
Sample Preparation Approach Influences PAM50 Risk of Recurrence Score in Early Breast Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
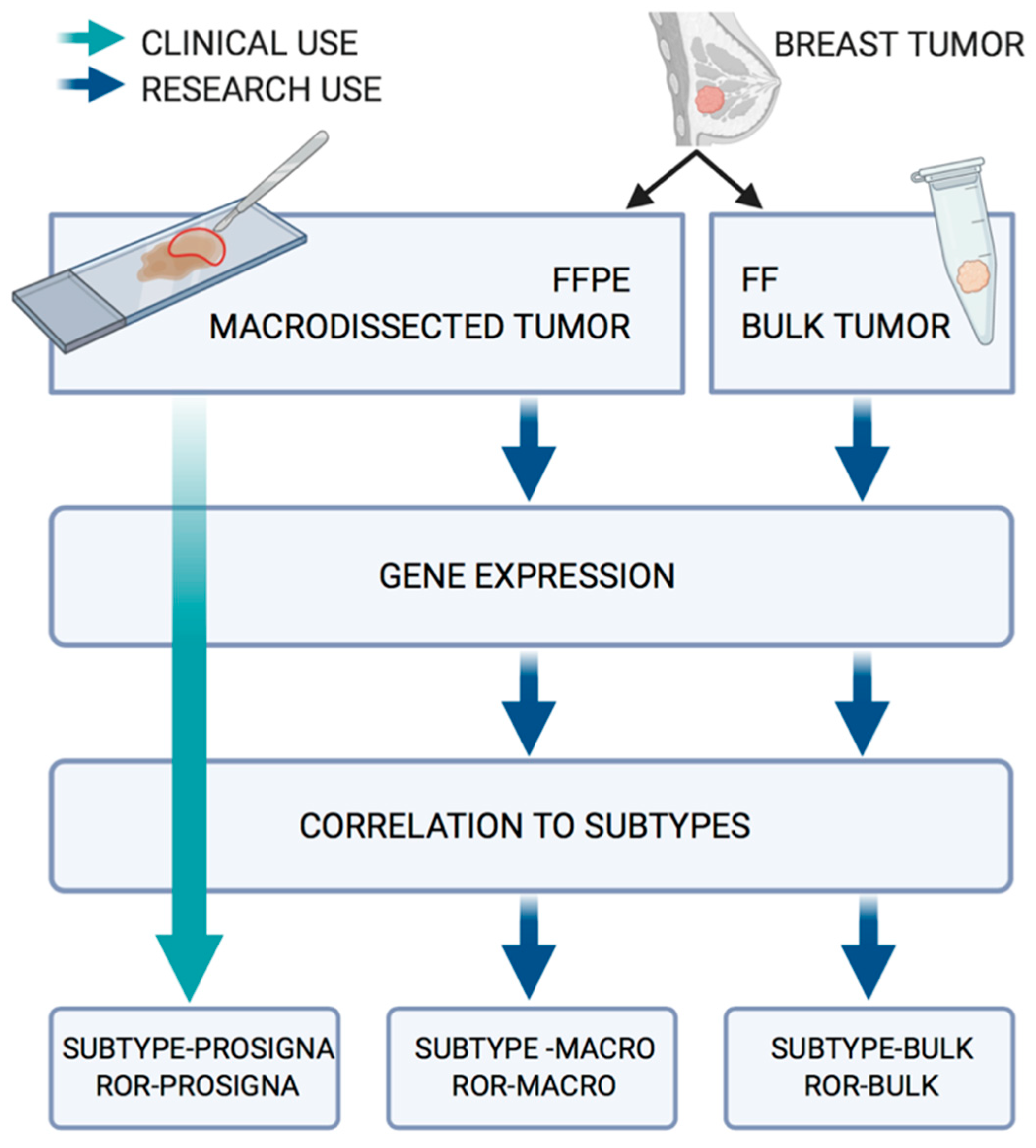
2. Materials and Methods
2.1. Ethics Statement
2.2. Patient and Tumor Characteristics
2.3. RNA Extraction and Gene Expression Analysis from FFPE Tissue
2.4. RNA Extraction and Gene Expression Analysis from FF Tissue
2.5. Gene Centering and Subtype Classification
2.6. Proliferation Score
2.7. Risk of Recurrence Score
2.8. Treatment Recommendation
3. Results
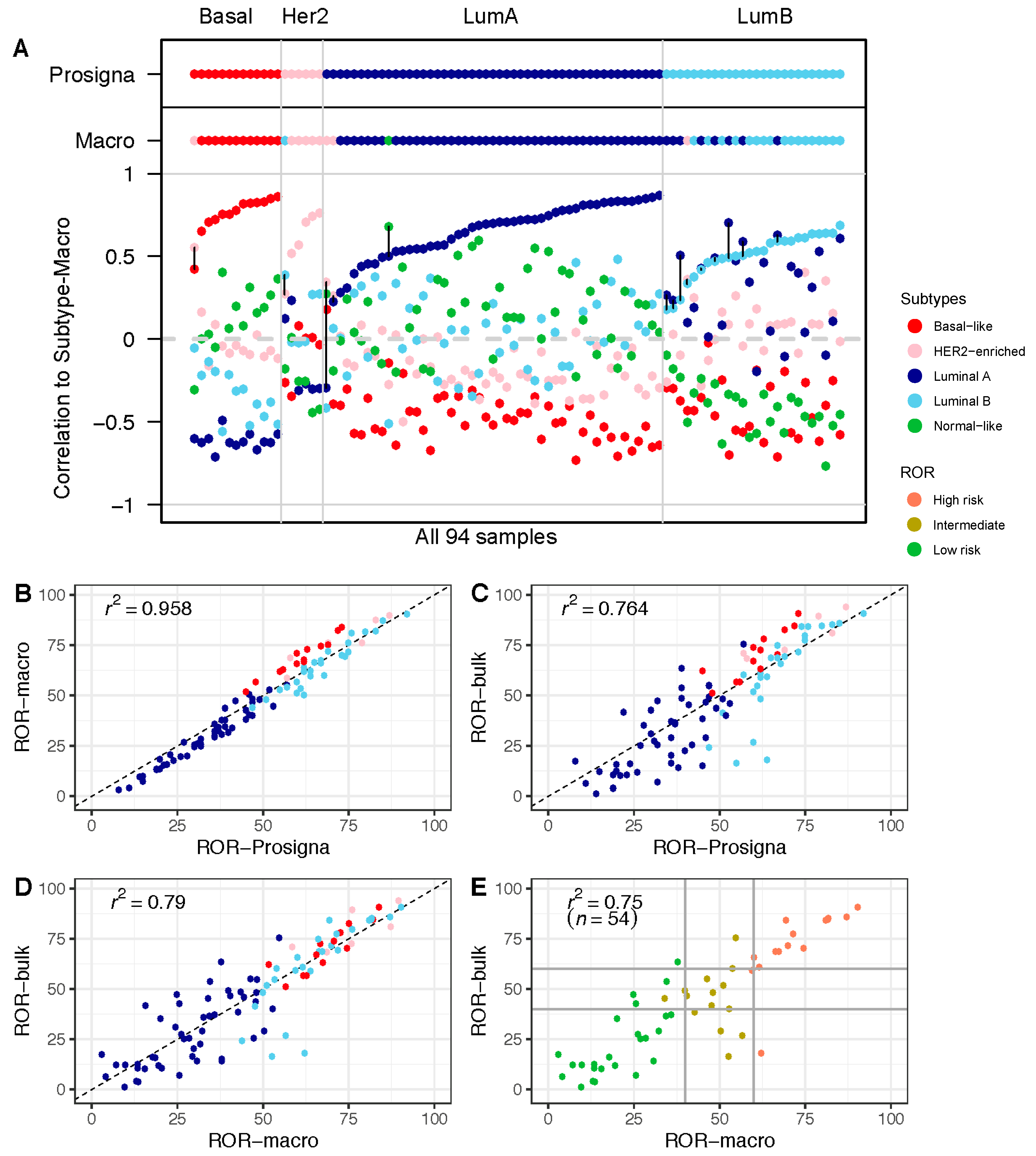
3.1. The Research-Based ROR-Macro Recapitulates the Approved ROR-Prosigna in FFPE Tumor Tissue
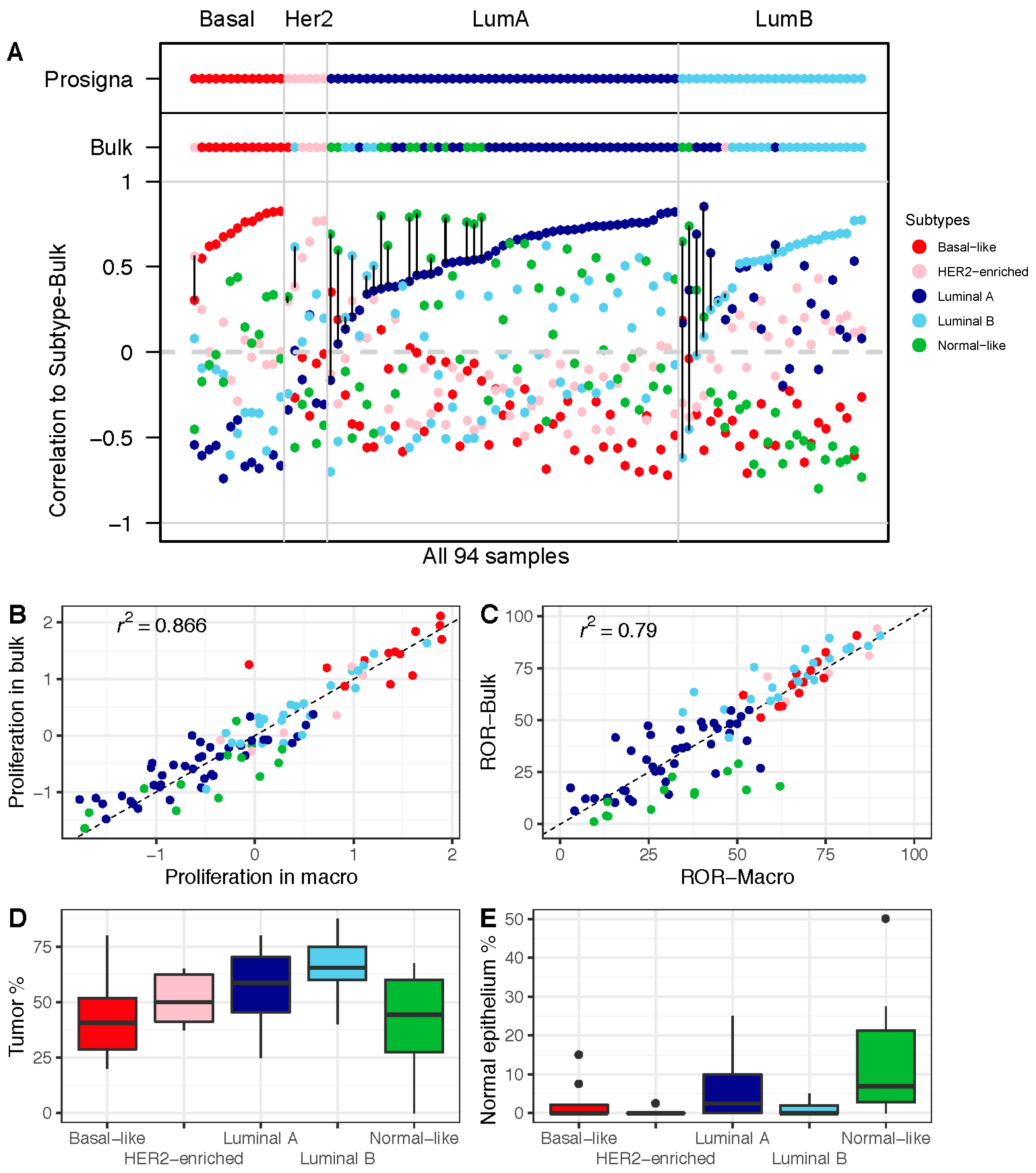
3.2. Comparison of ROR Scores Obtained from Macrodissected FFPE and FF Bulk Tumor Tissue
3.3. Higher Proportion of the Normal-Like Subtype in Data from FF Bulk Tumor Tissue Impacts ROR Score
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gnant, M.; Filipits, M.; Greil, R.; Stoeger, H.; Rudas, M.; Bago-Horvath, Z.; Mlineritsch, B.; Kwasny, W.; Knauer, M.; Singer, C.; et al. Predicting Distant Recurrence in Receptor-Positive Breast Cancer Patients with Limited Clinicopathological Risk: Using the PAM50 Risk of Recurrence Score in 1478 Postmenopausal Patients of the ABCSG-8 Trial Treated with Adjuvant Endocrine Therapy Alone. Ann. Oncol. 2014, 25, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Wallden, B.; Storhoff, J.; Nielsen, T.; Dowidar, N.; Schaper, C.; Ferree, S.; Liu, S.; Leung, S.; Geiss, G.; Snider, J.; et al. Development and Verification of the PAM50-Based Prosigna Breast Cancer Gene Signature Assay. BMC Med. Genom. 2015, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Ohnstad, H.O.; Borgen, E.; Falk, R.S.; Lien, T.G.; Aaserud, M.; Sveli, M.A.T.; Kyte, J.A.; Kristensen, V.N.; Geitvik, G.A.; Schlichting, E.; et al. Prognostic Value of PAM50 and Risk of Recurrence Score in Patients with Early-Stage Breast Cancer with Long-Term Follow-Up. Breast Cancer Res. 2017, 19, 120. [Google Scholar] [CrossRef]
- Jensen, M.B.; Lænkholm, A.V.; Balslev, E.; Buckingham, W.; Ferree, S.; Glavicic, V.; Dupont Jensen, J.; Søegaard Knoop, A.; Mouridsen, H.T.; Nielsen, D.; et al. The Prosigna 50-Gene Profile and Responsiveness to Adjuvant Anthracycline-Based Chemotherapy in High-Risk Breast Cancer Patients. NPJ Breast Cancer 2020, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørile, T.; Eisen, M.B.; van De Rijn, M.; Jeffrey, S.S.; Ress, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Reis, P.P.; Waldron, L.; Goswami, R.S.; Xu, W.; Xuan, Y.; Perez-Ordonez, B.; Gullane, P.; Irish, J.; Jurisica, I.; Kamel-Reid, S. MRNA Transcript Quantification in Archival Samples Using Multiplexed, Color-Coded Probes. BMC Biotechnol. 2011, 11, 46. [Google Scholar] [CrossRef] [Green Version]
- Cieślik, M.; Chinnaiyan, A.M. Cancer Transcriptome Profiling at the Juncture of Clinical Translation. Nat. Rev. Genet. 2018, 19, 93–109. [Google Scholar] [CrossRef]
- Carey, L.A. Through a Glass Darkly: Advances in Understanding Breast Cancer Biology, 2000–2010. Clin. Breast Cancer 2010, 10, 188–195. [Google Scholar] [CrossRef]
- Aure, M.R.; Jernström, S.; Krohn, M.; Vollan, H.K.M.; Due, E.U.; Rødland, E.; Kåresen, R.; Ram, P.; Lu, Y.; Mills, G.B.; et al. Integrated Analysis Reveals MicroRNA Networks Coordinately Expressed with Key Proteins in Breast Cancer. Genome Med. 2015, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekpli, X.; Lien, T.; Røssevold, A.H.; Nebdal, D.; Borgen, E.; Ohnstad, H.O.; Kyte, J.A.; Vallon-Christersson, J.; Fongaard, M.; Due, E.U.; et al. An Independent Poor-Prognosis Subtype of Breast Cancer Defined by a Distinct Tumor Immune Microenvironment. Nat. Commun. 2019, 10, 5499. [Google Scholar] [CrossRef] [Green Version]
- Saal, L.H.; Vallon-Christersson, J.; Häkkinen, J.; Hegardt, C.; Grabau, D.; Winter, C.; Brueffer, C.; Tang, E.H.E.; Reuterswärd, C.; Schulz, R.; et al. The Sweden Cancerome Analysis Network—Breast (SCAN-B) Initiative: A Large-Scale Multicenter Infrastructure towards Implementation of Breast Cancer Genomic Analyses in the Clinical Routine. Genome Med. 2015, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Norsk Bryst Cancer Gruppe. Nasjonalt Handlingsprogram 15. Utgave. Available online: https://www.helsebiblioteket.no/retningslinjer/brystkreft/adjuvant-og-neoadjuvant-systemisk-behandling/oversikt-over-anbefalt-adjuvant-behandling (accessed on 28 December 2020).
- Gendoo, D.M.A.; Ratanasirigulchai, N.; Schroder, M.S.; Pare, L.; Parker, J.S.; Prat, A.; Haibe-Kains, B. Genefu: Computation of Gene Expression-Based Signatures in Breast Cancer. R Package Version 2.6.0. Available online: http://www.bioconductor.org/packages/devel/bioc/html/genefu.html (accessed on 30 October 2021).
- Prat, A.; Perou, C.M. Deconstructing the Molecular Portraits of Breast Cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef]
- Bastien, R.R.; Rodríguez-Lescure, Á.; Ebbert, M.T.; Prat, A.; Munárriz, B.; Rowe, L.; Miller, P.; Ruiz-Borrego, M.; Anderson, D.; Lyons, B.; et al. PAM50 Breast Cancer Subtyping by RT-qPCR and Concordance with Standard Clinical Molecular Markers. BMC Med. Genom. 2012, 5, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elloumi, F.; Hu, Z.; Li, Y.; Parker, J.S.; Gulley, M.L.; Amos, K.D.; Troester, M.A. Systematic Bias in Genomic Classification Due to Contaminating Non-Neoplastic Tissue in Breast Tumor Samples. BMC Med. Genom. 2011, 4, 54. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, T.; Wallden, B.; Schaper, C.; Ferree, S.; Liu, S.; Gao, D.; Barry, G.; Dowidar, N.; Maysuria, M.; Storhoff, J. Analytical Validation of the PAM50-Based Prosigna Breast Cancer Prognostic Gene Signature Assay and NCounter Analysis System Using Formalin-Fixed Paraffin-Embedded Breast Tumor Specimens. BMC Cancer 2014, 14, 177. [Google Scholar] [CrossRef] [Green Version]
- Guedj, M.; Marisa, L.; De Reynies, A.; Orsetti, B.; Schiappa, R.; Bibeau, F.; MacGrogan, G.; Lerebours, F.; Finetti, P.; Longy, M.; et al. A Refined Molecular Taxonomy of Breast Cancer. Oncogene 2012, 31, 1196–1206. [Google Scholar] [CrossRef] [Green Version]
- Picornell, A.C.; Echavarria, I.; Alvarez, E.; López-Tarruella, S.; Jerez, Y.; Hoadley, K.; Parker, J.S.; Del Monte-Millán, M.; Ramos-Medina, R.; Gayarre, J.; et al. Breast Cancer PAM50 Signature: Correlation and Concordance between RNA-Seq and Digital Multiplexed Gene Expression Technologies in a Triple Negative Breast Cancer Series. BMC Genom. 2019, 20, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallon-Christersson, J.; Häkkinen, J.; Hegardt, C.; Saal, L.H.; Larsson, C.; Ehinger, A.; Lindman, H.; Olofsson, H.; Sjöblom, T.; Wärnberg, F.; et al. Cross Comparison and Prognostic Assessment of Breast Cancer Multigene Signatures in a Large Population-Based Contemporary Clinical Series. Sci. Rep. 2019, 9, 12184. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| All Patients | 94 (%) | 54 (%) |
|---|---|---|
| Prosigna subtype | ||
| Basal-like | 13 (14%) | 1 (2%) |
| HER2-enriched | 6 (6%) | |
| Luminal A | 49 (52%) | 34 (63%) |
| Luminal B | 26 (28%) | 19 (35%) |
| T status | ||
| T1b | 12 (13%) | 9 (17%) |
| T1c | 46 (49%) | 28 (52%) |
| T2 | 32 (34%) | 15 (28%) |
| T3 | 3 (3%) | 2 (4%) |
| T4 | 1 (1%) | |
| N status | ||
| pN0 | 64 (68%) | 54 (100%) |
| pN1 | 25 (27%) | |
| pN2 | 5 (5%) | |
| Histological grade | ||
| I | 17 (18%) | 12 (22%) |
| II | 46 (49%) | 31 (57%) |
| III | 31 (33%) | 11 (20%) |
| HER2 status | ||
| Positive | 6 (6%) | |
| Negative | 82 (87%) | 50 (93%) |
| Missing | 6 (6%) | 4 (7%) * |
| Ki67 | ||
| < 15% | 12 (13%) | 11 (20%) |
| 15–30% | 25 (27%) | 16 (30%) |
| ≥ 30% | 56 (60%) | 26 (48%) |
| Missing | 1 (1%) | 1 (2%) |
| Histological subtype | ||
| Ductal | 61 (65%) | 34 (63%) |
| Lobular | 12 (13%) | 9 (17%) |
| Other | 21 (22%) | 11 (20%) |
| Sample ID | Subtype | Prosigna | Prosigna (Cat) | ROR- Macro (Cont.) | ROR- Macro (Cat) | ROR- Bulk (Cont.) | ROR- Bulk (Cat) | Systemic Treatment Recommendation; Macro → Bulk | pT | Grade | Ki67 | Histological Subtype |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BC-34 | Luminal A | 39 | Low | 37.87 | Low | 63.56 | High | No adjuvant → Endo | T1b | II | >=30% | Ductal |
| BC-30 | Luminal A | 32 | Low | 24.9 | Low | 47.3 | Inter | No adjuvant → Endo | T1c | I | 15–30% | Ductal |
| BC-35 | Luminal A | 57 | Inter | 54.86 | Inter | 75.53 | High | Endo → Chemo | T2 | II | >=30% | Ductal |
| BC-20 | Luminal A | 39 | Low | 34.69 | Low | 53.77 | Inter | No change | T1c | II | Missing | Ductal |
| BC-85 | Luminal A | 30 | Low | 25.65 | Low | 42.83 | Inter | No change | T1b | II | 15–30% | Ductal |
| BC-72 | Luminal A | 41 | Inter | 34.1 | Low | 45.41 | Inter | No change | T2 | I | 15–30% | Ductal |
| BC-17 | Luminal A | 47 | Inter | 39.94 | Low | 49.07 | Inter | No change | T2 | II | >=30% | Ductal |
| BC-88 | Luminal B | 57 | Inter | 53.93 | Inter | 60.06 | High | Endo → Chemo | T1c | II | 15–30% | Ductal |
| BC-23 | Luminal A | 45 | Inter | 42.71 | Inter | 38.43 | Low | Endo → no adjuvant | T1c | I | 15–30% | Ductal |
| BC-58 | Luminal A | 46 | Inter | 50.42 | Inter | 28.99 | Low | No change | T1c | II | >=30% | Ductal |
| BC-47 | Luminal B | 60 | Inter | 56.66 | Inter | 26.85 | Low | No change | T1c | II | >=30% | Lobular |
| BC-38 | Luminal B | 55 | Inter | 52.69 | Inter | 16.35 | Low | No change | T1c | II | >=30% | Ductal |
| BC-70 | Luminal B | 64 | High | 62.18 | High | 18.12 | Low | Endo → no adjuvant | T1b | II | >=30% | Ductal |
| Subtype Macro | Subtype-Bulk | ||||
|---|---|---|---|---|---|
| Basal-Like | HER2-Enriched | Luminal A | Luminal B | Normal-Like | |
| Basal-like | 12 | ||||
| HER2-enriched | 1 | 6 | 2 | ||
| Luminal A | 39 | 6 | 9 | ||
| Luminal B | 17 | 1 | |||
| Normal-like | 1 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lien, T.G.; Ohnstad, H.O.; Lingjærde, O.C.; Vallon-Christersson, J.; Aaserud, M.; Sveli, M.A.T.; Borg, Å.; OSBREAC, o.b.o.; Garred, Ø.; Borgen, E.; et al. Sample Preparation Approach Influences PAM50 Risk of Recurrence Score in Early Breast Cancer. Cancers 2021, 13, 6118. https://doi.org/10.3390/cancers13236118
Lien TG, Ohnstad HO, Lingjærde OC, Vallon-Christersson J, Aaserud M, Sveli MAT, Borg Å, OSBREAC obo, Garred Ø, Borgen E, et al. Sample Preparation Approach Influences PAM50 Risk of Recurrence Score in Early Breast Cancer. Cancers. 2021; 13(23):6118. https://doi.org/10.3390/cancers13236118
Chicago/Turabian StyleLien, Tonje G., Hege Oma Ohnstad, Ole Christian Lingjærde, Johan Vallon-Christersson, Marit Aaserud, My Anh Tu Sveli, Åke Borg, on behalf of OSBREAC, Øystein Garred, Elin Borgen, and et al. 2021. "Sample Preparation Approach Influences PAM50 Risk of Recurrence Score in Early Breast Cancer" Cancers 13, no. 23: 6118. https://doi.org/10.3390/cancers13236118
APA StyleLien, T. G., Ohnstad, H. O., Lingjærde, O. C., Vallon-Christersson, J., Aaserud, M., Sveli, M. A. T., Borg, Å., OSBREAC, o. b. o., Garred, Ø., Borgen, E., Naume, B., Russnes, H., & Sørlie, T. (2021). Sample Preparation Approach Influences PAM50 Risk of Recurrence Score in Early Breast Cancer. Cancers, 13(23), 6118. https://doi.org/10.3390/cancers13236118