
The Hippo Signaling Pathway in Cancer: A Cell Cycle Perspective



Abstract
:Simple Summary
Abstract
1. Introduction
1.1. The Cell Cycle and Its Dysregulation in Cancer
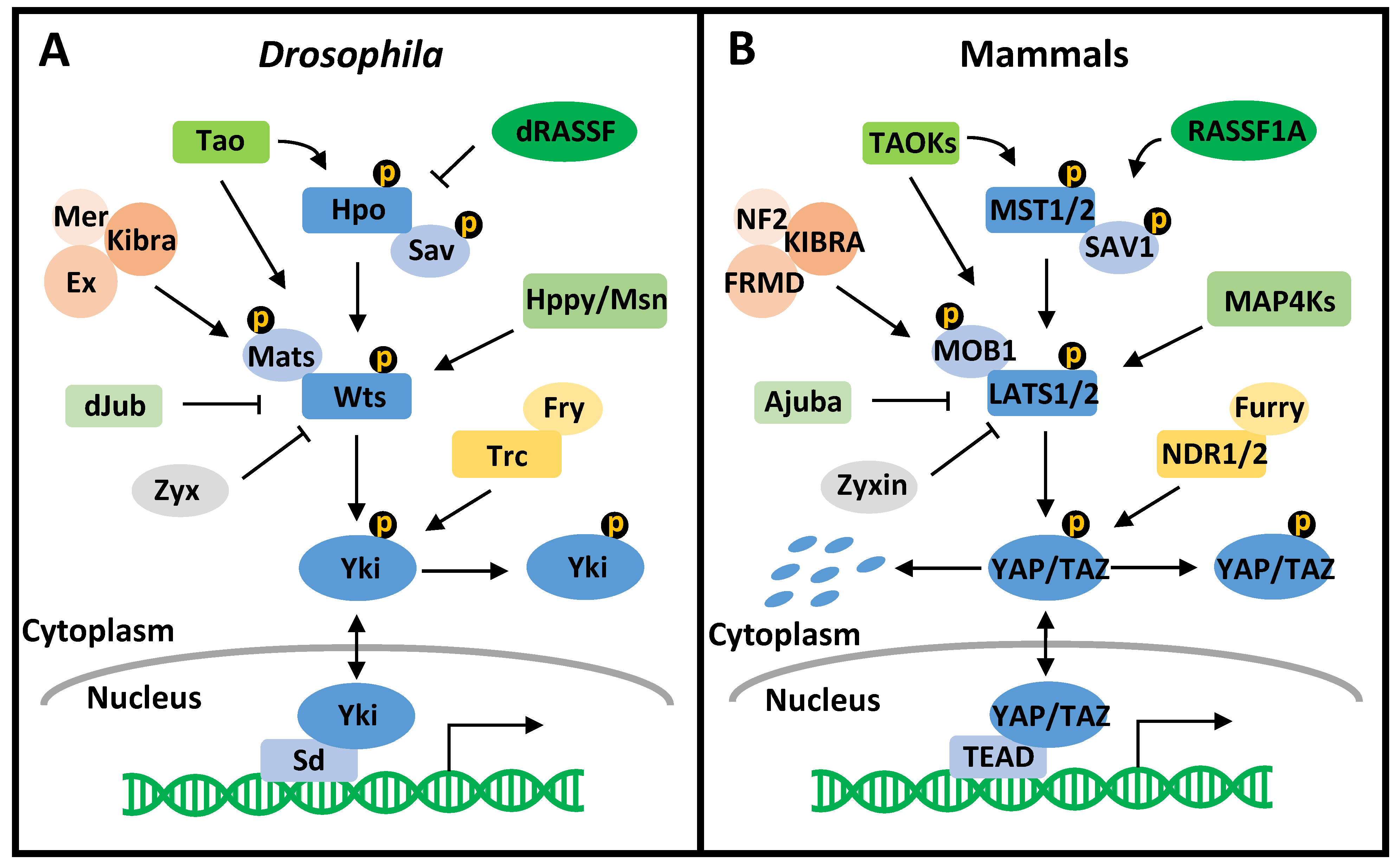
1.2. The Hippo Pathway and Its Dysregulation in Cancer
2. The Hippo Pathway in Mitosis: From Yeast and Fly to Mammal
2.1. The Yeast Hippo Pathway in the Regulation of Mitotic Exit and Cytokinesis
2.1.1. MEN and RAM: The Hippo Pathway of Saccharomyces Cerevisiae
2.1.2. SIN: The Hippo Pathway of Schizosaccharomyces Pombe
2.2. The Conservation of the Drosophila and Mammalian Hippo Pathway in Mitosis
2.2.1. The Drosophila Hippo Pathway in Mitosis
2.2.2. The Mammalian Hippo Pathway in Mitosis
3. The Core of Mammalian Hippo Pathway in Other Phases of the Cell Cycle
3.1. MST1/2 in G1 Tetraploidy Checkpoint, DNA Damage Checkpoint, and Centrosome Dynamics
3.2. SAV1 in G1 Tetraploidy Checkpoint, DNA Damage Checkpoint, and Centrosome Dynamics
3.3. LATS1/2 in E2F Activity, G1 Tetraploidy Checkpoint, DNA Synthesis, DNA Damage Checkpoint, and Centrosome Dynamics
3.4. MOB1A/B in Centrosome Dynamics
3.5. YAP/TAZ in E2F Activity, G1 Tetraploidy Checkpoint, DNA Synthesis, and DNA Damage Checkpoint
4. The Mammalian Hippo Pathway Regulators in Cell Cycle
4.1. TAOKs in DNA Damage Checkpoint and Mitosis
4.2. RASSF1A in E2F Activity, DNA Damage Checkpoint, Centrosome Dynamics, and Mitosis
4.3. KIBRA in DNA Damage Checkpoint and Mitosis
4.4. NF2 in E2F Activity, DNA Damage Checkpoint, Centrosome Dynamics, and Mitosis
4.5. Ajuba in DNA Damage Checkpoint and Mitosis
4.6. Zyxin in DNA Damage Checkpoint and Mitosis
4.7. NDR1/2 in E2F Activity, DNA Damage Checkpoint, and Centrosome Dynamics
5. Conclusions and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Howard, A. Synthesis of deoxyribonucleic acid in normal and irradiated cells and its relation to chromosome breakage. Hered. Suppl. 1953, 6, 261–273. [Google Scholar]
- Ford, H.L.; Pardee, A.B. Cancer and the cell cycle. J. Cell. Biochem. 1999, 75 (Suppl. 32), 166–172. [Google Scholar] [CrossRef]
- Assoian, R.K.; Zhu, X. Cell anchorage and the cytoskeleton as partners in growth factor dependent cell cycle progression. Curr. Opin. Cell Biol. 1997, 9, 93–98. [Google Scholar] [CrossRef]
- Sherr, C.J. D-type cyclins. Trends Biochem. Sci. 1995, 20, 187–190. [Google Scholar] [CrossRef]
- Kato, J.; Matsushime, H.; Hiebert, S.W.; Ewen, M.E.; Sherr, C.J. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993, 7, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Brehm, A.; Miska, E.; McCance, D.J.; Reid, J.L.; Bannister, A.; Kouzarides, T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nat. Cell Biol. 1998, 391, 597–601. [Google Scholar] [CrossRef]
- Pardee, A.B. Molecules involved in proliferation of normal and cancer cells: Presidential address. Cancer Res. 1987, 47, 1488–1491. [Google Scholar]
- Bracken, A.; Ciro, M.; Cocito, A.; Helin, K. E2F target genes: Unraveling the biology. Trends Biochem. Sci. 2004, 29, 409–417. [Google Scholar] [CrossRef]
- Harbour, J.W.; Luo, R.X.; Santi, A.D.; Postigo, A.A.; Dean, D.C. Cdk Phosphorylation Triggers Sequential Intramolecular Interactions that Progressively Block Rb Functions as Cells Move through G1. Cell 1999, 98, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Diffley, J.F. CDKs Promote DNA Replication Origin Licensing in Human Cells by Protecting Cdc6 from APC/C-Dependent Proteolysis. Cell 2005, 122, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, T.N.; Bakkenist, C.J. Regulation of the initiation of DNA replication in human cells. DNA Repair 2018, 72, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Woo, R.A.; Poon, R. Cyclin-Dependent Kinases and S Phase Control in Mammalian Cells. Cell Cycle 2003, 2, 315–323. [Google Scholar] [CrossRef]
- Pagano, M.; Pepperkok, R.; Verde, F.; Ansorge, W.; Draetta, G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992, 11, 961–971. [Google Scholar] [CrossRef]
- Fung, T.K.; Ma, H.T.; Poon, R.Y. Specialized Roles of the Two Mitotic Cyclins in Somatic Cells: Cyclin A as an Activator of M Phase–promoting Factor. Mol. Biol. Cell 2007, 18, 1861–1873. [Google Scholar] [CrossRef]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Molinari, M. Cell cycle checkpoints and their inactivation in human cancer. Cell Prolif. 2000, 33, 261–274. [Google Scholar] [CrossRef]
- Sherr, C.J.; Bartek, J. Cell Cycle–Targeted Cancer Therapies. Annu. Rev. Cancer Biol. 2017, 1, 41–57. [Google Scholar] [CrossRef]
- Bharadwaj, R.; Yu, H. The spindle checkpoint, aneuploidy, and cancer. Oncogene 2004, 23, 2016–2027. [Google Scholar] [CrossRef] [Green Version]
- Margolis, R.L.; Lohez, O.D.; Andreassen, P.R. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J. Cell. Biochem. 2003, 88, 673–683. [Google Scholar] [CrossRef]
- Malumbres, M.; Carnero, A. Cell cycle deregulation: A common motif in cancer. Prog. Cell Cycle Res. 2003, 5, 5–18. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- McDonald, E.R., 3rd; El-Deiry, W.S. Checkpoint genes in cancer. Ann. Med. 2001, 33, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Haraoka, S.; Yoshioka, S.; Hamasaki, M.; Fujiki, T.; Suzumiya, J.; Kawasaki, C.; Kanda, M.; Kikuchi, M. Mutation analysis of mitotic checkpoint genes (hBUB1 and hBUBR1) and microsatellite instability in adult T-cell leukemia/lymphoma. Cancer Lett. 2000, 158, 141–150. [Google Scholar] [CrossRef]
- Suski, J.M.; Braun, M.; Strmiska, V.; Sicinski, P. Targeting cell-cycle machinery in cancer. Cancer Cell 2021, 39, 759–778. [Google Scholar] [CrossRef]
- Whittaker, S.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 879. [Google Scholar] [CrossRef]
- Yu, F.-X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Huang, J.; Dong, J.; Pan, D. hippo Encodes a Ste-20 Family Protein Kinase that Restricts Cell Proliferation and Promotes Apoptosis in Conjunction with salvador and warts. Cell 2003, 114, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Tapon, N.; Harvey, K.; Bell, D.W.; Wahrer, D.C.; Schiripo, T.A.; Haber, D.A.; Hariharan, I.K. salvador Promotes Both Cell Cycle Exit and Apoptosis in Drosophila and Is Mutated in Human Cancer Cell Lines. Cell 2002, 110, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Z.-C.; Wei, X.; Shimizu, T.; Ramos, E.; Rohrbaugh, M.; Nikolaidis, N.; Ho, L.-L.; Li, Y. Control of Cell Proliferation and Apoptosis by Mob as Tumor Suppressor, Mats. Cell 2005, 120, 675–685. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo Signaling Pathway Coordinately Regulates Cell Proliferation and Apoptosis by Inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callus, B.A.; Verhagen, A.M.; Vaux, D. Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C-terminal coiled-coil domains, leads to its stabilization and phosphorylation. FEBS J. 2006, 273, 4264–4276. [Google Scholar] [CrossRef]
- Furth, N.; Aylon, Y. The LATS1 and LATS2 tumor suppressors: Beyond the Hippo pathway. Cell Death Differ. 2017, 24, 1488–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabuta, N.; Fujii, T.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Nishiguchi, H.; Endo, Y.; Toji, S.; Tanaka, H.; Nishimune, Y.; et al. Structure, Expression, and Chromosome Mapping of LATS2, a Mammalian Homologue of the Drosophila Tumor Suppressor Gene lats/warts. Genomics 2000, 63, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Praskova, M.; Xia, F.; Avruch, J. MOBKL1A/MOBKL1B Phosphorylation by MST1 and MST2 Inhibits Cell Proliferation. Curr. Biol. 2008, 18, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a Universal Size-Control Mechanism in Drosophila and Mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Lei, Q.-Y.; Zhang, H.; Zhao, B.; Zha, Z.; Bai, F.; Pei, X.-H.; Zhao, S.; Xiong, Y.; Guan, K.-L. TAZ Promotes Cell Proliferation and Epithelial-Mesenchymal Transition and Is Inhibited by the Hippo Pathway. Mol. Cell. Biol. 2008, 28, 2426–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.-X.; Guan, K.-L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Moroishi, T.; Guan, K.-L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.-L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [Green Version]
- Poon, C.L.; Lin, J.I.; Zhang, X.; Harvey, K. The Sterile 20-like Kinase Tao-1 Controls Tissue Growth by Regulating the Salvador-Warts-Hippo Pathway. Dev. Cell 2011, 21, 896–906. [Google Scholar] [CrossRef] [Green Version]
- Plouffe, S.W.; Meng, Z.; Lin, K.C.; Lin, B.; Hong, A.W.; Chun, J.V.; Guan, K.-L. Characterization of Hippo Pathway Components by Gene Inactivation. Mol. Cell 2016, 64, 993–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.J.; Lee, K.-K.; Song, S.J.; Jin, M.S.; Song, M.S.; Lee, J.H.; Im, C.R.; Lee, J.-O.; Yonehara, S.; Lim, D.-S. Role of the Tumor Suppressor RASSF1A in Mst1-Mediated Apoptosis. Cancer Res. 2006, 66, 2562–2569. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Tommasi, S.; Liu, L.; Yee, J.-K.; Dammann, R.; Pfeifer, G.P. RASSF1A Is Part of a Complex Similar to the Drosophila Hippo/Salvador/Lats Tumor-Suppressor Network. Curr. Biol. 2007, 17, 700–705. [Google Scholar] [CrossRef] [Green Version]
- Polesello, C.; Huelsmann, S.; Brown, N.H.; Tapon, N. The Drosophila RASSF Homolog Antagonizes the Hippo Pathway. Curr. Biol. 2006, 16, 2459–2465. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Moroishi, T.; Mottier-Pavie, V.; Plouffe, S.W.; Hansen, C.G.; Hong, A.W.; Park, H.W.; Mo, J.-S.; Lu, W.; Lu, S.; et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 2015, 6, 8357. [Google Scholar] [CrossRef]
- Yu, J.; Zheng, Y.; Dong, J.; Klusza, S.; Deng, W.-M.; Pan, D. Kibra Functions as a Tumor Suppressor Protein that Regulates Hippo Signaling in Conjunction with Merlin and Expanded. Dev. Cell 2010, 18, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Chen, Y.; Ji, M.; Dong, J. KIBRA Regulates Hippo Signaling Activity via Interactions with Large Tumor Suppressor Kinases. J. Biol. Chem. 2011, 286, 7788–7796. [Google Scholar] [CrossRef] [Green Version]
- Angus, L.; Moleirinho, S.; Herron, L.; Sinha, A.; Zhang, X.; Niestrata, M.; Dholakia, K.; Prystowsky, M.B.; Harvey, K.F.; Reynolds, P.A.; et al. Willin/FRMD6 expression activates the Hippo signaling pathway kinases in mammals and antagonizes oncogenic YAP. Oncogene 2012, 31, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.; Yu, J.; Zheng, Y.; Chen, Q.; Zhang, N.; Pan, D. Spatial Organization of Hippo Signaling at the Plasma Membrane Mediated by the Tumor Suppressor Merlin/NF2. Cell 2013, 154, 1342–1355. [Google Scholar] [CrossRef] [Green Version]
- Das Thakur, M.; Feng, Y.; Jagannathan, R.; Seppa, M.J.; Skeath, J.B.; Longmore, G.D. Ajuba LIM Proteins Are Negative Regulators of the Hippo Signaling Pathway. Curr. Biol. 2010, 20, 657–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Cheng, H.; Gao, R.; Mu, C.; Chen, L.; Wu, S.; Chen, Q.; Zhu, Y. Zyxin-Siah2–Lats2 axis mediates cooperation between Hippo and TGF-β signalling pathways. Nat. Commun. 2016, 7, 11123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauskolb, C.; Pan, G.; Reddy, B.V.V.G.; Oh, H.; Irvine, K.D. Zyxin Links Fat Signaling to the Hippo Pathway. PLoS Biol. 2011, 9, e1000624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Tang, F.; Terracciano, L.; Hynx, D.; Kohler, R.; Bichet, S.; Hess, D.; Cron, P.; Hemmings, B.A.; Hergovich, A.; et al. NDR Functions as a Physiological YAP1 Kinase in the Intestinal Epithelium. Curr. Biol. 2015, 25, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Irie, K.; Nagai, T.; Mizuno, K. Furry protein suppresses nuclear localization of yes-associated protein (YAP) by activating NDR kinase and binding to YAP. J. Biol. Chem. 2020, 295, 3017–3028. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.-L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [Green Version]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, A.; Varelas, X.; Guan, K.-L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.L. Mitotic Exit and Separation of Mother and Daughter Cells. Genetics 2012, 192, 1165–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visintin, R.; Amon, A. Regulation of the Mitotic Exit Protein Kinases Cdc15 and Dbf2. Mol. Biol. Cell 2001, 12, 2961–2974. [Google Scholar] [CrossRef] [Green Version]
- Rock, J.M.; Lim, D.; Stach, L.; Ogrodowicz, R.W.; Keck, J.M.; Jones, M.H.; Wong, C.C.; Yates, J.R., 3rd; Winey, M.; Smerdon, S.J.; et al. Activation of the yeast Hippo pathway by phosphorylation-dependent assembly of signaling complexes. Science 2013, 340, 871–875. [Google Scholar] [CrossRef]
- Mah, A.S.; Jang, J.; Deshaies, R.J. Protein kinase Cdc15 activates the Dbf2-Mob1 kinase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 7325–7330. [Google Scholar] [CrossRef] [Green Version]
- Mohl, D.A.; Huddleston, M.J.; Collingwood, T.S.; Annan, R.S.; Deshaies, R.J. Dbf2–Mob1 drives relocalization of protein phosphatase Cdc14 to the cytoplasm during exit from mitosis. J. Cell Biol. 2009, 184, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Visintin, R.; Craig, K.; Hwang, E.S.; Prinz, S.; Tyers, M.; Amon, A. The Phosphatase Cdc14 Triggers Mitotic Exit by Reversal of Cdk-Dependent Phosphorylation. Mol. Cell 1998, 2, 709–718. [Google Scholar] [CrossRef]
- Tzeng, Y.-W.; Huang, J.N.; Schuyler, S.C.; Wu, C.-H.; Juang, Y.-L. Functions of the mitotic B-type cyclins CLB1, CLB2, and CLB3 at mitotic exit antagonized by the CDC14 phosphatase. Fungal Genet. Biol. 2011, 48, 966–978. [Google Scholar] [CrossRef]
- Bremmer, S.C.; Hall, H.; Martinez, J.S.; Eissler, C.L.; Hinrichsen, T.H.; Rossie, S.; Parker, L.L.; Hall, M.C.; Charbonneau, H. Cdc14 Phosphatases Preferentially Dephosphorylate a Subset of Cyclin-dependent kinase (Cdk) Sites Containing Phosphoserine. J. Biol. Chem. 2012, 287, 1662–1669. [Google Scholar] [CrossRef] [Green Version]
- Jaspersen, S.L.; Morgan, D. Cdc14 activates Cdc15 to promote mitotic exit in budding yeast. Curr. Biol. 2000, 10, 615–618. [Google Scholar] [CrossRef] [Green Version]
- Mehellou, Y.; Alessi, D.R.; Macartney, T.J.; Szklarz, M.; Knapp, S.; Elkins, J.M. Structural insights into the activation of MST3 by MO25. Biochem. Biophys. Res. Commun. 2013, 431, 604–609. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.; Weiss, E.L. Cell cycle regulated interaction of a yeast Hippo kinase and its activator MO25/Hym1. PLoS ONE 2013, 8, e78334. [Google Scholar] [CrossRef] [Green Version]
- Nelson, B.; Kurischko, C.; Horecka, J.; Mody, M.; Nair, P.; Pratt, L.; Zougman, A.; McBroom, L.D.; Hughes, T.R.; Boone, C.; et al. RAM: A Conserved Signaling Network That Regulates Ace2p Transcriptional Activity and Polarized Morphogenesis. Mol. Biol. Cell 2003, 14, 3782–3803. [Google Scholar] [CrossRef] [Green Version]
- Stark, C.; Breitkreutz, B.-J.; Chatr-Aryamontri, A.; Boucher, L.; Oughtred, R.; Livstone, M.S.; Nixon, J.; Van Auken, K.; Wang, X.; Shi, X.; et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011, 39, D698–D704. [Google Scholar] [CrossRef] [Green Version]
- Weiss, E.L.; Kurischko, C.; Zhang, C.; Shokat, K.M.; Drubin, D.G.; Luca, F.C. The Saccharomyces cerevisiae Mob2p–Cbk1p kinase complex promotes polarized growth and acts with the mitotic exit network to facilitate daughter cell–specific localization of Ace2p transcription factor. J. Cell Biol. 2002, 158, 885–900. [Google Scholar] [CrossRef]
- Colman-Lerner, A.; Chin, T.E.; Brent, R. Yeast Cbk1 and Mob2 Activate Daughter-Specific Genetic Programs to Induce Asymmetric Cell Fates. Cell 2001, 107, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Mazanka, E.; Alexander, J.; Yeh, B.J.; Charoenpong, P.; Lowery, D.M.; Yaffe, M.; Weiss, E.L. The NDR/LATS Family Kinase Cbk1 Directly Controls Transcriptional Asymmetry. PLoS Biol. 2008, 6, e203. [Google Scholar] [CrossRef] [PubMed]
- Doolin, M.-T.; Johnson, A.L.; Johnston, L.H.; Butler, G. Overlapping and distinct roles of the duplicated yeast transcription factors Ace2p and Swi5p. Mol. Microbiol. 2001, 40, 422–432. [Google Scholar] [CrossRef]
- Ray, S.; Kume, K.; Gupta, S.; Ge, W.; Balasubramanian, M.; Hirata, D.; McCollum, D. The mitosis-to-interphase transition is coordinated by cross talk between the SIN and MOR pathways in Schizosaccharomyces pombe. J. Cell Biol. 2010, 190, 793–805. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Mccollum, D. Crosstalk between NDR kinase pathways coordinates cell cycle dependent actin rearrangements. Cell Div. 2011, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krapp, A.; Simanis, V. An overview of the fission yeast septation initiation network (SIN). Biochem. Soc. Trans. 2008, 36, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.; Sohrmann, M.; Hofmann, K.; Woollard, A.; Simanis, V. The Spg1p GTPase is an essential, dosage-dependent inducer of septum formation in Schizosaccharomyces pombe. Genes Dev. 1997, 11, 1519–1534. [Google Scholar] [CrossRef] [Green Version]
- Bardin, A.J.; Amon, A. Men and sin: What’s the difference? Nature reviews. Mol. Cell Biol. 2001, 2, 815–826. [Google Scholar]
- Sohrmann, M.; Schmidt, S.; Hagan, I.; Simanis, V. Asymmetric segregation on spindle poles of the Schizosaccharomyces pombe septum-inducing protein kinase Cdc7p. Genes Dev. 1998, 12, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, J.A.; Tomlin, G.C.; McDonald, W.H.; Snydsman, B.E.; Muller, E.G.; Yates, J.R., 3rd; Gould, K.L. Ppc89 links multiple proteins, including the septation initiation network, to the core of the fission yeast spindle-pole body. Mol. Biol. Cell 2006, 17, 3793–3805. [Google Scholar] [CrossRef] [Green Version]
- Krapp, A.; Schmidt, S.; Cano, E.; Simanis, V.S. pombe cdc11p, together with sid4p, provides an anchor for septation initiation network proteins on the spindle pole body. Curr. Biol. 2001, 11, 1559–1568. [Google Scholar] [CrossRef] [Green Version]
- Morrell-Falvey, J.; Tomlin, G.C.; Rajagopalan, S.; Venkatram, S.; Feoktistova, A.S.; Tasto, J.J.; Mehta, S.; Jennings, J.L.; Link, A.; Balasubramanian, M.; et al. Sid4p-Cdc11p Assembles the Septation Initiation Network and Its Regulators at the S. pombe SPB. Curr. Biol. 2004, 14, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guertin, D.A.; Chang, L.; Irshad, F.; Gould, K.L.; Mccollum, D. The role of the Sid1p kinase and Cdc14p in regulating the onset of cytokinesis in fission yeast. EMBO J. 2000, 19, 1803–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, C.A.; Morphew, M.; Mccollum, D. Sid2p, a Spindle Pole Body Kinase That Regulates the Onset of Cytokinesis. J. Cell Biol. 1999, 146, 777–790. [Google Scholar] [CrossRef] [Green Version]
- Hou, M.-C.; Salek, J.; McCollum, D. Mob1p interacts with the Sid2p kinase and is required for cytokinesis in fission yeast. Curr. Biol. 2000, 10, 619–622. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-T.; Feoktistova, A.; Chen, J.-S.; Shim, Y.-S.; Clifford, D.M.; Gould, K.L.; McCollum, D. The SIN Kinase Sid2 Regulates Cytoplasmic Retention of the S. pombe Cdc14-like Phosphatase Clp1. Curr. Biol. 2008, 18, 1594–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trautmann, S.; Wolfe, B.A.; Jorgensen, P.; Tyers, M.; Gould, K.L.; McCollum, D. Fission yeast Clp1p phosphatase regulates G2/M transition and coordination of cytokinesis with cell cycle progression. Curr. Biol. 2001, 11, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, B.A.; Gould, K.L. Fission yeast Clp1p phosphatase affects G2/M transition and mitotic exit through Cdc25p inactivation. EMBO J. 2004, 23, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Willet, A.; DeWitt, A.K.; Beckley, J.R.; Clifford, D.M.; Gould, K.L. NDR Kinase Sid2 Drives Anillin-like Mid1 from the Membrane to Promote Cytokinesis and Medial Division Site Placement. Curr. Biol. 2019, 29, 1055–1063.e2. [Google Scholar] [CrossRef] [Green Version]
- Clifford, D.M.; Wolfe, B.A.; Roberts-Galbraith, R.H.; McDonald, W.H.; Yates, J.R., 3rd; Gould, K.L. The Clp1/Cdc14 phosphatase contributes to the robustness of cytokinesis by association with anillin-related Mid1. J. Cell Biol. 2008, 181, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knockleby, J.; Kim, B.J.; Mehta, A.; Lee, H. Cdk1-mediated phosphorylation of Cdc7 suppresses DNA re-replication. Cell Cycle 2016, 15, 1494–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feoktistova, A.; Morrell-Falvey, J.; Chen, J.-S.; Singh, N.S.; Balasubramanian, M.K.; Gould, K.L. The fission yeast septation initiation network (SIN) kinase, Sid2, is required for SIN asymmetry and regulates the SIN scaffold, Cdc11. Mol. Biol. Cell 2012, 23, 1636–1645. [Google Scholar] [CrossRef]
- Park, B.H.; Lee, Y.H. Phosphorylation of SAV1 by mammalian ste20-like kinase promotes cell death. BMB Rep. 2011, 44, 584–589. [Google Scholar] [CrossRef] [Green Version]
- Bettencourt-Dias, M.; Giet, R.; Sinka, R.; Mazumdar, A.; Lock, W.G.; Balloux, F.; Zafiropoulos, P.J.; Yamaguchi, S.; Winter, S.; Carthew, R.W.; et al. Genome-wide survey of protein kinases required for cell cycle progression. Nat. Cell Biol. 2004, 432, 980–987. [Google Scholar] [CrossRef]
- Rieder, C.L.; Maiato, H. Stuck in Division or Passing through: What Happens When Cells Cannot Satisfy the Spindle Assembly Checkpoint. Dev. Cell 2004, 7, 637–651. [Google Scholar] [CrossRef] [Green Version]
- Dewey, E.B.; Sanchez, D.; Johnston, C.A. Warts Phosphorylates Mud to Promote Pins-Mediated Mitotic Spindle Orientation in Drosophila, Independent of Yorkie. Curr. Biol. 2015, 25, 2751–2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simcox, A.; Mitra, S.; Truesdell, S.; Paul, L.; Chen, T.; Butchar, J.P.; Justiniano, S. Efficient Genetic Method for Establishing Drosophila Cell Lines Unlocks the Potential to Create Lines of Specific Genotypes. PLoS Genet. 2008, 4, e1000142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2018, 19, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Ho, L.-L.; Lai, Z.-C. The mob as tumor suppressor Gene Is Essential for Early Development and Regulates Tissue Growth in Drosophila. Genetics 2008, 178, 957–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, S.L.; Shandala, T.; O’Keefe, L.; Jones, L.; Murray, M.J.; Saint, R. A Drosophila overexpression screen for modifiers of Rho signalling in cytokinesis. Fly 2007, 1, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Jahanshahi, M.; Hsiao, K.; Jenny, A.; Pfleger, C.M. The Hippo Pathway Targets Rae1 to Regulate Mitosis and Organ Size and to Feed Back to Regulate Upstream Components Merlin, Hippo, and Warts. PLoS Genet. 2016, 12, e1006198. [Google Scholar] [CrossRef] [Green Version]
- Blower, M.D.; Nachury, M.; Heald, R.; Weis, K. A Rae1-Containing Ribonucleoprotein Complex Is Required for Mitotic Spindle Assembly. Cell 2005, 121, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Nagai, T.; Mizuno, K. Multifaceted roles of Furry proteins in invertebrates and vertebrates. J. Biochem. 2014, 155, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Manning, S.; Dent, L.G.; Kondo, S.; Zhao, Z.W.; Plachta, N.; Harvey, K.F. Dynamic Fluctuations in Subcellular Localization of the Hippo Pathway Effector Yorkie In Vivo. Curr. Biol. 2018, 28, 1651–1660.e4. [Google Scholar] [CrossRef] [Green Version]
- Galli, G.; Carrara, M.; Yuan, W.-C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; de Laat, W.; et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol. Cell 2015, 60, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Liang, K.; Woodfin, A.R.; Slaughter, B.D.; Unruh, J.; Box, A.C.; Rickels, R.A.; Gao, X.; Haug, J.S.; Jaspersen, S.L.; Shilatifard, A. Mitotic Transcriptional Activation: Clearance of Actively Engaged Pol II via Transcriptional Elongation Control in Mitosis. Mol. Cell 2015, 60, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.J.; Kim, M.J.; Song, S.J.; Kim, T.; Lee, D.; Kwon, S.-H.; Choi, E.-J.; Lim, D.-S. MST1 Limits the Kinase Activity of Aurora B to Promote Stable Kinetochore-Microtubule Attachment. Curr. Biol. 2010, 20, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S.; Ikeda, M.; Katsunuma, K.; Ohashi, K.; Mizuno, K. MST2- and Furry-Mediated Activation of NDR1 Kinase Is Critical for Precise Alignment of Mitotic Chromosomes. Curr. Biol. 2009, 19, 675–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Chen, Y.; Dong, J. MST2 phosphorylation at serine 385 in mitosis inhibits its tumor suppressing activity. Cell. Signal. 2016, 28, 1826–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardin, B.R.; Lange, C.; Baxter, J.E.; Hardy, T.; Scholz, S.R.; Fry, A.M.; Schiebel, E. Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nat. Cell Biol. 2010, 12, 1166–1176. [Google Scholar] [CrossRef] [Green Version]
- Iida, S.-I.; Hirota, T.; Morisaki, T.; Marumoto, T.; Hara, T.; Kuninaka, S.; Honda, S.; Kosai, K.-I.; Kawasuji, M.; Pallas, D.C.; et al. Tumor suppressor WARTS ensures genomic integrity by regulating both mitotic progression and G1 tetraploidy checkpoint function. Oncogene 2004, 23, 5266–5274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aylon, Y.; Michael, D.; Shmueli, A.; Yabuta, N.; Nojima, H.; Oren, M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006, 20, 2687–2700. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Li, D.M.; Chen, W.; Xu, T. Human homologue of Drosophila lats, LATS1, negatively regulate growth by inducing G(2)/M arrest or apoptosis. Oncogene 2001, 20, 6516–6523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Qi, H.; Li, Y.; Pei, J.; Barton, J.; Blackstad, M.; Xu, T.; Tao, W. LATS1 tumor suppressor regulates G2/M transition and apoptosis. Oncogene 2002, 21, 1233–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiyoda, T.; Sugiyama, N.; Shimizu, T.; Naoe, H.; Kobayashi, Y.; Ishizawa, J.; Arima, Y.; Tsuda, H.; Ito, M.; Kaibuchi, K.; et al. LATS1/WARTS phosphorylates MYPT1 to counteract PLK1 and regulate mammalian mitotic progression. J. Cell Biol. 2012, 197, 625–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamikubo, Y.; Takaori-Kondo, A.; Uchiyama, T.; Hori, T. Inhibition of Cell Growth by Conditional Expression of kpm, a Human Homologue of Drosophila warts/lats Tumor Suppressor. J. Biol. Chem. 2003, 278, 17609–17614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, W.; Zhang, S.; Turenchalk, G.S.; Stewart, R.A.; John, M.A.R.S.; Chen, W.; Xu, T. Human homologue of the Drosophila melanogaster lats tumour suppressor modulates CDC2 activity. Nat. Genet. 1999, 21, 177–181. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Hirota, T.; Morisaki, T.; Hara, T.; Marumoto, T.; Iida, S.-I.; Makino, K.; Yamamoto, H.; Hiraoka, T.; Kitamura, N.; et al. A human homolog ofDrosophilawarts tumor suppressor, h-warts, localized to mitotic apparatus and specifically phosphorylated during mitosis. FEBS Lett. 1999, 459, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Morisaki, T.; Hirota, T.; Iida, S.; Marumoto, T.; Hara, T.; Nishiyama, Y.; Kawasuzi, M.; Hiraoka, T.; Mimori, T.; Araki, N.; et al. WARTS tumor suppressor is phosphorylated by Cdc2/cyclin B at spindle poles during mitosis. FEBS Lett. 2002, 529, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Yeung, B.; Khanal, P.; Mehta, V.; Trinkle-Mulcahy, L.; Yang, X. Identification of Cdk1–LATS–Pin1 as a Novel Signaling Axis in Anti-tubulin Drug Response of Cancer Cells. Mol. Cancer Res. 2018, 16, 1035–1045. [Google Scholar] [CrossRef] [Green Version]
- Yabuta, N.; Mukai, S.; Okada, N.; Aylon, Y.; Nojima, H. The tumor suppressor Lats2 is pivotal in Aurora A and Aurora B signaling during mitosis. Cell Cycle 2011, 10, 2724–2736. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Pei, J.; Xia, H.; Ke, H.; Wang, H.; Tao, W. Lats2, a putative tumor suppressor, inhibits G1/S transition. Oncogene 2003, 22, 4398–4405. [Google Scholar] [CrossRef] [Green Version]
- Bothos, J.; Tuttle, R.L.; Ottey, M.; Luca, F.C.; Halazonetis, T.D. Human LATS1 Is a Mitotic Exit Network Kinase. Cancer Res. 2005, 65, 6568–6575. [Google Scholar] [CrossRef] [Green Version]
- Yabuta, N.; Mukai, S.; Okamoto, A.; Okuzaki, D.; Suzuki, H.; Torigata, K.; Yoshida, K.; Okada, N.; Miura, D.; Ito, A.; et al. N-terminal truncation of Lats1 causes abnormal cell growth control and chromosomal instability. J. Cell Sci. 2013, 126, 508–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabuta, N.; Yoshida, K.; Mukai, S.; Kato, Y.; Torigata, K.; Nojima, H. Large tumor suppressors 1 and 2 regulate Aurora-B through phosphorylation of INCENP to ensure completion of cytokinesis. Heliyon 2016, 2, e00131. [Google Scholar] [CrossRef] [Green Version]
- McPherson, J.P.; Tamblyn, L.; Elia, A.; Migon, E.; Shehabeldin, A.; Matysiak-Zablocki, E.; Lemmers, B.; Salmena, L.; Hakem, A.; Fish, J.; et al. Lats2/Kpm is required for embryonic development, proliferation control and genomic integrity. EMBO J. 2004, 23, 3677–3688. [Google Scholar] [CrossRef] [Green Version]
- Yabuta, N.; Okada, N.; Ito, A.; Hosomi, T.; Nishihara, S.; Sasayama, Y.; Fujimori, A.; Okuzaki, D.; Zhao, H.; Ikawa, M.; et al. Lats2 Is an Essential Mitotic Regulator Required for the Coordination of Cell Division. J. Biol. Chem. 2007, 282, 19259–19271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, A.; Yabuta, N.; Mukai, S.; Torigata, K.; Nojima, H. Phosphorylation of CHO1 by Lats1/2 regulates the centrosomal activation of LIMK1 during cytokinesis. Cell Cycle 2015, 14, 1568–1582. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yu, K.; Hao, Y.; Li, D.-M.; Stewart, R.; Insogna, K.L.; Xu, T. LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat. Cell Biol. 2004, 6, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Chu, L.; Qin, B.; Wang, Z.; Liu, X.; Jin, C.; Zhang, G.; Gomez, M.; Hergovich, A.; Chen, Z.; et al. Regulation of NDR1 activity by PLK1 ensures proper spindle orientation in mitosis. Sci. Rep. 2015, 5, 10449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilmeth, L.J.; Shrestha, S.; Montaño, G.; Rashe, J.; Shuster, C.B. Mutual Dependence of Mob1 and the Chromosomal Passenger Complex for Localization during Mitosis. Mol. Biol. Cell 2010, 21, 380–392. [Google Scholar] [CrossRef] [Green Version]
- Adriaans, I.; Hooikaas, P.J.; Aher, A.; Vromans, M.J.; van Es, R.M.; Grigoriev, I.; Akhmanova, A.; Lens, S.M. MKLP2 Is a Motile Kinesin that Transports the Chromosomal Passenger Complex during Anaphase. Curr. Biol. 2020, 30, 2628–2637.e9. [Google Scholar] [CrossRef]
- Florindo, C.; Perdigão, J.; Fesquet, D.; Schiebel, E.; Pines, J.; Tavares, Á.A. Human Mob1 proteins are required for cytokinesis by controlling microtubule stability. J. Cell Sci. 2012, 125, 3085–3090. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Chiba, S.; Ohashi, K.; Mizuno, K. Furry Protein Promotes Aurora A-mediated Polo-like Kinase 1 Activation. J. Biol. Chem. 2012, 287, 27670–27681. [Google Scholar] [CrossRef] [Green Version]
- Nagai, T.; Ikeda, M.; Chiba, S.; Kanno, S.-I.; Mizuno, K. Furry promotes acetylation of microtubules in the mitotic spindle by inhibition of SIRT2 tubulin deacetylase. J. Cell Sci. 2013, 126, 4369–4380. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.; Dubois, F.; Teulier, S.; Martin, A.P.; Levallet, J.; Maille, E.; Brosseau, S.; Elie, N.; Hergovich, A.; Bergot, E.; et al. NDR2 kinase contributes to cell invasion and cytokinesis defects induced by the inactivation of RASSF1A tumor-suppressor gene in lung cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 158. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Monroe, T.; Hill, M.; Morikawa, Y.; Leach, J.; Heallen, T.; Cao, S.; Krijger, P.; de Laat, W.; Wehrens, X.; Rodney, G.; et al. YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In Vivo. Dev. Cell 2019, 48, 765–779.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschuor, C.; Kachaylo, E.; Ungethüm, U.; Song, Z.; Lehmann, K.; Sánchez-Velázquez, P.; Linecker, M.; Kambakamba, P.; Raptis, D.A.; Limani, P.; et al. Yes-associated protein promotes early hepatocyte cell cycle progression in regenerating liver after tissue loss. FASEB BioAdv. 2019, 1, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef] [Green Version]
- Pattschull, G.; Walz, S.; Gründl, M.; Schwab, M.; Rühl, E.; Baluapuri, A.; Cindric-Vranesic, A.; Kneitz, S.; Wolf, E.; Ade, C.P.; et al. The Myb-MuvB Complex Is Required for YAP-Dependent Transcription of Mitotic Genes. Cell Rep. 2019, 27, 3533–3546.e7. [Google Scholar] [CrossRef] [Green Version]
- Weiler, S.M.; Pinna, F.; Wolf, T.; Lutz, T.; Geldiyev, A.; Sticht, C.; Knaub, M.; Thomann, S.; Bissinger, M.; Wan, S.; et al. Induction of Chromosome Instability by Activation of Yes-Associated Protein and Forkhead Box M1 in Liver Cancer. Gastroenterology 2017, 152, 2037–2051.e22. [Google Scholar] [CrossRef]
- Eisinger-Mathason, T.S.K.; Mucaj, V.; Biju, K.; Nakazawa, M.S.; Gohil, M.; Cash, T.P.; Yoon, S.S.; Skuli, N.; Park, K.M.; Gerecht, S.; et al. Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E3402–E3411. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.-J.; Sadanandam, A.; Hu, B.; et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Cho, Y.S.; Wang, X.; Park, O.; Ma, X.; Kim, H.; Gan, W.; Jho, E.H.; Cha, B.; Jeung, Y.J.; et al. Hippo signaling is intrinsically regulated during cell cycle progression by APC/C(Cdh1). Proc. Natl. Acad. Sci. USA 2019, 116, 9423–9432. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Sun, Y.; Wan, G.; Sun, J.; Sun, J.; Pan, C. Knockdown of YAP inhibits growth in Hep-2 laryngeal cancer cells via epithelial-mesenchymal transition and the Wnt/β-catenin pathway. BMC Cancer 2019, 19, 654. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, L.; Liu, M.; Chong, R.; Ding, S.-J.; Chen, Y.; Dong, J. CDK1 Phosphorylation of YAP Promotes Mitotic Defects and Cell Motility and Is Essential for Neoplastic Transformation. Cancer Res. 2013, 73, 6722–6733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Chen, X.; Stauffer, S.; Yang, S.; Chen, Y.; Dong, J. CDK1 phosphorylation of TAZ in mitosis inhibits its oncogenic activity. Oncotarget 2015, 6, 31399–31412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhang, L.; Chen, X.; Chen, Y.; Dong, J. Oncoprotein YAP Regulates the Spindle Checkpoint Activation in a Mitotic Phosphorylation-dependent Manner through Up-regulation of BubR1. J. Biol. Chem. 2015, 290, 6191–6202. [Google Scholar] [CrossRef] [Green Version]
- Bui, D.A.; Lee, W.; White, A.E.; Harper, J.W.; Schackmann, R.C.J.; Overholtzer, M.; Selfors, L.M.; Brugge, J.S. Cytokinesis involves a nontranscriptional function of the Hippo pathway effector YAP. Sci. Signal. 2016, 9, ra23. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Ernsting, B.R.; Wishart, M.J.; Lohse, D.L.; Dixon, J.E. A Family of Putative Tumor Suppressors Is Structurally and Functionally Conserved in Humans and Yeast. J. Biol. Chem. 1997, 272, 29403–29406. [Google Scholar] [CrossRef] [Green Version]
- Rosso, L.; Marques, A.C.; Weier, M.; Lambert, N.; Lambot, M.-A.; Vanderhaeghen, P.; Kaessmann, H. Birth and Rapid Subcellular Adaptation of a Hominoid-Specific CDC14 Protein. PLoS Biol. 2008, 6, e140. [Google Scholar] [CrossRef]
- Cho, H.P.; Liu, Y.; Gomez, M.; Dunlap, J.; Tyers, M.; Wang, Y. The Dual-Specificity Phosphatase CDC14B Bundles and Stabilizes Microtubules. Mol. Cell. Biol. 2005, 25, 4541–4551. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Novelle, M.D.; Esteban, V.; Bueno, A.; Sacristán, M.P. Functional homology among human and fission yeast Cdc14 phosphatases. J. Biol. Chem. 2005, 280, 29144–29150. [Google Scholar] [CrossRef] [Green Version]
- Bembenek, J.; Yu, H. Regulation of the Anaphase-promoting Complex by the Dual Specificity Phosphatase Human Cdc14a. J. Biol. Chem. 2001, 276, 48237–48242. [Google Scholar] [CrossRef] [Green Version]
- Mishima, M.; Pavicic, V.; Gruneberg, U.; Nigg, E.; Glotzer, M. Cell cycle regulation of central spindle assembly. Nat. Cell Biol. 2004, 430, 908–913. [Google Scholar] [CrossRef]
- Zhu, C.; Bossy-Wetzel, E.; Jiang, W. Recruitment of MKLP1 to the spindle midzone/midbody by INCENP is essential for midbody formation and completion of cytokinesis in human cells. Biochem. J. 2005, 389, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, B.K.; Zimmerman, Z.A.; Charbonneau, H.; Jackson, P.K. Disruption of Centrosome Structure, Chromosome Segregation, and Cytokinesis by Misexpression of Human Cdc14A Phosphatase. Mol. Biol. Cell 2002, 13, 2289–2300. [Google Scholar] [CrossRef] [Green Version]
- Tumurbaatar, I.; Cizmecioglu, O.; Hoffmann, I.; Grummt, I.; Voit, R. Human Cdc14B promotes progression through mitosis by dephosphorylating Cdc25 and regulating Cdk1/cyclin B activity. PLoS ONE 2011, 6, e14711. [Google Scholar] [CrossRef] [Green Version]
- Berdougo, E.; Nachury, M.V.; Jackson, P.K.; Jallepalli, P.V. The nucleolar phosphatase Cdc14B is dispensable for chromosome segregation and mitotic exit in human cells. Cell Cycle 2008, 7, 1184–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partscht, P.; Uddin, B.; Schiebel, E. Human cells lacking CDC14A and CDC14B show differences in ciliogenesis but not in mitotic progression. J. Cell Sci. 2021, 134, jcs255950. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Yang, S.; Chen, Y.; Xiao, L.; Zhang, L.; Dong, J. Phospho-regulation of KIBRA by CDK1 and CDC14 phosphatase controls cell-cycle progression. Biochem. J. 2012, 447, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chen, Q.; Liu, Q.; Li, Y.; Sun, X.; Hong, L.; Ji, S.; Liu, C.; Geng, J.; Zhang, W.; et al. Hippo Signaling Suppresses Cell Ploidy and Tumorigenesis through Skp2. Cancer Cell 2017, 31, 669–684.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyberg, K.A.; Michelson, R.J.; Putnam, C.W.; Weinert, T.A. Toward Maintaining the Genome: DNA Damage and Replication Checkpoints. Annu. Rev. Genet. 2002, 36, 617–656. [Google Scholar] [CrossRef] [Green Version]
- Bakhoum, S.F.; Kabeche, L.; Compton, D.A.; Powell, S.N.; Bastians, H. Mitotic DNA Damage Response: At the Crossroads of Structural and Numerical Cancer Chromosome Instabilities. Trends Cancer 2017, 3, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pefani, D.-E.; Latusek, R.; Pires, I.; Grawenda, A.M.; Yee, K.S.; Hamilton, G.; Van Der Weyden, L.; Esashi, F.; Hammond, E.M.; O’Neill, E. RASSF1A–LATS1 signalling stabilizes replication forks by restricting CDK2-mediated phosphorylation of BRCA2. Nat. Cell Biol. 2014, 16, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Pefani, D.E.; Tognoli, M.L.; Ercan, D.P.; Gorgoulis, V.; O’Neill, E. MST2 kinase suppresses rDNA transcription in response to DNA damage by phosphorylating nucleolar histone H2B. EMBO J. 2018, 37, 98760. [Google Scholar] [CrossRef]
- Hamilton, G.; Yee, K.S.; Scrace, S.; O’Neill, E. ATM Regulates a RASSF1A-Dependent DNA Damage Response. Curr. Biol. 2009, 19, 2020–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, F.; Xie, Q.; Wu, J.; Bai, Y.; Mao, B.; Dong, Y.; Bi, W.; Ji, G.; Tao, W.; Wang, Y.; et al. MST1 Promotes Apoptosis through Regulating Sirt1-dependent p53 Deacetylation. J. Biol. Chem. 2011, 286, 6940–6945. [Google Scholar] [CrossRef] [Green Version]
- Bettencourt-Dias, M.; Glover, D. Centrosome biogenesis and function: Centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol. 2007, 8, 451–463. [Google Scholar] [CrossRef]
- Gönczy, P. Centrosomes and cancer: Revisiting a long-standing relationship. Nat. Rev. Cancer 2015, 15, 639–652. [Google Scholar] [CrossRef]
- Hergovich, A.; Kohler, R.S.; Schmitz, D.; Vichalkovski, A.; Cornils, H.; Hemmings, B.A. The MST1 and hMOB1 Tumor Suppressors Control Human Centrosome Duplication by Regulating NDR Kinase Phosphorylation. Curr. Biol. 2009, 19, 1692–1702. [Google Scholar] [CrossRef] [Green Version]
- Karchugina, S.; Benton, D.; Chernoff, J. Regulation of MST complexes and activity via SARAH domain modifications. Biochem. Soc. Trans. 2021, 49, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Donninger, H.; Allen, N.; Henson, A.; Pogue, J.; Williams, A.; Gordon, L.; Kassler, S.; Dunwell, T.; Latif, F.; Clark, G.J. Salvador Protein Is a Tumor Suppressor Effector of RASSF1A with Hippo Pathway-independent Functions. J. Biol. Chem. 2011, 286, 18483–18491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschöp, K.; Conery, A.R.; Litovchick, L.; DeCaprio, J.A.; Settleman, J.; Harlow, E.; Dyson, N. A kinase shRNA screen links LATS2 and the pRB tumor suppressor. Genes Dev. 2011, 25, 814–830. [Google Scholar] [CrossRef] [Green Version]
- Litovchick, L.; Florens, L.A.; Swanson, S.K.; Washburn, M.P.; DeCaprio, J.A. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011, 25, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Lit, L.C.; Scott, S.; Zhang, H.; Stebbing, J.; Photiou, A.; Giamas, G. LATS2 is a modulator of estrogen receptor alpha. Anticancer Res. 2013, 33, 53–63. [Google Scholar]
- Ganem, N.J.; Cornils, H.; Chiu, S.-Y.; O’Rourke, K.P.; Arnaud, J.; Yimlamai, D.; Théry, M.; Camargo, F.D.; Pellman, D. Cytokinesis Failure Triggers Hippo Tumor Suppressor Pathway Activation. Cell 2014, 158, 833–848. [Google Scholar] [CrossRef] [Green Version]
- Aylon, Y.; Ofir-Rosenfeld, Y.; Yabuta, N.; Lapi, E.; Nojima, H.; Lu, X.; Oren, M. The Lats2 tumor suppressor augments p53-mediated apoptosis by promoting the nuclear proapoptotic function of ASPP1. Genes Dev. 2010, 24, 2420–2429. [Google Scholar] [CrossRef] [Green Version]
- Lavado, A.; Park, J.Y.; Paré, J.; Finkelstein, D.; Pan, H.; Xu, B.; Fan, Y.; Kumar, R.P.; Neale, G.; Kwak, Y.D.; et al. The Hippo Pathway Prevents YAP/TAZ-Driven Hypertranscription and Controls Neural Progenitor Number. Dev. Cell 2018, 47, 576–591.e8. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Aylon, Y.; Yabuta, N.; Besserglick, H.; Buganim, Y.; Rotter, V.; Nojima, H.; Oren, M. Silencing of the Lats2 tumor suppressor overrides a p53-dependent oncogenic stress checkpoint and enables mutant H-Ras-driven cell transformation. Oncogene 2009, 28, 4469–4479. [Google Scholar] [CrossRef] [Green Version]
- Toji, S.; Yabuta, N.; Hosomi, T.; Nishihara, S.; Kobayashi, T.; Suzuki, S.; Tamai, K.; Nojima, H. The centrosomal protein Lats2 is a phosphorylation target of Aurora-A kinase. Genes Cells 2004, 9, 383–397. [Google Scholar] [CrossRef]
- Mukai, S.; Yabuta, N.; Yoshida, K.; Okamoto, A.; Miura, D.; Furuta, Y.; Abe, T.; Nojima, H. Lats1 suppresses centrosome overduplication by modulating the stability of Cdc25B. Sci. Rep. 2015, 5, 16173. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zheng, Q.; Li, Y.; Wang, G.; Gao, S.; Zhang, X.; Yan, X.; Zhang, X.; Xie, J.; Wang, Y.; et al. Metformin targets a YAP1-TEAD4 complex via AMPKα to regulate CCNE1/2 in bladder cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 376. [Google Scholar] [CrossRef]
- Cao, J.-J.; Zhao, X.-M.; Wang, D.-L.; Chen, K.-H.; Sheng, X.; Li, W.-B.; Li, M.-C.; Liu, W.-J.; He, J. YAP is overexpressed in clear cell renal cell carcinoma and its knockdown reduces cell proliferation and induces cell cycle arrest and apoptosis. Oncol. Rep. 2014, 32, 1594–1600. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wang, J.; Yao, S.-F.; Zhao, Y.; Liu, L.; Li, L.-W.; Xu, T.; Gan, L.-G.; Xiao, C.-L.; Shan, Z.-L.; et al. Effect of YAP Inhibition on Human Leukemia HL-60 Cells. Int. J. Med. Sci. 2017, 14, 902–910. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Huang, M.; Tan, J.; Hou, J.; He, J.; Wang, F.; Cui, H.; Yi, L. Transcriptional co-activator with PDZ-binding motif overexpression promotes cell proliferation and transcriptional co-activator with PDZ-binding motif deficiency induces cell cycle arrest in neuroblastoma. Oncol. Lett. 2017, 13, 4295–4301. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Tong, R.; Yang, B.; Lv, Z.; Du, C.; Peng, C.; Ding, C.; Cheng, S.; Zhou, L.; Xie, H.; et al. TAZ regulates cell proliferation and sensitivity to vitamin D3 in intrahepatic cholangiocarcinoma. Cancer Lett. 2016, 381, 370–379. [Google Scholar] [CrossRef]
- Fang-Liang, Y.; Yang, F.; Zhu, C.; Tang, L. Effect and mechanism of RNAi targeting WWTR1 on biological activity of gastric cancer cells SGC7901. Mol. Med. Rep. 2017, 17, 2853–2860. [Google Scholar] [CrossRef]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Chen, Y.; Wan, Y.; Liu, T.; Wang, J.; Zhang, Y.; Wei, L.; Hu, Q.; Xu, B.; Chernov, M.; et al. Identification of TAZ-Dependent Breast Cancer Vulnerabilities Using a Chemical Genomics Screening Approach. Front. Cell Dev. Biol. 2021, 9, 673374. [Google Scholar] [CrossRef]
- Xie, K.; Xu, C.; Zhang, M.; Wang, M.; Min, L.; Qian, C.; Wang, Q.; Ni, Z.; Mou, S.; Dai, H.; et al. Yes-associated protein regulates podocyte cell cycle re-entry and dedifferentiation in adriamycin-induced nephropathy. Cell Death Dis. 2019, 10, 915. [Google Scholar] [CrossRef]
- Xie, Q.; Chen, J.; Feng, H.; Peng, S.; Adams, U.; Bai, Y.; Huang, L.; Li, J.; Huang, J.; Meng, S.; et al. YAP/TEAD–Mediated Transcription Controls Cellular Senescence. Cancer Res. 2013, 73, 3615–3624. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Kim, S.; Lee, M.-W.; Jeon, H.-J.; Ryu, H.; Kim, J.-M.; Lee, H.-J. MITF Promotes Cell Growth, Migration and Invasion in Clear Cell Renal Cell Carcinoma by Activating the RhoA/YAP Signal Pathway. Cancers 2021, 13, 2920. [Google Scholar] [CrossRef]
- Yang, R.; Wu, Y.; Zou, J.; Zhou, J.; Wang, M.; Hao, X.; Cui, H. The Hippo transducer TAZ promotes cell proliferation and tumor formation of glioblastoma cells through EGFR pathway. Oncotarget 2016, 7, 36255–36265. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, T.; Imoto, I.; Matsui, T.; Kozaki, K.-I.; Haruki, S.; Sudol, M.; Shimada, Y.; Tsuda, H.; Kawano, T.; Inazawa, J. YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis 2010, 32, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Hsueh, Y.-J.; Chen, H.-C.; Wu, S.-E.; Wang, T.-K.; Chen, J.-K.; Ma, D.H.-K. Lysophosphatidic acid induces YAP-promoted proliferation of human corneal endothelial cells via PI3K and ROCK pathways. Mol. Ther.-Methods Clin. Dev. 2015, 2, 15014. [Google Scholar] [CrossRef]
- Hoxha, S.; Shepard, A.; Troutman, S.; Diao, H.; Doherty, J.R.; Janiszewska, M.; Witwicki, R.M.; Pipkin, M.E.; Ja, W.W.; Kareta, M.S.; et al. YAP-Mediated Recruitment of YY1 and EZH2 Represses Transcription of Key Cell-Cycle Regulators. Cancer Res. 2020, 80, 2512–2522. [Google Scholar] [CrossRef]
- Böttcher, R.T.; Sun, Z.; Fässler, R. A forceful connection: Mechanoregulation of oncogenic YAP. EMBO J. 2017, 36, 2467–2469. [Google Scholar] [CrossRef]
- Jang, W.; Kim, T.; Koo, J.S.; Kim, S.K.; Lim, D.S. Mechanical cue-induced YAP instructs Skp2-dependent cell cycle exit and oncogenic signaling. EMBO J. 2017, 36, 2510–2528. [Google Scholar] [CrossRef]
- Wang, I.-C.; Chen, Y.-J.; Hughes, D.; Petrovic, V.; Major, M.L.; Park, H.J.; Tan, Y.; Ackerson, T.; Costa, R.H. Forkhead Box M1 Regulates the Transcriptional Network of Genes Essential for Mitotic Progression and Genes Encoding the SCF (Skp2-Cks1) Ubiquitin Ligase. Mol. Cell. Biol. 2005, 25, 10875–10894. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Stanger, B.Z. YAP Regulates S-Phase Entry in Endothelial Cells. PLoS ONE 2015, 10, e0117522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elaimy, A.L.; Amante, J.; Zhu, L.J.; Wang, M.; Walmsley, C.S.; FitzGerald, T.J.; Goel, H.L.; Mercurio, A.M. The VEGF receptor neuropilin 2 promotes homologous recombination by stimulating YAP/TAZ-mediated Rad51 expression. Proc. Natl. Acad. Sci. USA 2019, 116, 14174–14180. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Wang, J.; Li, R.; Zhao, X.; Zhang, Y.; Liu, B.; Lei, Y.; Hu, Y. A New Regulatory Mechanism Between P53 And YAP Crosstalk By SIRT1 Mediated Deacetylation To Regulate Cell Cycle And Apoptosis In A549 Cell Lines. Cancer Manag. Res. 2019, 11, 8619–8633. [Google Scholar] [CrossRef] [Green Version]
- Miyajima, C.; Kawarada, Y.; Inoue, Y.; Suzuki, C.; Mitamura, K.; Morishita, D.; Ohoka, N.; Imamura, T.; Hayashi, H. Transcriptional Coactivator TAZ Negatively Regulates Tumor Suppressor p53 Activity and Cellular Senescence. Cells 2020, 9, 171. [Google Scholar] [CrossRef] [Green Version]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2015, 17, 188–201. [Google Scholar] [CrossRef]
- Bai, N.; Zhang, C.; Liang, N.; Zhang, Z.; Chang, A.; Yin, J.; Li, Z.; Li, N.; Tan, X.; Luo, N.; et al. Yes-associated protein (YAP) increases chemosensitivity of hepatocellular carcinoma cells by modulation of p53. Cancer Biol. Ther. 2013, 14, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Strano, S.; Munarriz, E.; Rossi, M.; Castagnoli, L.; Shaul, Y.; Sacchi, A.; Oren, M.; Sudol, M.; Cesareni, G.; Blandino, G. Physical Interaction with Yes-associated Protein Enhances p73 Transcriptional Activity. J. Biol. Chem. 2001, 276, 15164–15173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The Transcriptional Coactivator Yes-Associated Protein Drives p73 Gene-Target Specificity in Response to DNA Damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2006, 14, 743–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.Y.; Zha, Z.Y.; Zhou, X.; Zhang, H.; Huang, W.; Zhao, D.; Li, T.; Chan, S.W.; Lim, C.J.; Hong, W.; et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J. Biol. Chem. 2010, 285, 37159–37169. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 Phosphorylation by c-Abl Is a Critical Step in Selective Activation of Proapoptotic Genes in Response to DNA Damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Lapi, E.; Di Agostino, S.; Donzelli, S.; Gal, H.; Domany, E.; Rechavi, G.; Pandolfi, P.P.; Givol, D.; Strano, S.; Lu, X.; et al. PML, YAP, and p73 Are Components of a Proapoptotic Autoregulatory Feedback Loop. Mol. Cell 2008, 32, 803–814. [Google Scholar] [CrossRef]
- Raman, M.; Earnest, S.; Zhang, K.; Zhao, Y.; Cobb, M.H. TAO kinases mediate activation of p38 in response to DNA damage. EMBO J. 2007, 26, 2005–2014. [Google Scholar] [CrossRef]
- Wojtala, R.L.; Tavares, I.A.; Morton, P.E.; Valderrama, F.; Thomas, N.S.B.; Morris, J.D. Prostate-derived Sterile 20-like Kinases (PSKs/TAOKs) Are Activated in Mitosis and Contribute to Mitotic Cell Rounding and Spindle Positioning. J. Biol. Chem. 2011, 286, 30161–30170. [Google Scholar] [CrossRef] [Green Version]
- King, I.; Heberlein, U. Tao kinases as coordinators of actin and microtubule dynamics in developing neurons. Commun. Integr. Biol. 2011, 4, 554–556. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.L.; Tamura, N.; Fries, A.; Levin, N.; Clark, J.; Draviam, V.M. TAO1 kinase maintains chromosomal stability by facilitating proper congression of chromosomes. Open Biol. 2014, 4, 130108. [Google Scholar] [CrossRef] [Green Version]
- Garg, R.; Koo, C.Y.; Infante, E.; Giacomini, C.; Ridley, A.; Morris, J.D.H. Rnd3 interacts with TAO kinases and contributes to mitotic cell rounding and spindle positioning. J. Cell Sci. 2020, 133, jcs235895. [Google Scholar] [CrossRef] [PubMed]
- Koo, C.Y.; Giacomini, C.; Corral, M.R.; Olmos, Y.; Tavares, I.A.; Marson, C.; Linardopoulos, S.; Tutt, A.N.; Morris, J.D. Targeting TAO Kinases Using a New Inhibitor Compound Delays Mitosis and Induces Mitotic Cell Death in Centrosome Amplified Breast Cancer Cells. Mol. Cancer Ther. 2017, 16, 2410–2421. [Google Scholar] [CrossRef] [Green Version]
- Whang, Y.M.; Kim, Y.H.; Kim, J.S.; Yoo, Y.D. RASSF1A Suppresses the c-Jun-NH2-Kinase Pathway and Inhibits Cell Cycle Progression. Cancer Res. 2005, 65, 3682–3690. [Google Scholar] [CrossRef] [Green Version]
- Shivakumar, L.; Minna, J.; Sakamaki, T.; Pestell, R.; White, M.A. The RASSF1A Tumor Suppressor Blocks Cell Cycle Progression and Inhibits Cyclin D1 Accumulation. Mol. Cell. Biol. 2002, 22, 4309–4318. [Google Scholar] [CrossRef] [Green Version]
- Liao, A.; Tan, G.; Chen, L.; Zhou, W.; Hu, H. RASSF1A inhibits gastric cancer cell proliferation by miR-711- mediated downregulation of CDK4 expression. Oncotarget 2016, 7, 5842–5851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, Y.; Na, A.; Lee, M.-S.; Yoon, S.; Kim, J. RASSF1A suppresses oncogenic H-Ras-induced c-Jun N-terminal kinase activation. Int. J. Oncol. 2006, 29, 1541–1547. [Google Scholar] [CrossRef] [Green Version]
- Donninger, H.; Clark, J.; Rinaldo, F.; Nelson, N.; Barnoud, T.; Schmidt, M.L.; Hobbing, K.R.; Vos, M.D.; Sils, B.; Clark, G.J. The RASSF1A Tumor Suppressor Regulates XPA-Mediated DNA Repair. Mol. Cell. Biol. 2015, 35, 277–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.S.; Song, S.J.; Kim, S.Y.; Oh, H.J.; Lim, D.-S. The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2–DAXX–HAUSP complex. EMBO J. 2008, 27, 1863–1874. [Google Scholar] [CrossRef]
- Dallol, A.; Agathanggelou, A.; Fenton, S.L.; Ahmed-Choudhury, J.; Hesson, L.; Vos, M.D.; Clark, G.J.; Downward, J.; Maher, E.R.; Latif, F. RASSF1A Interacts with Microtubule-Associated Proteins and Modulates Microtubule Dynamics. Cancer Res. 2004, 64, 4112–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnette, C.; Efimova, N.; Zhu, X.; Clark, G.J.; Kaverina, I. Microtubule segment stabilization by RASSF1A is required for proper microtubule dynamics and Golgi integrity. Mol. Biol. Cell 2014, 25, 800–810. [Google Scholar] [CrossRef]
- Liu, L.; Tommasi, S.; Lee, D.-H.; Dammann, R.; Pfeifer, G.P. Control of microtubule stability by the RASSF1A tumor suppressor. Oncogene 2003, 22, 8125–8136. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Guo, C.; Dammann, R.; Tommasi, S.; Pfeifer, G.P. RASSF1A interacts with and activates the mitotic kinase Aurora-A. Oncogene 2008, 27, 6175–6186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.S.; Song, S.J.; Ayad, N.G.; Chang, J.S.; Lee, J.-H.; Hong, H.K.; Lee, H.; Choi, N.; Kim, J.; Kim, H.; et al. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC–Cdc20 complex. Nat. Cell Biol. 2004, 6, 129–137. [Google Scholar] [CrossRef]
- Rong, R.; Jiang, L.Y.; Sheikh, M.S.; Huang, Y. Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates RASSF1A-mediated microtubule interaction and M-phase cell cycle regulation. Oncogene 2007, 26, 7700–7708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.J.; Song, M.S.; Kim, S.J.; Kim, S.Y.; Kwon, S.H.; Kim, J.G.; Calvisi, D.F.; Kang, D.; Lim, D.-S. Aurora A Regulates Prometaphase Progression by Inhibiting the Ability of RASSF1A to Suppress APC-Cdc20 Activity. Cancer Res. 2009, 69, 2314–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, C.; Wong, N.; Pagano, M.; Lun, S.W.-M.; Nakayama, K.-I.; Lo, K.-W. Regulation of APC/CCdc20 activity by RASSF1A–APC/CCdc20 circuitry. Oncogene 2011, 31, 1975–1987. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Rong, R.; Sheikh, M.S.; Huang, Y. Cullin-4A·DNA Damage-binding Protein 1 E3 Ligase Complex Targets Tumor Suppressor RASSF1A for Degradation during Mitosis. J. Biol. Chem. 2011, 286, 6971–6978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Rong, R.; Sheikh, M.S.; Huang, Y. Mitotic Arrest by Tumor Suppressor RASSF1A Is Regulated via CHK1 Phosphorylation. Mol. Cancer Res. 2013, 12, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Song, S.J.; Kim, S.J.; Song, M.S.; Lim, D.-S. Aurora B-Mediated Phosphorylation of RASSF1A Maintains Proper Cytokinesis by Recruiting Syntaxin16 to the Midzone and Midbody. Cancer Res. 2009, 69, 8540–8544. [Google Scholar] [CrossRef] [Green Version]
- Ahmed-Choudhury, J.; Agathanggelou, A.; Fenton, S.L.; Ricketts, C.; Clark, G.J.; Maher, E.; Latif, F. Transcriptional Regulation of Cyclin A2 by RASSF1A through the Enhanced Binding of p120E4F to the Cyclin A2 Promoter. Cancer Res. 2005, 65, 2690–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavuluri, J.; Beesetti, S.; Surabhi, R.; Kremerskothen, J.; Venkatraman, G.; Rayala, S.K. Phosphorylation-Dependent Regulation of the DNA Damage Response of Adaptor Protein KIBRA in Cancer Cells. Mol. Cell. Biol. 2016, 36, 1354–1365. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Chen, Y.; Ji, M.; Volle, D.J.; Lewis, R.E.; Tsai, M.-Y.; Dong, J. KIBRA Protein Phosphorylation Is Regulated by Mitotic Kinase Aurora and Protein Phosphatase. J. Biol. Chem. 2011, 286, 36304–36315. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Iyer, J.; Chowdhury, A.; Ji, M.; Xiao, L.; Yang, S.; Chen, Y.; Tsai, M.-Y.; Dong, J. KIBRA Regulates Aurora Kinase Activity and Is Required for Precise Chromosome Alignment During Mitosis. J. Biol. Chem. 2012, 287, 34069–34077. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lim, J.Y.; Kim, Y.H.; Kim, H.; Park, S.-H.; Lee, K.-H.; Han, H.; Jeun, S.-S.; Lee, J.H.; Rha, H.K. Inhibition of ras-mediated activator protein 1 activity and cell growth by merlin. Mol. Cells 2002, 14, 108–114. [Google Scholar]
- Xiao, G.-H.; Gallagher, R.; Shetler, J.; Skele, K.; Altomare, D.A.; Pestell, R.G.; Jhanwar, S.; Testa, J.R. The NF2 Tumor Suppressor Gene Product, Merlin, Inhibits Cell Proliferation and Cell Cycle Progression by Repressing Cyclin D1 Expression. Mol. Cell. Biol. 2005, 25, 2384–2394. [Google Scholar] [CrossRef] [Green Version]
- Beltrami, S.; Kim, R.; Gordon, J. Neurofibromatosis type 2 protein, NF2: An uncoventional cell cycle regulator. Anticancer Res. 2013, 33, 1–11. [Google Scholar] [PubMed]
- Kim, H.; Kwak, N.-J.; Lee, J.Y.; Choi, B.H.; Lim, Y.; Ko, Y.J.; Kim, Y.-H.; Huh, P.-W.; Lee, K.-H.; Rha, H.K.; et al. Merlin Neutralizes the Inhibitory Effect of Mdm2 on p53. J. Biol. Chem. 2004, 279, 7812–7818. [Google Scholar] [CrossRef] [Green Version]
- Hebert, A.M.; DuBoff, B.; Casaletto, J.B.; Gladden, A.B.; McClatchey, A.I. Merlin/ERM proteins establish cortical asymmetry and centrosome position. Genes Dev. 2012, 26, 2709–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandati, V.; Del Maestro, L.; Dingli, F.; Lombard, B.; Loew, D.; Molinie, N.; Romero, S.; Bouvard, D.; Gautreau, A.M.; Pasmant, E.; et al. Phosphorylation of Merlin by Aurora A kinase appears necessary for mitotic progression. J. Biol. Chem. 2019, 294, 12992–13005. [Google Scholar] [CrossRef]
- Smole, Z.; Thoma, C.R.; Applegate, K.T.; Duda, M.; Gutbrodt, K.L.; Danuser, G.; Krek, W. Tumor suppressor NF2/Merlin is a microtubule stabilizer. Cancer Res. 2014, 74, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Grönholm, M.; Muranen, T.; Toby, G.G.; Utermark, T.; Hanemann, C.O.; Golemis, E.; Carpén, O. A functional association between merlin and HEI10, a cell cycle regulator. Oncogene 2006, 25, 4389–4398. [Google Scholar] [CrossRef] [Green Version]
- Fowler, S.; Maguin, P.; Kalan, S.; Loayza, D. LIM Protein Ajuba associates with the RPA complex through direct cell cycle-dependent interaction with the RPA70 subunit. Sci. Rep. 2018, 8, 9536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalan, S.; Matveyenko, A.; Loayza, D. LIM Protein Ajuba Participates in the Repression of the ATR-Mediated DNA Damage Response. Front. Genet. 2013, 4, 95. [Google Scholar] [CrossRef] [Green Version]
- Ferrand, A.; Chevrier, V.; Chauvin, J.-P.; Birnbaum, D. Ajuba: A new microtubule-associated protein that interacts with BUBR1 and Aurora B at kinetochores in metaphase. Biol. Cell 2009, 101, 221–240. [Google Scholar] [CrossRef]
- Hirota, T.; Kunitoku, N.; Sasayama, T.; Marumoto, T.; Zhang, D.; Nitta, M.; Hatakeyama, K.; Saya, H. Aurora-A and an Interacting Activator, the LIM Protein Ajuba, Are Required for Mitotic Commitment in Human Cells. Cell 2003, 114, 585–598. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y.; Ohsugi, M.; Haraguchi, K.; Fujimoto, J.; Yamamoto, T. LATS2-Ajuba complex regulates gamma-tubulin recruitment to centrosomes and spindle organization during mitosis. FEBS Lett. 2006, 580, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Stauffer, S.; Chen, Y.; Dong, J. Ajuba Phosphorylation by CDK1 Promotes Cell Proliferation and Tumorigenesis. J. Biol. Chem. 2016, 291, 14761–14772. [Google Scholar] [CrossRef] [Green Version]
- Crone, J.; Glas, C.; Schultheiss, K.; Moehlenbrink, J.; Krieghoff-Henning, E.; Hofmann, T.G. Zyxin Is a Critical Regulator of the Apoptotic HIPK2-p53 Signaling Axis. Cancer Res. 2011, 71, 2350–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, T.; Morisaki, T.; Nishiyama, Y.; Marumoto, T.; Tada, K.; Hara, T.; Masuko, N.; Inagaki, M.; Hatakeyama, K.; Saya, H. Zyxin, a Regulator of Actin Filament Assembly, Targets the Mitotic Apparatus by Interacting with H-Warts/Lats1 Tumor Suppressor. J. Cell Biol. 2000, 149, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zeng, Y.; Cui, L.; Chen, X.; Stauffer, S.; Wang, Z.; Yu, F.; Lele, S.M.; Talmon, G.A.; Black, A.R.; et al. Zyxin promotes colon cancer tumorigenesis in a mitotic phosphorylation-dependent manner and through CDK8-mediated YAP activation. Proc. Natl. Acad. Sci. USA 2018, 115, E6760–E6769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornils, H.; Kohler, R.S.; Hergovich, A.; Hemmings, B.A. Human NDR kinases control G(1)/S cell cycle transition by directly regulating p21 stability. Mol. Cell Biol. 2011, 31, 1382–1395. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Rohmer, D.; Probst, S.; Yang, Z.-Z.; Laurent, F.; Stadler, M.B.; Zúñiga, A.; Zeller, R.; Hynx, D.; Hemmings, B.A.; Hergovich, A. NDR Kinases Are Essential for Somitogenesis and Cardiac Looping during Mouse Embryonic Development. PLoS ONE 2015, 10, e0136566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-M.; Choi, J.Y.; Yi, J.M.; Chung, J.W.; Leem, S.-H.; Koh, S.S.; Kang, T.-H. NDR1 modulates the UV-induced DNA-damage checkpoint and nucleotide excision repair. Biochem. Biophys. Res. Commun. 2015, 461, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Yu, J.; Nowsheen, S.; Zhao, F.; Wang, L.; Lou, Z. STK38 promotes ATM activation by acting as a reader of histone H4 ufmylation. Sci. Adv. 2020, 6, eaax8214. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, T.; Enomoto, A.; Miyagawa, K. Serine-Threonine Kinase 38 regulates CDC25A stability and the DNA damage-induced G2/M checkpoint. Cell. Signal. 2015, 27, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Hergovich, A.; Lamla, S.; Nigg, E.; Hemmings, B.A. Centrosome-Associated NDR Kinase Regulates Centrosome Duplication. Mol. Cell 2007, 25, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Fernández, M.; Malumbres, M. Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell 2020, 37, 514–529. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| S. cerevisiae (MEN) | S. cerevisiae (RAM) | S. pombe (SIN) | D. melanogaster | Mammals | |
|---|---|---|---|---|---|
| The Ste20-like protein kinase | Cdc15 | Kic1 | Cdc7 | Hpo | MST1/2 |
| The scaffold protein | Nud1 | Tao3 | Cdc11-Sid4-Ppc89 complex | Sav | SAV1 or Furry |
| The NDR protein kinase | Dbf2/20 | Cbk1 | Sid2 | Wts | LATS1/2 or NDR1/2 |
| The adaptor protein | Mob1 | Mob2 | Mob1 | Mats | MOB1A/B |
| The effector protein | Cdc14 | Ace2 | Clp1 | Yki | YAP/TAZ |
| Function in mitosis | Mitotic exit | Cytokinesis | Mitotic exit and cytokinesis | Mitotic progression and cytokinesis | Mitotic progression and cytokinesis |
| E2F Activity | G1 Tetraploidy Checkpoint | DNA Synthesis | DNA Damage Checkpoint | Centrosome Dynamics | Mitosis | |
|---|---|---|---|---|---|---|
| MST1/2 | Prevents polyploidization | Promotes DNA repair and DNA damage-induced apoptosis | Centrosome duplication and centrosome separation | Chromosome alignment and spindle formation | ||
| SAV1 | Prevents polyploidization | Promotes DNA damage-induced apoptosis | Facilitates centrosome separation | Spindle formation | ||
| LATS1/2 | Promotes DREAM complex assembly to repress E2F | Prevents polyploidization, enhances G1 checkpoint | Affects DNA synthesis initiation | Promotes DNA repair and DNA damage-induced apoptosis | Prevents centrosome overduplication | SAC, chromosome alignment and segregation, cytokinesis |
| MOB1A/B | Centrosome duplication, separation | Spindle orientation and cytokinesis | ||||
| YAP/TAZ | Activates E2F and CDK2/4/6, suppresses p21/p27 | Stimulates polyploidization | Promotes transcription of DNA synthesis genes | Enhances DNA repair, promotes or suppresses DNA damage-induced apoptosis | SAC, chromosome alignment and segregation, spindle orientation, and cytokinesis | |
| TAOKs | Triggers DNA damage-induced G2/M arrest | Mitotic cell rounding, chromosome alignment and segregation, spindle orientation, cytokinesis | ||||
| RASSF1A | Decreases Cyclin D-CDK4, increases p27 | Promotes DNA repair and DNA damage-induced apoptosis | Inhibits centrosome separation | Chromosome alignment and segregation, cytokinesis | ||
| KIBRA | Promotes DNA repair | Chromosome alignment and segregation | ||||
| NF2 | Decreases Cyclin D/E, increases p21/p27 | Promotes DNA damage-induced apoptosis | Centrosome position | Regulates spindle positioning | ||
| Ajuba | Represses DNA damage-induced apoptosis/arrest | Regulates spindle formation | ||||
| Zyxin | Promotes DNA damage-induced apoptosis | |||||
| Furry | Chromosome alignment and spindle formation | |||||
| NDR1/2 | Centrosome duplication | Chromosome alignment, spindle orientation, and cytokinesis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Y.; Dong, J. The Hippo Signaling Pathway in Cancer: A Cell Cycle Perspective. Cancers 2021, 13, 6214. https://doi.org/10.3390/cancers13246214
Xiao Y, Dong J. The Hippo Signaling Pathway in Cancer: A Cell Cycle Perspective. Cancers. 2021; 13(24):6214. https://doi.org/10.3390/cancers13246214
Chicago/Turabian StyleXiao, Yi, and Jixin Dong. 2021. "The Hippo Signaling Pathway in Cancer: A Cell Cycle Perspective" Cancers 13, no. 24: 6214. https://doi.org/10.3390/cancers13246214
APA StyleXiao, Y., & Dong, J. (2021). The Hippo Signaling Pathway in Cancer: A Cell Cycle Perspective. Cancers, 13(24), 6214. https://doi.org/10.3390/cancers13246214