg. Res. 2013, 184, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; AlTahan, A.; Jones, D.T.; Buffa, F.M.; Bridges, E.; Interiano, R.B.; Qu, C.; Vogt, N.; Li, J.L.; Baban, D.; et al. Estrogen receptor-α directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15172–15177. [Google Scholar] [CrossRef] [Green Version]
- Carr, J.R.; Kiefer, M.M.; Park, H.J.; Li, J.; Wang, Z.; Fontanarosa, J.; DeWaal, D.; Kopanja, D.; Benevolenskaya, E.V.; Guzman, G.; et al. FoxM1 regulates mammary luminal cell fate. Cell Rep. 2012, 1, 715–729. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Ning, Z.; Zhang, H.; Liu, F. Corepressor metastasis-associated protein 3 modulates epithelial-to-mesenchymal transition and metastasis. Chin. J. Cancer 2017, 36, 28. [Google Scholar] [CrossRef] [Green Version]
- Thiruchelvam, P.T.R.; Lai, C.F.; Hua, H.; Thomas, R.S.; Hurtado, A.; Hudson, W.; Bayly, A.R.; Kyle, F.J.; Periyasamy, M.; Photiou, A.; et al. The liver receptor homolog-1 regulates estrogen receptor expression in breast cancer cells. Breast Cancer Res. Treat. 2011, 127, 385–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfahl, M. Nuclear receptor/AP-1 interaction. Endocr. Rev. 1993, 14, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Puig, I.; Caretti, E.; Bonaventure, J.; Nelles, L.; van Roy, F.; Dargemont, C.; de Herreros, A.G.; Bellacosa, A.; Larue, L. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 2007, 26, 7445–7456. [Google Scholar] [CrossRef] [PubMed]
- Woodfield, G.W.; Chen, Y.; Bair, T.B.; Domann, F.E.; Weigel, R.J. Identification of primary gene targets of TFAP2C in hormone responsive breast carcinoma cells. Genes Chromosom. Cancer 2010, 49, 948–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizamtsidi, M.; Nastos, C.; Mastorakos, G.; Dina, R.; Vassiliou, I.; Gazouli, M.; Palazzo, F. Diagnosis, management, histology and genetics of sporadic primary hyperparathyroidism: Old knowledge with new tricks. Endocr. Connect. 2018, 7, R56–R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, P.; Pramesh, C.; Aggarwal, R. Common pitfalls in statistical analysis: Measures of agreement. Perspect. Clin. Res. 2017, 8. [Google Scholar] [CrossRef]
- Ahmed, M.; Kim, D.R. pcr: An R package for quality assessment, analysis and testing of qPCR data. PeerJ 2018, 6, e4473. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2021. [Google Scholar]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- Ruiz Castro, P.A.; Yepiskoposyan, H.; Gubian, S.; Calvino-Martin, F.; Kogel, U.; Renggli, K.; Peitsch, M.C.; Hoeng, J.; Talikka, M. Systems biology approach highlights mechanistic differences between Crohn’s disease and ulcerative colitis. Sci. Rep. 2021, 11, 11519. [Google Scholar] [CrossRef]
- Lo Sasso, G.; Phillips, B.W.; Sewer, A.; Battey, J.N.; Kondylis, A.; Talikka, M.; Titz, B.; Guedj, E.; Peric, D.; Bornand, D.; et al. The reduction of DSS-induced colitis severity in mice exposed to cigarette smoke is linked to immune modulation and microbial shifts. Sci. Rep. 2020, 10, 3829. [Google Scholar] [CrossRef] [Green Version]
- Martin, T.A.; Goyal, A.; Watkins, G.; Jiang, W.G. Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann. Surg. Oncol. 2005, 12, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Barberà, M.J.; Puig, I.; Domínguez, D.; Julien-Grille, S.; Guaita-Esteruelas, S.; Peiró, S.; Baulida, J.; Francí, C.; Dedhar, S.; Larue, L.; et al. Regulation of Snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene 2004, 23, 7345–7354. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Bonavida, B. The activated NF-kappaB-Snail-RKIP circuitry in cancer regulates both the metastatic cascade and resistance to apoptosis by cytotoxic drugs. Crit. Rev. Immunol. 2009, 29, 241–254. [Google Scholar] [CrossRef] [PubMed]
- You, D.J.; Park, C.R.; Lee, H.B.; Moon, M.J.; Kang, J.H.; Lee, C.; Oh, S.H.; Ahn, C.; Seong, J.Y.; Hwang, J.I. A splicing variant of NME1 negatively regulates NF-κB signaling and inhibits cancer metastasis by interacting with IKKβ. J. Biol. Chem. 2014, 289, 17709–17720. [Google Scholar] [CrossRef] [PubMed] [Green Version]








| Category | Genes |
|---|---|
| Cell-cell adhesion | Epican (CD44), Tetraspanin 27 (CD82), Cadherin 11 (CDH11), Cadherin 2 (CDH2), Cadherin 1 (CDH1) and Gelsolin (GSN) |
| Scaffolding | Gravin/a-kinase anchor protein 12 (AKAP12) |
| MAPK | Dual specificity mitogen-activated protein kinase kinase 6 (MAP2K6), 4 (MAP2K4), 7 (MAP2K7) and Mitogen-activated protein kinase 14 (MAPK14) |
| Transcription | NME/NM23 Nucleoside Diphosphate Kinase 1 (NME1) and breast cancer metastasis-suppressor (BRMS1) |
| GTP-binding | Rho GDP Dissociation Inhibitor Beta (ARGHDIB) and Developmentally-regulated GTP-biding protein 1 (DRG1) |
| Other | Ribonucleotide Reductase Catalytic Subunit M1 (RRM1) and Phosphatidylethanolamine-binding protein 1 (PEBP1) |
| TF | Name | Dataset ID | Ref. |
|---|---|---|---|
| ESR1 | Estrogen receptor 1 | GSE10061 | [17] |
| FOS | Fos Proto-Oncogene AP-1 Transcription Factor Subunit | GSE36586 | [18] |
| FOXM1 | Forkhead Box M1 | GSE55204 | [19] |
| GATA3 | GATA Binding Protein 3 | GSE39623 | [20] |
| HIF1A | Hypoxia Inducible Factor 1 Subunit Alpha | GSE3188 | [21] |
| NR5A2 | Nuclear Receptor Subfamily 5 Group A Member 2 | GSE47803 | [22] |
| RARA | Retinoic Acid Receptor Alpha | GSE26298 | [23] |
| SPDEF | SAM Pointed Domain Containing ETS Transcription Factor | GSE40985 | [24] |
| TFAP2C | Transcription Factor AP-2 Gamma | GSE26740 | [25] |
| YBX1 | Y-Box Binding Protein 1 | GSE28433 | [26] |
| ZFX | Zinc Finger Protein X-Linked | ENCSR005AHI | [27] |
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| ESR1 | 5′-TGGAGTCTGGTCCTGTGAGG-3′ | 5′-GGTCTTTTCGTATCCCACCTTTC-3′ |
| SNAI1 | 5′-CCAGTGCCTCGACCACTATG-3′ | 5′-CTGCTGGAAGGTAAACTCTGG-3′ |
| RELA | 5′-CCTATAGAAGAGCAGCGTGGG-3′ | 5′-AGATCTTGAGCTCGGCAGTG-3′ |
| NME1 | 5′-ACTAAGTCAGCCTGGTGTGC-3′ | 5′-CGCCTTGAAAGACGATCCCT-3′ |
| PEBP1 | 5′-GTCACACTTTAGCGGCCTGT-3′ | 5′-CTCTCCGATTATGTGGGCTC-3′ |
| GAPDH | 5′-TGCACCACCAACTGCTTAGC-3′ | 5′-GGCATGGACTGTGGTCATGAG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, M.; Lai, T.H.; Kim, W.; Kim, D.R. A Functional Network Model of the Metastasis Suppressor PEBP1/RKIP and Its Regulators in Breast Cancer Cells. Cancers 2021, 13, 6098. https://doi.org/10.3390/cancers13236098
Ahmed M, Lai TH, Kim W, Kim DR. A Functional Network Model of the Metastasis Suppressor PEBP1/RKIP and Its Regulators in Breast Cancer Cells. Cancers. 2021; 13(23):6098. https://doi.org/10.3390/cancers13236098
Chicago/Turabian StyleAhmed, Mahmoud, Trang Huyen Lai, Wanil Kim, and Deok Ryong Kim. 2021. "A Functional Network Model of the Metastasis Suppressor PEBP1/RKIP and Its Regulators in Breast Cancer Cells" Cancers 13, no. 23: 6098. https://doi.org/10.3390/cancers13236098
APA StyleAhmed, M., Lai, T. H., Kim, W., & Kim, D. R. (2021). A Functional Network Model of the Metastasis Suppressor PEBP1/RKIP and Its Regulators in Breast Cancer Cells. Cancers, 13(23), 6098. https://doi.org/10.3390/cancers13236098