
Combined Treatment with Immunotherapy-Based Strategies for MSS Metastatic Colorectal Cancer

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
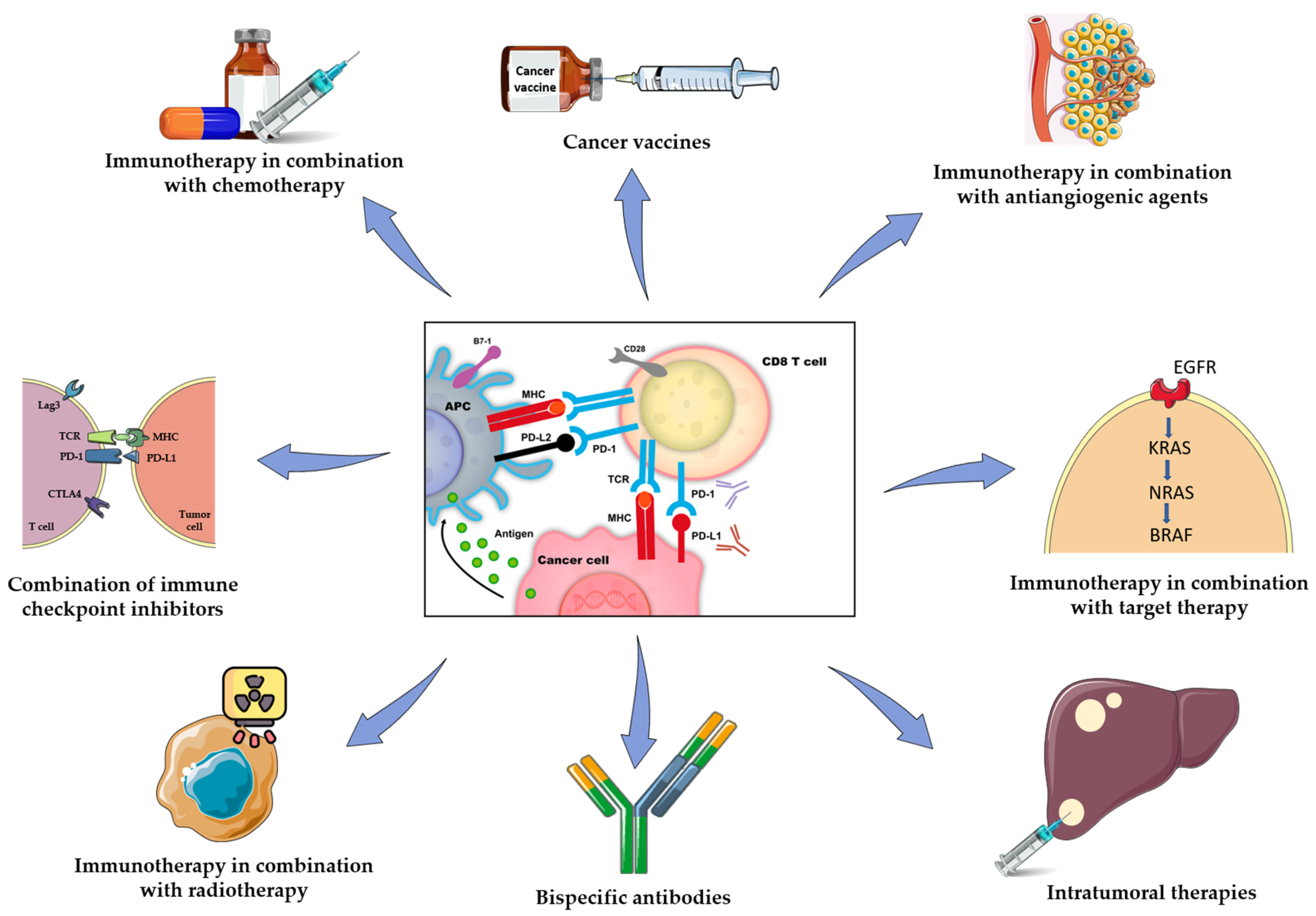
2. Immunotherapy-Based Combinations in pMMR/MSS mCCR
2.1. Combination of Immune Checkpoint Inhibitors
2.2. Immunotherapy in Combination with Chemotherapy
2.2.1. Immunotherapy in Combination with Chemotherapy and Anti-VEGF Agents
2.2.2. Immunotherapy in Combination with Chemotherapy and Anti-EGFR Agents
2.2.3. Immunotherapy in Combination with Temozolomide
2.3. Immunotherapy in Combination with Antiangiogenic Agents
2.4. Immunotherapy in Combination with Radiotherapy
2.5. Immunotherapy in Combination with Target Therapy
2.5.1. Immunotherapy in Combination with MAPK Signaling Inhibitors
Immunotherapy in Combination with KRAS Inhibitors
Immunotherapy in Combination with BRAF Inhibitors
Immunotherapy in Combination with MEK Inhibitors
2.5.2. Immunotherapy in Combination with Inhibition of the PI3K/AKT/mTOR Pathway
2.6. Bispecific Antibodies
2.7. Cancer Vaccines
2.8. Intratumoral Therapies
3. Conclusions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, S.M.; Banerjea, A.; Feakins, R.; Li, S.R.; Bustin, S.A.; Dorudi, S. Tumour-infiltrating lymphocytes in colorectal cancer with microsatellite instability are activated and cytotoxic. Br. J. Surg. 2004, 91, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Wang, C.; Lee, P.P.; Chu, P.; Fakih, M. Response to PD-1 Blockade in Microsatellite Stable Metastatic Colorectal Cancer Harboring aPOLEMutation. J. Natl. Compr. Cancer Netw. 2017, 15, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo, E.; Freeman-Mills, L.; Rayner, E.; Glaire, M.; Briggs, S.; Vermeulen, L.; Fessler, E.; Medema, J.P.; Boot, A.; Morreau, H.; et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroenterol. Hepatol. 2016, 1, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.X.; Jonker, D.J.; Loree, J.; Kennecke, H.F.; Berry, S.R.; Couture, F.; Ahmad, C.E.; Goffin, J.R.; Kavan, P.; Harb, M.; et al. Effect of Combined Immune Checkpoint Inhibition vs. Best Supportive Care Alone in Patients with Advanced Colorectal Cancer. JAMA Oncol. 2020, 6, 831–838. [Google Scholar] [CrossRef]
- Garralda, E.; Sukari, A.; Lakhani, N.J.; Patnaik, A.; Lou, Y.; Im, S.-A.; Golan, T.; Geva, R.; Wermke, M.; De Miguel, M.; et al. A phase 1 first-in-human study of the anti-LAG-3 antibody MK4280 (favezelimab) plus pembrolizumab in previously treated, advanced microsatellite stable colorectal cancer. J. Clin. Oncol. 2021, 39, 3584. [Google Scholar] [CrossRef]
- Grothey, A.; Tabernero, J.; Arnold, D.; De Gramont, A.; Ducreux, M.; O’Dwyer, P.; Van Cutsem, E.; Bosanac, I.; Srock, S.; Mancao, C.; et al. Fluoropyrimidine (FP) + bevacizumab (BEV) + atezolizumab vs. FP/BEV in BRAFwt metastatic colorectal cancer (mCRC): Findings from Cohort 2 of MODUL—A multicentre, randomized trial of biomarker-driven maintenance treatment following first-line induction therapy. Ann. Oncol. 2018, 29, viii714–viii715. [Google Scholar] [CrossRef]
- Cremolini, C.; Rossini, D.; Antoniotti, C.; Pietrantonio, F.; Lonardi, S.; Salvatore, L.; Marmorino, F.; Borelli, B.; Ambrosini, M.; Barsotti, G.; et al. LBA20 FOLFOXIRI plus bevacizumab (bev) plus atezolizumab (atezo) versus FOLFOXIRI plus bev as first-line treatment of unresectable metastatic colorectal cancer (mCRC) patients: Results of the phase II randomized AtezoTRIBE study by GONO. Ann. Oncol. 2021, 32, S1294–S1295. [Google Scholar] [CrossRef]
- Mettu, N.; Twohy, E.; Ou, F.-S.; Halfdanarson, T.; Lenz, H.; Breakstone, R.; Boland, P.; Crysler, O.; Wu, C.; Grothey, A.; et al. BACCI: A phase II randomized, double-blind, multicenter, placebo-controlled study of capecitabine (C) bevacizumab (B) plus atezolizumab (A) or placebo (P) in refractory metastatic colorectal cancer (mCRC): An ACCRU network study. Ann. Oncol. 2019, 30, v203. [Google Scholar] [CrossRef]
- Stein, A.; Binder, M.; Goekkurt, E.; Lorenzen, S.; Riera-Knorrenschild, J.; Depenbusch, R.; Ettrich, T.J.; Doerfel, S.; Al-Batran, S.-E.; Karthaus, M.; et al. Avelumab and cetuximab in combination with FOLFOX in patients with previously untreated metastatic colorectal cancer (MCRC): Final results of the phase II AVETUX trial (AIO-KRK-0216). J. Clin. Oncol. 2020, 38, 96. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Morano, F.; Lonardi, S.; Raimondi, A.; Salvatore, L.; Marmorino, F.; Murgioni, S.; Pella, N.; Antonuzzo, L.; Ritorto, G.; et al. 383O MAYA trial: Temozolomide (TMZ) priming followed by combination with low-dose ipilimumab and nivolumab in patients with microsatellite stable (MSS), MGMT silenced metastatic colorectal cancer (mCRC). Ann. Oncol. 2021, 32, S530–S531. [Google Scholar] [CrossRef]
- Gomez-Roca, C.A.; Yanez, E.; Im, S.-A.; Castanon-Alvarez, E.; Senellart, H.; Doherty, M.; Garcia-Corbacho, J.; Lopez, J.S.; Basu, B.; Maurice-Dror, C.; et al. LEAP-005: A phase 2 multicohort study of lenvatinib plus pembrolizumab in patients with previously treated selected solid tumors—Results from the colorectal cancer cohort. J. Clin. Oncol. 2021, 39, 3564. [Google Scholar] [CrossRef]
- Kim, R.; Imanirad, I.; Carballido, E.; Strosberg, J.; Kim, Y.; Kim, D. O-20 Phase I/IB study of regorafenib and nivolumab in mismatch repair (MMR) proficient advanced refractory colorectal cancer. Ann. Oncol. 2020, 31, 239. [Google Scholar] [CrossRef]
- Cousin, S.; Cantarel, C.; Guegan, J.-P.; Gomez-Roca, C.; Metges, J.-P.; Adenis, A.; Pernot, S.; Bellera, C.A.; Kind, M.; Auzanneau, C.; et al. Regorafenib-Avelumab Combination in Patients with Microsatellite Stable Colorectal Cancer (REGOMUNE): A Single-arm, Open-label, Phase II Trial. Clin. Cancer Res. 2021, 27, 2139–2147. [Google Scholar] [CrossRef]
- Segal, N.H.; Cercek, A.; Ku, G.; Wu, A.J.; Rimner, A.; Khalil, D.N.; Reidy-Lagunes, D.; Cuaron, J.; Yang, T.J.; Weiser, M.R.; et al. Phase II Single-arm Study of Durvalumab and Tremelimumab with Concurrent Radiotherapy in Patients with Mismatch Repair–proficient Metastatic Colorectal Cancer. Clin. Cancer Res. 2021, 27, 2200–2208. [Google Scholar] [CrossRef]
- Parikh, A.R.; Clark, J.W.; Wo, J.Y.-L.; Yeap, B.Y.; Allen, J.N.; Blaszkowsky, L.S.; Ryan, D.P.; Giantonio, B.J.; Weekes, C.D.; Zhu, A.X.; et al. A phase II study of ipilimumab and nivolumab with radiation in microsatellite stable (MSS) metastatic colorectal adenocarcinoma (mCRC). J. Clin. Oncol. 2019, 37, 3514. [Google Scholar] [CrossRef]
- Hellmann, M.; Kim, T.-W.; Lee, C.; Goh, B.-C.; Miller, W.; Oh, D.-Y.; Jamal, R.; Chee, C.-E.; Chow, L.; Gainor, J.; et al. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann. Oncol. 2019, 30, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.; Kim, T.W.; Bendell, J.; Argilés, G.; Tebbutt, N.C.; Di Bartolomeo, M.; Falcone, A.; Fakih, M.; Kozloff, M.; Segal, N.H.; et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): A multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019, 20, 849–861. [Google Scholar] [CrossRef]
- Jakubowski, C.; Collins, N.B.; Sugar, E.A.; Berg, M.; Cao, H.; Giannakis, M.; Jaffee, E.M.; Azad, N.S. A phase I/II study of PI3Kinase inhibition with copanlisib combined with the anti-PD-1 antibody nivolumab in relapsed/refractory solid tumors with expansions in MSS colorectal cancer. J. Clin. Oncol. 2020, 38, TPS4114. [Google Scholar] [CrossRef]
- Tabernero, J.; Melero, I.; Ros, W.; Argiles, G.; Marabelle, A.; Rodriguez-Ruiz, M.E.; Albanell, J.; Calvo, E.; Moreno, V.; Cleary, J.M.; et al. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: Preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2017, 35, 3002. [Google Scholar] [CrossRef]
- Caballero-Baños, M.; Benitez-Ribas, D.; Tabera, J.; Varea, S.; Vilana, R.; Bianchi, L.; Ayuso, J.R.; Pagés, M.; Carrera, G.; Cuatrecasas, M.; et al. Phase II randomised trial of autologous tumour lysate dendritic cell plus best supportive care compared with best supportive care in pre-treated advanced colorectal cancer patients. Eur. J. Cancer 2016, 64, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Hazama, S.; Nakamura, Y.; Tanaka, H.; Hirakawa, K.; Tahara, K.; Shimizu, R.; Ozasa, H.; Etoh, R.; Sugiura, F.; Okuno, K.; et al. A phase ΙI study of five peptides combination with oxaliplatin-based chemotherapy as a first-line therapy for advanced colorectal cancer (FXV study). J. Transl. Med. 2014, 12, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geevarghese, S.K.; Geller, D.A.; De Haan, H.A.; Hörer, M.; Knoll, A.E.; Mescheder, A.; Nemunaitis, J.; Reid, T.R.; Sze, D.Y.; Tanabe, K.K.; et al. Phase I/II Study of Oncolytic Herpes Simplex Virus NV1020 in Patients with Extensively Pretreated Refractory Colorectal Cancer Metastatic to the Liver. Hum. Gene Ther. 2010, 21, 1119–1128. [Google Scholar] [CrossRef] [Green Version]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rébé, C.; Ghiringhelli, F. 5-Fluorouracil Selectively Kills Tumor-Associated Myeloid-Derived Suppressor Cells Resulting in Enhanced T Cell–Dependent Antitumor Immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef] [Green Version]
- Kanterman, J.; Sade-Feldman, M.; Biton, M.; Ish-Shalom, E.; Lasry, A.; Goldshtein, A.; Hubert, A.; Baniyash, M. Adverse Immunoregulatory Effects of 5FU and CPT11 Chemotherapy on Myeloid-Derived Suppressor Cells and Colorectal Cancer Outcomes. Cancer Res. 2014, 74, 6022–6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Aparicio, M.; Alzuguren, P.; Mauleon, I.; Medina-Echeverz, J.; Hervas-Stubbs, S.; Mancheno, U.; Berraondo, P.; Crettaz, J.; Gonzalez-Aseguinolaza, G.; Prieto, J.; et al. Oxaliplatin in combination with liver-specific expression of interleukin 12 reduces the immunosuppressive microenvironment of tumours and eradicates metastatic colorectal cancer in mice. Gut 2011, 60, 341–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galaine, J.; Turco, C.; Vauchy, C.; Royer, B.; Mercier-Letondal, P.; Queiroz, L.; Loyon, R.; Mouget, V.; Boidot, R.; Laheurte, C.; et al. CD4 T cells target colorectal cancer antigens upregulated by oxaliplatin. Int. J. Cancer 2019, 145, 3112–3125. [Google Scholar] [CrossRef] [PubMed]
- Dosset, M.; Rivera-Vargas, T.; Lagrange, A.; Boidot, R.; Vegran, F.; Roussey, A.; Chalmin, F.; Dondaine, L.; Paul, C.; Marie-Joseph, E.L.; et al. PD-1/PD-L1 pathway: An adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. OncoImmunology 2018, 7, e1433981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- di Blasio, S.; Wortel, I.M.N.; Van Bladel, D.A.G.; de Vries, L.E.; Duiveman-de Boer, T.; Worah, K.; de Haas, N.; Buschow, S.I.; De Vries, I.J.M.; Figdor, C.G.; et al. Human CD1c(+) DCs are critical cellular mediators of immune responses induced by immunogenic cell death. OncoImmunology 2016, 5, e1192739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wu, L.; Zhang, J.; Wu, H.; Han, E.; Guo, Q. Chemoimmunotherapy by combining oxaliplatin with immune checkpoint blockades reduced tumor burden in colorectal cancer animal model. Biochem. Biophys. Res. Commun. 2017, 487, 1–7. [Google Scholar] [CrossRef]
- Limagne, E.; Euvrard, R.; Thibaudin, M.; Rébé, C.; Derangère, V.; Chevriaux, A.; Boidot, R.; Vegran, F.; Bonnefoy, N.; Vincent, J.; et al. Accumulation of MDSC and Th17 Cells in Patients with Metastatic Colorectal Cancer Predicts the Efficacy of a FOLFOX–Bevacizumab Drug Treatment Regimen. Cancer Res. 2016, 76, 5241–5252. [Google Scholar] [CrossRef] [Green Version]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR Pathway Blockade Inhibits Tumor-Induced Regulatory T-cell Proliferation in Colorectal Cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Gavalas, N.G.; Tsiatas, M.; Tsitsilonis, O.; Politi, E.; Ioannou, K.; Ziogas, A.C.; Rodolakis, A.; Vlahos, G.; Thomakos, N.; Haidopoulos, D.; et al. VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type. Br. J. Cancer 2012, 107, 1869–1875. [Google Scholar] [CrossRef] [Green Version]
- Osada, T.; Chong, G.; Tansik, R.; Hong, T.; Spector, N.; Kumar, R.; Hurwitz, H.I.; Dev, I.; Nixon, A.B.; Lyerly, H.K.; et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol. Immunother. 2008, 57, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Trotta, A.M.; Ottaiano, A.; Romano, C.; Nasti, G.; Nappi, A.; De Divitiis, C.; Napolitano, M.; Zanotta, S.; Casaretti, R.; D’Alterio, C.; et al. Prospective Evaluation of Cetuximab-Mediated Antibody-Dependent Cell Cytotoxicity in Metastatic Colorectal Cancer Patients Predicts Treatment Efficacy. Cancer Immunol. Res. 2016, 4, 366–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H.; Sakai, K.; Arao, T.; Shimoyama, T.; Tamura, T.; Nishio, K. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci. 2007, 98, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wei, Y.; Fang, W.; Lu, C.; Chen, J.; Cui, G.; Diao, H. Cetuximab Enhanced the Cytotoxic Activity of Immune Cells during Treatment of Colorectal Cancer. Cell. Physiol. Biochem. 2017, 44, 1038–1050. [Google Scholar] [CrossRef] [Green Version]
- Cremolini, C.; Rossini, D.; Dell’Aquila, E.; Lonardi, S.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Danesi, R.; Aprile, G.; et al. Rechallenge for Patients With RAS and BRAF Wild-Type Metastatic Colorectal Cancer With Acquired Resistance to First-line Cetuximab and Irinotecan. JAMA Oncol. 2019, 5, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, E.; Martini, G.; Famiglietti, V.; Troiani, T.; Napolitano, S.; Pietrantonio, F.; Ciardiello, D.; Terminiello, M.; Borrelli, C.; Vitiello, P.P.; et al. Cetuximab Rechallenge Plus Avelumab in Pretreated Patients With RAS Wild-type Metastatic Colorectal Cancer: The Phase 2 Single-Arm Clinical CAVE Trial. JAMA Oncol. 2021, 7, 1529. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMTGene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.; Szeto, C.; Tian, Y.; Cecchi, F.; Corallo, S.; Calegari, M.A.; Di Bartolomeo, M.; Morano, F.; Raimondi, A.; Fucà, G.; et al. Refining the selection of patients with metastatic colorectal cancer for treatment with temozolomide using proteomic analysis of O6-methylguanine-DNA-methyltransferase. Eur. J. Cancer 2019, 107, 164–174. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Pietrantonio, F.; Amatu, A.; Milione, M.; Cassingena, A.; Ghezzi, S.; Caporale, M.; Berenato, R.; Falcomatà, C.; Pellegrinelli, A.; et al. Digital PCR assessment of MGMT promoter methylation coupled with reduced protein expression optimises prediction of response to alkylating agents in metastatic colorectal cancer patients. Eur. J. Cancer 2017, 71, 43–50. [Google Scholar] [CrossRef]
- Calegari, M.A.; Inno, A.; Monterisi, S.; Orlandi, A.; Santini, D.; Basso, M.; Cassano, A.; Martini, M.; Cenci, T.; De Pascalis, I.; et al. A phase 2 study of temozolomide in pretreated metastatic colorectal cancer with MGMT promoter methylation. Br. J. Cancer 2017, 116, 1279–1286. [Google Scholar] [CrossRef]
- Yip, S.; Miao, J.; Cahill, D.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 Mutations Arise in Glioblastomas during Temozolomide Therapy and Mediate Temozolomide Resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [Green Version]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A Hypermutation Phenotype and Somatic MSH6 Mutations in Recurrent Human Malignant Gliomas after Alkylator Chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germano, G.; Lamba, S.E.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Tabata, K.; Kimura, T.; Yachie-Kinoshita, A.; Ozawa, Y.; Yamada, K.; Ito, J.; Tachino, S.; Hori, Y.; Matsuki, M.; et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS ONE 2019, 14, e0212513. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kato, Y.; Ozawa, Y.; Kodama, K.; Ito, J.; Ichikawa, K.; Yamada, K.; Hori, Y.; Tabata, K.; Takase, K.; et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1-6 hepatocellular carcinoma model. Cancer Sci. 2018, 109, 3993–4002. [Google Scholar] [CrossRef]
- Abou-Elkacem, L.; Arns, S.; Brix, G.; Gremse, F.; Zopf, D.; Kiessling, F.; Lederle, W. Regorafenib Inhibits Growth, Angiogenesis, and Metastasis in a Highly Aggressive, Orthotopic Colon Cancer Model. Mol. Cancer Ther. 2013, 12, 1322–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-W.; Ou, D.-L.; Hsu, C.-L.; Lin, L.; Cheng, A.-L.; Hsu, C. Regorafenib may enhance efficacy of anti-program cell death-1 therapy in hepatocellular carcinoma through modulation of macrophage polarization. J. Hepatol. 2019, 70, e605–e606. [Google Scholar] [CrossRef]
- Wu, R.-Y.; Kong, P.-F.; Xia, L.-P.; Huang, Y.; Li, Z.-L.; Tang, Y.-Y.; Chen, Y.-H.; Li, X.; Senthilkumar, R.; Zhang, H.-L.; et al. Regorafenib Promotes Antitumor Immunity via Inhibiting PD-L1 and IDO1 Expression in Melanoma. Clin. Cancer Res. 2019, 25, 4530–4541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuodeh, Y.; Venkat, P.; Kim, S. Systematic review of case reports on the abscopal effect. Curr. Probl. Cancer 2016, 40, 25–37. [Google Scholar] [CrossRef]
- Rodriguez-Ruiz, M.E.; Rodriguez, I.; Barbes, B.; Mayorga, L.; Sanchez-Paulete, A.R.; Ponz-Sarvise, M.; Pérez-Gracia, J.L.; Melero, I. Brachytherapy attains abscopal effects when combined with immunostimulatory monoclonal antibodies. Brachytherapy 2017, 16, 1246–1251. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.-X. Irradiation and anti–PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Aryankalayil, M.; Pilones, K.; Formenti, S.; Coleman, C.N.; Demaria, S. Fractionated but not single dose radiation releases key signals of in situ tumor vaccination. J. Immunother. Cancer 2014, 2, P164. [Google Scholar] [CrossRef] [Green Version]
- Young, K.H.; Baird, J.R.; Savage, T.; Cottam, B.; Friedman, D.; Bambina, S.; Messenheimer, D.J.; Fox, B.; Newell, P.; Bahjat, K.S.; et al. Optimizing Timing of Immunotherapy Improves Control of Tumors by Hypofractionated Radiation Therapy. PLoS ONE 2016, 11, e0157164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, K.; Almoguera, C.; Han, K.; Grizzle, W.E.; Perucho, M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nat. Cell Biol. 1987, 327, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Kumar, S.; Principe, D.R.; Singh, S.K.; Viswakarma, N.; Sondarva, G.; Rana, B.; Rana, A. Mitogen-Activated Protein Kinase Inhibitors and T-Cell-Dependent Immunotherapy in Cancer. Pharmaceuticals 2020, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, W.N.; Chang, C.-F.; Fischer, A.M.; Li, M.; Hedrick, S.M. The Erk2 MAPK Regulates CD8 T Cell Proliferation and Survival. J. Immunol. 2008, 181, 7617–7629. [Google Scholar] [CrossRef]
- Vella, L.; Pasam, A.; Dimopoulos, N.; Andrews, M.; Knights, A.; Puaux, A.-L.; Louahed, J.; Chen, W.; Woods, K.; Cebon, J. MEK Inhibition, Alone or in Combination with BRAF Inhibition, Affects Multiple Functions of Isolated Normal Human Lymphocytes and Dendritic Cells. Cancer Immunol. Res. 2014, 2, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, N.; White, B.S.; Goussous, G.; Pickles, O.; Mason, M.; Beggs, A.; Taniere, P.; Willcox, B.E.; Guinney, J.; Middleton, G.W. KRAS Mutation and Consensus Molecular Subtypes 2 and 3 Are Independently Associated with Reduced Immune Infiltration and Reactivity in Colorectal Cancer. Clin. Cancer Res. 2018, 24, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Blaj, C.; Schmidt, E.M.; Lamprecht, S.; Hermeking, H.; Jung, A.; Kirchner, T.; Horst, D. Oncogenic Effects of High MAPK Activity in Colorectal Cancer Mark Progenitor Cells and Persist Irrespective of RAS Mutations. Cancer Res. 2017, 77, 1763–1774. [Google Scholar] [CrossRef] [Green Version]
- Becht, E.; De Reyniès, A.; Giraldo, N.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautes-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Sorbye, H.; Dragomir, A.; Sundström, M.; Pfeiffer, P.; Thunberg, U.; Bergfors, M.; Aasebø, K.; Eide, G.E.; Ponten, F.; Qvortrup, C.; et al. High BRAF Mutation Frequency and Marked Survival Differences in Subgroups According to KRAS/BRAF Mutation Status and Tumor Tissue Availability in a Prospective Population-Based Metastatic Colorectal Cancer Cohort. PLoS ONE 2015, 10, e0131046. [Google Scholar] [CrossRef] [Green Version]
- Tran, B.; Kopetz, S.; Tie, J.; Gibbs, P.; Jiang, Z.-Q.; Lieu, C.H.; Agarwal, A.; Maru, D.M.; Sieber, O.; Desai, J. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011, 117, 4623–4632. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-Mediated Reactivation of MAPK Signaling Contributes to Insensitivity of BRAF-Mutant Colorectal Cancers to RAF Inhibition with Vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, R.; Giannakis, M.; Allen, J.; Chen, J.; Pelka, K.; Chao, S.; Meyerhardt, J.; Enzinger, A.; Enzinger, P.; McCleary, N.; et al. SO-26 Clinical efficacy of combined BRAF, MEK, and PD-1 inhibition in BRAFV600E colorectal cancer patients. Ann. Oncol. 2020, 31, S226–S227. [Google Scholar] [CrossRef]
- Ebert, P.J.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.-Y.; Hopson, C.; et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef] [Green Version]
- Nusrat, M.; Roszik, J.; Katkhuda, R.; Menter, D.; Raghav, K.P.S.; Morris, V.K.; Sharma, P.; Allison, J.P.; Blando, J.M.; Maru, D.M.; et al. Association of phosphatidylinositol 3-kinase (PI3K) pathway activation with increased immune checkpoint expression in colorectal cancer (CRC) patients. J. Clin. Oncol. 2018, 36, 653. [Google Scholar] [CrossRef]
- Collins, N.B.; Abosy, R.A.; Miller, B.; Bi, K.; Manguso, R.; Yates, K.; Haining, W.N. PI3K activated tumors evade tumor immunity by promoting an inhibitory myeloid microenvironment. J. Immunol. 2019, 202, 58.17. [Google Scholar]
- Ahmad, S.; Abu Eid, R.; Shrimali, R.; Webb, M.; Verma, V.; Doroodchi, A.; Berrong, Z.; Samara, R.; Rodriguez, P.C.; Mkrtichyan, M.; et al. Differential PI3Kδ Signaling in CD4+ T-cell Subsets Enables Selective Targeting of T Regulatory Cells to Enhance Cancer Immunotherapy. Cancer Res. 2017, 77, 1892–1904. [Google Scholar] [CrossRef] [Green Version]
- Carnevalli, L.S.; Sinclair, C.; Taylor, M.A.; Morentin-Gutierrez, P.; Langdon, S.; Coenen-Stass, A.M.L.; Mooney, L.; Hughes, A.; Jarvis, L.; Staniszewska, A.; et al. PI3Kα/δ inhibition promotes anti-tumor immunity through direct enhancement of effector CD8+ T-cell activity. J. Immunother. Cancer 2018, 6, 158. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Chang, C.-H.; Wang, Y.; Li, R.; Rossi, D.L.; Liu, D.; Rossi, E.A.; Cardillo, T.M.; Goldenberg, D.M. Combination Therapy with Bispecific Antibodies and PD-1 Blockade Enhances the Antitumor Potency of T Cells. Cancer Res. 2017, 77, 5384–5394. [Google Scholar] [CrossRef] [Green Version]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.F.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osada, T.; Patel, S.P.; Hammond, S.A.; Osada, K.; Morse, M.A.; Lyerly, H. CEA/CD3-bispecific T cell-engaging (BiTE) antibody-mediated T lymphocyte cytotoxicity maximized by inhibition of both PD1 and PD-L1. Cancer Immunol. Immunother. 2015, 64, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual Blockade of PD-1 and CTLA-4 Combined with Tumor Vaccine Effectively Restores T-Cell Rejection Function in Tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chon, H.J.; Kim, H.; Noh, J.H.; Yang, H.; Lee, W.S.; Kong, S.J.; Lee, S.J.; Lee, Y.S.; Kim, W.R.; Kim, J.H.; et al. STING signaling is a potential immunotherapeutic target in colorectal cancer. J. Cancer 2019, 10, 4932–4938. [Google Scholar] [CrossRef]


{kind=link}
| Study | Treatment | Phase | Setting | Sample Size | ORR | Median PFS | Median OS |
|---|---|---|---|---|---|---|---|
| Combination of immune checkpoint inhibitors | |||||||
| CCTG CO.26 [11] | Durvalumab + Tremelimumab vs. BSC | II | Refractory | 119 vs. 61 | 0.5% vs. 0% | 1.8 vs. 1.9 months | 6.6 vs. 4.1 months |
| NCT02720068 [12] | Pembrolizumab + Favezelimab | I | Refractory | 80 | 6.3% | 2.1 months | 8.3 months |
| Immunotherapy in combination with chemotherapy | |||||||
| Immunotherapy in combination with chemotherapy and anti-VEGF agents | |||||||
| MODUL [13] | FOLFOX + Bevacizumab followed by 5-FU + Bevacizumab + Atezolizumab vs. 5-FU + Bevacizumab | II | First line | 297 vs. 148 | NA | 7.2 vs. 7.39 months | 22 vs. 21.9 months |
| ATEZOTRIBE [14] | FOLFOX + Bevacizumab + Atezolizumab vs. FOLFOX + Atezolizumab | III | First line | 132 vs. 67 | 59% vs. 64% | 12.9 vs. 11.4 months | NA |
| CA2099 × 8 | FOLFOX + Bevacizumab + Nivolumab vs. FOLFOX + Bevacizumab | II/III | First line | Active | NA | NA | NA |
| COLUMBIA-1 | FOLFOX + Bevacizumab + Durvalumab + Oleclumab vs. FOLFOX + Bevacizumab | II | First line | Active | NA | NA | NA |
| NIVACOR (NCT04072198) | FOLFOXIRI + Bevacizumab + Nivolumab | II | First line | Recruiting | NA | NA | NA |
| BACCI (NCT0287319) [15] | Capecitabine + Bevacizumab + Atezolizumab vs. Capecitabine + Bevacizumab | II | Refractory | 82 vs. 46 | 8.5% vs. 4.3% | 4.4 vs. 3.3 months | NA |
| Immunotherapy in combination with chemotherapy and anti-EGFR agents | |||||||
| AVETRIC (NCT04513951) | FOLFOXIRI + Cetuximab + Avelumab | II | First line | Recruiting | NA | NA | NA |
| AVETUX (NCT03174405) [16] | FOLFOX + Cetuximab + Avelumab | II | First line | 43 | 79.5% | 11.5 months | NA |
| Immunotherapy in combination with temozolomide | |||||||
| ARETHUSA (NCT03519412) | Temozolomide followed by Pembrolizumab | II | Refractory | Recruiting | NA | NA | NA |
| MAYA [17] | Temozolomide + Nivolumab + Ipilimumab | II | Refractory | 33 | 42% | 7.1 months | 18.4 months |
| Immunotherapy in combination with antiangiogenic agents | |||||||
| LEAP-005 [18] | Pembrolizumab + Lenvatinib | II | Refractory | 32 | 22% | 2.3 months | 7.5 months |
| REGONIVO [19] | Nivolumab + Regorafenib | Ib | Refractory | 25 | 36% | 7.9 months | NA |
| REGOMUNE [20] | Avelumab + Regorafenib | II | ≥2 lines | 48 | 0% | 3.6 months | 10.8 months |
| Immunotherapy in combination with radiotherapy | |||||||
| NCT03122509 [21] | Radiotherapy + Durvalumab + Tremelimumab | II | >2 lines | 24 | 8.3% | 1.8 months | 11.4 months |
| NCT03104439 [22] | SBRT + Nivolumab + Ipilimumab | II | >2 lines | 40 | 12,5% | NA | NA |
| NCT02992912 | SABR + Atezolizumab | II | Refractory | Recruiting | NA | NA | NA |
| Immunotherapy in combination with MAPK signaling inhibitors | |||||||
| Immunotherapy in combination with KRAS inhibitors | |||||||
| CodeBreaK 100 | AMG 510 +/− AntiPD-1/L1 | I/II | Refractory | Recruiting | NA | NA | NA |
| NCT04699188 | JDQ443 +/− TNO155 +/- Spartalizumab | I/II | Refractory | Recruiting | NA | NA | NA |
| Immunotherapy in combination with BRAF inhibitors | |||||||
| NCT03668431 | Dabrafenib + Trametinib (MEK) + Spartalizumab | II | Refractory | Recruiting | NA | NA | NA |
| NCT04294160 | Dabrafenib + LTT462 (ERK) + Spartalizumab | I | Refractory | Recruiting | NA | NA | NA |
| Immunotherapy in combination with MEK inhibitors | |||||||
| NCT01988896 [23] | Cobimetinib + Atezolizumab | I/Ib | Refractory | 84 | 8% | 1.9 months | 9.8 months |
| IMBlaze 370 [24] | Atezolizumab + Cobimetinib vs. Atezolizumab vs. Regorafenib | III | Refractory | 183 vs. 90 vs. 90 | 3% | 1.9 vs. 1.9 vs. 2 months | 8.9 vs. 7.1 vs. 8.5 months |
| Immunotherapy in combination with inhibition of the PI3K/AKT/mTOR pathway | |||||||
| NCT03711058 [25] | Copanlisib + Nivolumab | I/II | Refractory | Recruiting | NA | NA | NA |
| Biespecific antibodies | |||||||
| NCT02324257 [26] | Cibisatamab | I | Refractory | 68 | 6% | NA | NA |
| NCT02650713 [26] | Cibisatamab + Atezolizumab | I | Refractory | 38 | 12% | NA | NA |
| NCT04826003 | RO7122290 + Cibisatamab + Obinutuzumab | I | Refractory | Recruiting | NA | NA | NA |
| NCT04468607 | BLYG8824A | I | Refractory | Recruiting | NA | NA | NA |
| Vaccines | |||||||
| NCT01413295 [27] | Dendritic cell vaccine vs. BSC | II | >2 lines | 28 vs. 24 | 0% vs. 0% | 2.7 vs. 2.3 months | 6.2 vs. 4.7 months |
| FXV [28] | HLA-A*2402-restricted peptides + oxaliplatin-based chemotherapy | II | First line | 50 (HLA-A*2402-matched) | 62% | 7.2 months | 20.7 months |
| II | First line | 46 (HLA-A*2402-unmatched) | 60.9% | 8.7 months | 24.0 months | ||
| Intratumoral therapies | |||||||
| NCT00149396 [29] | Hepatic artery infusion of NV1020 followed by conventional chemotherapy | I/II | Refractory | 22 | 4.5% | 6.4 months | 11.8 months |
| NCT03256344 | Talimogene laherparepvec + Atezolizumab | Ib | Refractory | Active | NA | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baraibar, I.; Mirallas, O.; Saoudi, N.; Ros, J.; Salvà, F.; Tabernero, J.; Élez, E. Combined Treatment with Immunotherapy-Based Strategies for MSS Metastatic Colorectal Cancer. Cancers 2021, 13, 6311. https://doi.org/10.3390/cancers13246311
Baraibar I, Mirallas O, Saoudi N, Ros J, Salvà F, Tabernero J, Élez E. Combined Treatment with Immunotherapy-Based Strategies for MSS Metastatic Colorectal Cancer. Cancers. 2021; 13(24):6311. https://doi.org/10.3390/cancers13246311
Chicago/Turabian StyleBaraibar, Iosune, Oriol Mirallas, Nadia Saoudi, Javier Ros, Francesc Salvà, Josep Tabernero, and Elena Élez. 2021. "Combined Treatment with Immunotherapy-Based Strategies for MSS Metastatic Colorectal Cancer" Cancers 13, no. 24: 6311. https://doi.org/10.3390/cancers13246311
APA StyleBaraibar, I., Mirallas, O., Saoudi, N., Ros, J., Salvà, F., Tabernero, J., & Élez, E. (2021). Combined Treatment with Immunotherapy-Based Strategies for MSS Metastatic Colorectal Cancer. Cancers, 13(24), 6311. https://doi.org/10.3390/cancers13246311