
Targeting KRAS in NSCLC: Old Failures and New Options for “Non-G12c” Patients


 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. KRAS Structure and Signalling Pathway
3. KRAS Mutation Subtypes
4. KRAS Co-Mutations
5. Clinical Characteristics and Standard Treatment of KRAS-Mutant NSCLC Patients
6. Therapeutic Strategies: Recent Clinical Evidence
6.1. Direct Targeting of Mutant “Non-p.G12C” KRAS
6.2. Targeting KRAS Membrane Anchorage
6.3. Targeting KRAS Downstream Pathways
6.4. Targeting Co-Dependent Vulnerabilities or Synthetic Lethal Partners
6.5. Targeting KRAS Activity Regulators
6.6. Targeting Immune System
6.7. Epigenetic Approaches
7. Discussion
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Malapelle, U.; Passiglia, F.; Cremolini, C.; Reale, M.L.; Pepe, F.; Pisapia, P.; Avallone, A.; Cortinovis, D.; De Stefano, A.; Fassan, M.; et al. RAS as a positive predictive biomarker: Focus on lung and colorectal cancer patients. Eur. J. Cancer 2021, 146, 74–83. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reck, M.; Spira, A.; Besse, B.; Wolf, J.; Skoulidis, F.; Borghaei, H.; Goto, K.; Park, K.; Griesinger, F.; Felip, E.; et al. 1416TiP CodeBreak 200: A phase III multicenter study of sotorasib (AMG 510), a KRAS(G12C) inhibitor, versus docetaxel in patients with previously treated advanced non-small cell lung cancer (NSCLC) harboring KRAS p.G12C mutation. Ann Oncol. 2020, 31, S894–S895. [Google Scholar] [CrossRef]
- Passiglia, F.; Malapelle, U.; Del Re, M.; Righi, L.; Pagni, F.; Furlan, D.; Danesi, R.; Troncone, G.; Novello, S. KRAS inhibition in non–small cell lung cancer: Past failures, new findings and upcoming challenges. Eur. J. Cancer 2020, 137, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Gimple, R.C.; Wang, X. RAS: Striking at the Core of the Oncogenic Circuitry. Front. Oncol. 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer-Smith, R.; O’Bryan, J.P. Direct inhibition of RAS: Quest for the Holy Grail? Semin. Cancer Biol. 2019, 54, 138–148. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic targeting of RAS: New hope for drugging the “undruggable”. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118570. [Google Scholar] [CrossRef]
- Ferrer, I.; Zugazagoitia, J.; Herbertz, S.; John, W.; Paz-Ares, L.; Schmid-Bindert, G. KRAS-Mutant non-small cell lung cancer: From biology to therapy. Lung Cancer 2018, 124, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Bodemann, B.O.; White, M.A. Ral GTPases and cancer: Linchpin support of the tumorigenic platform. Nat. Rev. Cancer 2008, 8, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Kashatus, D.F. Ral GTPases in tumorigenesis: Emerging from the shadows. Exp. Cell Res. 2013, 319, 2337–2342. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef]
- Bellahcène, A.; Nokin, M.J.; Castronovo, V.; Schalkwijk, C. Methylglyoxal-derived stress: An emerging biological factor involved in the onset and progression of cancer. Semin. Cancer Biol. 2018, 49, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2011, 39, 945–950. [Google Scholar] [CrossRef] [Green Version]
- Timar, J.; Kashofer, K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metastasis Rev. 2020, 39, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- El Osta, B.; Hu, F.; Sadek, R.; Chintalapally, R.; Tang, S.C. Not all immune-checkpoint inhibitors are created equal: Meta-analysis and systematic review of immune-related adverse events in cancer trials. Crit. Rev. Oncol. Hematol. 2017, 119, 1–12. [Google Scholar] [CrossRef]
- Veluswamy, R.; Mack, P.C.; Houldsworth, J.; Elkhouly, E.; Hirsch, F.R. KRAS G12C–Mutant Non–Small Cell Lung Cancer: Biology, Developmental Therapeutics, and Molecular Testing. J. Mol. Diagn. 2021, 23, 507–520. [Google Scholar] [CrossRef]
- Friedlaender, A.; Drilon, A.; Weiss, G.J.; Banna, G.L.; Addeo, A. KRAS as a druggable target in NSCLC: Rising like a phoenix after decades of development failures. Cancer Treat. Rev. 2020, 85, 101978. [Google Scholar] [CrossRef]
- Lee, B.; Lee, T.; Lee, S.H.; Choi YLa Han, J. Clinicopathologic characteristics of EGFR, KRAS, and ALK alterations in 6595 lung cancers. Oncotarget 2016, 7, 23874–23884. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Xie, Z.; Liu, S.Y.; Tu, H.Y.; Chen, H.J.; Sun, Y.L.; Zhou, Q.; et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Daemen, A.; Nickles, D.; Jeon, S.M.; Foreman, O.; Sudini, K.; Gnad, F.; Lajoie, S.; Gour, N.; Mitzner, W.; et al. NRF2 Activation Promotes Aggressive Lung Cancer and Associates with Poor Clinical Outcomes. Clin. Cancer Res. 2021, 27, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Best, S.A.; Ding, S.; Kersbergen, A.; Dong, X.; Song, J.Y.; Xie, Y.; Reljic, B.; Li, K.; Vince, J.E.; Rathi, V.; et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat Commun. 2019, 13, 4190. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [Green Version]
- Sayin, V.I.; LeBoeuf, S.E.; Singh, S.X.; Davidson, S.M.; Biancur, D.; Guzelhan, B.S.; Alvarez, S.W.; Wu, W.L.; Karakousi, T.R.; Zavitsanou, A.M.; et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 2017, 6, e28083. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavan, A.; Bragadin, A.B.; Calvetti, L.; Ferro, A.; Zulato, E.; Attili, I.; Nardo, G.; Dal Maso, A.; Frega, S.; Menin, A.G.; et al. Role of next generation sequencing-based liquid biopsy in advanced non-small cell lung cancer patients treated with immune checkpoint inhibitors: Impact of STK11, KRAS and TP53 mutations and co-mutations on outcome. Transl. Lung Cancer Res. 2021, 10, 202–220. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Schuster, K.; Venkateswaran, N.; Rabellino, A.; Girard, L.; Peña-Llopis, S.; Scaglioni, P.P. Nullifying the CDKN2AB locus promotes mutant K-ras lung tumorigenesis. Mol. Cancer Res. 2014, 12, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Garrido, P.; Olmedo, M.E.; Gómez, A.; Paz Ares, L.; López-Ríos, F.; Rosa-Rosa, J.M.; Palacios, J. Treating KRAS -mutant NSCLC: Latest evidence and clinical consequences. Ther. Adv. Med. Oncol. 2017, 9, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular epidemiology of EGFR and KRAS mutations in 3026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [Green Version]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- NCCN. NCCN Guidelines for Patients® Non-Small Cell Lung Cancer-Metastatic. 2021. Available online: http://www.nccn.org/patients (accessed on 15 July 2021).
- Nadal, E.; Chen, G.; Prensner, J.R.; Shiratsuchi, H.; Sam, C.; Zhao, L.; Kalemkerian, G.P.; Brenner, D.; Lin, J.; Reddy, R.M.; et al. KRAS-G12C mutation is associated with poor outcome in surgically resected lung adenocarcinoma. J. Thorac. Oncol. 2014, 9, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- Rodenhuis, S.; Boerrigter, L.; Top, B.; Slebos, R.J.; Mooi, W.J.; van’t Veer, L.; van Zandwijk, N. Mutational activation of the K-ras oncogene and the effect of chemotherapy in advanced adenocarcinoma of the lung: A prospective study. J. Clin. Oncol. 1997, 15, 285–291. [Google Scholar] [CrossRef]
- Schiller, J.H.; Adak, S.; Feins, R.H.; Keller, S.M.; Fry, W.A.; Livingston, R.B.; Hammond, M.E.; Wolf, B.; Sabatini, L.; Jett, J.; et al. Lack of prognostic significance of p53 and K-ras mutations in primary resected non-small-cell lung cancer on E4592: A laboratory ancillary study on an eastern cooperative oncology group prospective randomized trial of postoperative adjuvant therapy. J. Clin. Oncol. 2001, 19, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, D.A.; Johnson, B.E.; Amler, L.C.; Goddard, A.D.; Heldens, S.L.; Herbst, R.S.; Ince, W.L.; Jänne, P.A.; Januario, T.; Johnson, D.H.; et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J. Clin. Oncol. 2005, 23, 5900–5909. [Google Scholar] [CrossRef] [PubMed]
- Hames, M.L.; Chen, H.; Iams, W.; Aston, J.; Lovly, C.M.; Horn, L. Correlation between KRAS mutation status and response to chemotherapy in patients with advanced non-small cell lung cancer. Lung Cancer 2016, 92, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Marabese, M.; Ganzinelli, M.; Garassino, M.C.; Shepherd, F.A.; Piva, S.; Caiola, E.; Macerelli, M.; Bettini, A.; Lauricella, C.; Floriani, I.; et al. KRAS mutations affect prognosis of non-small-cell lung cancer patients treated with first-line platinum containing chemotherapy. Oncotarget 2015, 6, 34014–34022. [Google Scholar] [CrossRef] [Green Version]
- Dingemans, A.-M.C.; Ernst, S.; Mellema, W.; Burgers, S.; Staal-van den Brekel, J.; Hendriks, L.E.L.; Hiltermann, T.J.N.; van Walree, N.; Maas, K.; Youssef-ElSoud, M.; et al. LBA50 A randomized phase III study comparing cisplatin-pemetrexed (cis-pem) with carboplatin (C)-paclitaxel (P)-bevacizumab (B) in chemotherapy naïve patients (pts) with advanced KRAS mutated non-small cell lung cancer (NSCLC): NVALT22. Ann. Oncol. 2021, 32, S1327. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Lacas, B.; Le Teuff, G.; Hainaut, P.; Jänne, P.A.; Pignon, J.P.; Le Chevalier, T.; Seymour, L.; Douillard, J.Y.; Graziano, S.; et al. Pooled analysis of the prognostic and predictive effects of TP53 comutation status combined with KRAS or EGFR mutation in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J. Clin. Oncol. 2017, 35, 2018–2027. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Lee, C.K.; Man, J.; Lord, S.; Cooper, W.; Links, M.; Gebski, V.; Herbst, R.S.; Gralla, R.J.; Mok, T.; Yang, J.C. Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non-small cell lung carcinoma: A systematic review and meta-analysis. JAMA Oncol. 2018, 4, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.S.; Kim, B.J. Prognostic value of KRAS mutation in advanced non-small-cell lung cancer treated with immune checkpoint inhibitors: A metaanalysis and review. Oncotarget 2017, 8, 48248–48252. [Google Scholar] [CrossRef] [Green Version]
- Passiglia, F.; Cappuzzo, F.; Alabiso, O.; Bettini, A.C.; Bidoli, P.; Chiari, R.; Defferrari, C.; Delmonte, A.; Finocchiaro, G.; Francini, G.; et al. Efficacy of nivolumab in pre-treated non-small-cell lung cancer patients harbouring KRAS mutations. Br. J. Cancer 2019, 120, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Nagasaka, M.; Li, Y.; Sukari, A.; Ou, S.H.I.; Al-Hallak, M.N.; Azmi, A.S. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat. Rev. 2020, 84, 101974. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, C.R.; Jamal-Hanjani, M.; Forster, M.; Blackhall, F. KRAS: Reasons for optimism in lung cancer. Eur. J. Cancer 2018, 99, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.C.; Gurbani, D.; Ficarro, S.B.; Carrasco, M.A.; Lim, S.M.; Choi, H.G.; Xie, T.; Marto, J.A.; Chen, Z.; Gray, N.S.; et al. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. USA 2014, 111, 8895–8900. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Solomon, M.; Li, L.S.; Hansen, R.; Rosen, N. Cancer therapeutics: Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.M.; Westover, K.D.; Ficarro, S.B.; Harrison, R.A.; Choi, H.G.; Pacold, M.E.; Carrasco, M.; Hunter, J.; Kim, N.D.; Xie, T.; et al. Therapeutic targeting of oncogenic K-ras by a covalent catalytic site inhibitor. Angew. Chemie. Int. Ed. 2014, 53, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 127, 578–589.e17. [Google Scholar] [CrossRef] [Green Version]
- Therapeutics, M. Mirati Therapeutics Reports Investigational Adagrasib (MRTX849) Preliminary Data Demonstrating Tolerability and Durable Anti-Tumor Activity as Well as Initial MRTX1133 Preclinical Data. 2020. Available online: https://ir.mirati.com/news-releases/news-details/2020/Mirati-Therapeutics-Reports-Investigational-Adagrasib-MRTX849-Preliminary-Data-Demonstrating-Tolerability-and-Durable-Anti-Tumor-Activity-aswell-as-Initial-MRTX1133-Preclinical-Data/default.aspx (accessed on 3 December 2021).
- Available online: https://www.revmed.com/pipeline/rason-inhibitors (accessed on 7 December 2021).
- End, D.W.; Smets, G.; Todd, A.V.; Applegate, T.L.; Fuery, C.J.; Angibaud, P.; Venet, M.; Sanz, G.; Poignet, H.; Skrzat, S.; et al. Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Res. 2001, 61, 131–137. [Google Scholar]
- Gunning, W.T.; Kramer, P.M.; Lubet, R.A.; Steele, V.E.; End, D.W.; Wouters, W.; Pereira, M.A. Chemoprevention of benzo(a)pyrene-induced lung tumors in mice by the farnesyltransferase inhibitor R115777. Clin. Cancer Res. 2003, 9, 1927–1930. [Google Scholar]
- Kim, E.S.; Kies, M.S.; Fossella, F.V.; Glisson, B.S.; Zaknoen, S.; Statkevich, P.; Munden, R.F.; Summey, C.; Pisters, K.M.; Papadimitrakopoulou, V.; et al. Phase II study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in patients with taxane-refractory/resistant nonsmall cell lung carcinoma. Cancer 2005, 104, 561–569. [Google Scholar] [CrossRef]
- Wong, N.S.; Meadows, K.L.; Rosen, L.S.; Adjei, A.A.; Kaufmann, S.H.; Morse, M.A.; Petros, W.P.; Zhu, Y.; Statkevich, P.; Cutler, D.L.; et al. A phase I multicenter study of continuous oral administration of lonafarnib (SCH 66336) and intravenous gemcitabine in patients with advanced cancer. Cancer Investig. 2011, 29, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Adjei, A.A.; Mauer, A.; Bruzek, L.; Marks, R.S.; Hillman, S.; Geyer, S.; Hanson, L.J.; Wright, J.J.; Erlichman, C.; Kaufmann, S.H.; et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2003, 21, 1760–1766. [Google Scholar] [CrossRef]
- Tanaka, A.; Radwan, M.O.; Hamasaki, A.; Ejima, A.; Obata, E.; Koga, R.; Tateishi, H.; Okamoto, Y.; Fujita, M.; Nakao, M.; et al. A novel inhibitor of farnesyltransferase with a zinc site recognition moiety and a farnesyl group. Bioorganic Med. Chem. Lett. 2017, 27, 3862–3866. [Google Scholar] [CrossRef]
- Kazi, A.; Xiang, S.; Yang, H.; Chen, L.; Kennedy, P.; Ayaz, M.; Fletcher, S.; Cummings, C.; Lawrence, H.R.; Beato, F.; et al. Dual farnesyl and geranylgeranyl transferase inhibitor thwarts mutant KRAS-driven patient-derived pancreatic tumors. Clin. Cancer Res. 2019, 25, 5984–5996. [Google Scholar] [CrossRef] [PubMed]
- Zundelevich, A.; Elad-Sfadia, G.; Haklai, R.; Kloog, Y. Suppression of lung cancer tumor growth in a nude mouse model by the Ras inhibitor salirasib (farnesylthiosalicylic acid). Mol. Cancer Ther. 2007, 6, 1765–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riely, G.J.; Johnson, M.L.; Medina, C.; Rizvi, N.A.; Miller, V.A.; Kris, M.G.; Pietanza, M.C.; Azzoli, C.G.; Krug, L.M.; Pao, W.; et al. A phase II trial of salirasib in patients with lung adenocarcinomas with KRAS mutations. J. Thorac. Oncol. 2011, 6, 1435–1437. [Google Scholar] [CrossRef] [Green Version]
- Kenessey, I.; Kói, K.; Horváth, O.; Cserepes, M.; Molnár, D.; Izsák, V.; Dobos, J.; Hegedűs, B.; Tóvári, J.; Tímár, J. KRAS-mutation status dependent effect of zoledronic acid in human non-small cell cancer preclinical models. Oncotarget 2016, 7, 79503–79514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS membrane association: Back to the future for anti-RAS drug discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [Google Scholar] [CrossRef] [Green Version]
- Takezawa, K.; Okamoto, I.; Yonesaka, K.; Hatashita, E.; Yamada, Y.; Fukuoka, M.; Nakagawa, K. Sorafenib inhibits non-small cell lung cancer cell growth by targeting B-RAF in KRAS wild-type cells and C-RAF in KRAS mutant cells. Cancer Res. 2009, 69, 6515–6521. [Google Scholar] [CrossRef] [Green Version]
- Dingemans, A.M.C.; Mellema, W.W.; Groen, H.J.M.; Van Wijk, A.; Burgers, S.A.; Kunst, P.W.A.; Thunnissen, E.; Heideman, D.A.; Smit, E.F. A phase II study of sorafenib in patients with platinum-pretreated, advanced (stage IIIb or IV) non-small cell lung cancer with a KRAS mutation. Clin. Cancer Res. 2013, 19, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Paz-Ares, L.; Hirsh, V.; Zhang, L.; De Marinis, F.; Yang, J.C.H.; Wakelee, H.A.; Seto, T.; Wu, Y.L.; Novello, S.; Juhász, E.; et al. Monotherapy Administration of Sorafenib in Patients with Non-Small Cell Lung Cancer (MISSION) Trial: A Phase III, Multicenter, Placebo-Controlled Trial of Sorafenib in Patients with Relapsed or Refractory Predominantly Nonsquamous Non-Small-Cell Lung Cancer. J. Thorac. Oncol. 2015, 10, 1745–1753. [Google Scholar]
- Jänne, P.A.; Shaw, A.T.; Pereira, J.R.; Jeannin, G.; Vansteenkiste, J.; Barrios, C.; Franke, F.A.; Grinsted, L.; Zazulina, V.; Smith, P.; et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: A randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013, 14, 38–47. [Google Scholar] [CrossRef]
- Jänne, P.A.; Van Den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crinò, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: The SELECT-1 randomized clinical trial. JAMA J. Am. Med. Assoc. 2017, 317, 1844–1853. [Google Scholar] [CrossRef] [Green Version]
- Carter, C.A.; Rajan, A.; Keen, C.; Szabo, E.; Khozin, S.; Thomas, A.; Brzezniak, C.; Guha, U.; Doyle, L.A.; Steinberg, S.M.; et al. Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced nonsmall-cell lung cancer. Ann. Oncol. 2016, 24, 693–699. [Google Scholar] [CrossRef]
- Blumenschein, G.R.; Smit, E.F.; Planchard, D.; Kim, D.W.; Cadranel, J.; De Pas, T.; Dunphy, F.; Udud, K.; Ahn, M.J.; Hanna, N.H.; et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC). Ann. Oncol. 2015, 26, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Gadgeel, S.M.; Miao, J.; Riess, J.W.; Mack, P.C.; Gerstner, G.J.; Burns, T.F.; Taj, A.; Akerley, W.L.; Dragnev, K.H.; Moon, J.; et al. S1507: Phase II study of docetaxel and trametinib in patients with G12C or non-G12C KRAS mutation positive (+) recurrent non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2019, 37, 9021. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, R.; Chen, H.; Wang, L.; Ren, C.; Pataer, A.; Wu, S.; Meng, Q.H.; Ha, M.J.; Morris, J.; et al. KRT-232 and navitoclax enhance trametinib’s anti-Cancer activity in non-small cell lung cancer patient-derived xenografts with KRAS mutations. Am. J. Cancer Res. 2020, 10, 4464–4475. [Google Scholar]
- Corcoran, R.B.; Do, K.T.; Cleary, J.M.; Parikh, A.R.; Yeku, O.O.; Weekes, C.D.; Veneris, J.; Ahronian, L.G.; Mauri, G.; Van Seventer, E.E.; et al. Phase I/II study of combined BCL-XL and MEK inhibition with navitoclax (N) and trametinib (T) in KRAS or NRAS mutant advanced solid tumours. Ann. Oncol. 2019, 30, v164. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Patnaik, A.; Papadopoulos, K.P.; Rasco, D.W.; Becerra, C.R.; Allred, A.J.; Orford, K.; Aktan, G.; Ferron-Brady, G.; Ibrahim, N.; et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother Pharmacol. 2015, 75, 183–189. [Google Scholar] [CrossRef]
- Mita, M.; Fu, S.; Piha-Paul, S.A.; Janku, F.; Mita, A.; Natale, R.; Guo, W.; Zhao, C.; Kurzrock, R.; Naing, A. Phase I trial of MEK 1/2 inhibitor pimasertib combined with mTOR inhibitor temsirolimus in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Gandhi, L.; Mita, M.M.; Damstrup, L.; Campana, F.; Hidalgo, M.; Grande, E.; Hyman, D.M.; Heist, R.S. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br. J. Cancer 2018, 119, 1471–1476. [Google Scholar] [CrossRef]
- Dual RAF-MEK Inhibitor Assessed. Cancer Discov. 2021, 11, 5–6. [CrossRef]
- Ishii, N.; Harada, N.; Joseph, E.W.; Ohara, K.; Miura, T.; Sakamoto, H.; Matsuda, Y.; Tomii, Y.; Tachibana-Kondo, Y.; Iikura, H.; et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of raf activity. Cancer Res. 2013, 73, 4050–4060. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Saborowski, A.; Yue, J.; Solomon, M.; Joseph, E.; Gadal, S.; Saborowski, M.; Kastenhuber, E.; Fellmann, C.; Ohara, K.; et al. Disruption of CRAF-Mediated MEK activation is required for effective mek inhibition in KRAS mutant tumors. Cancer Cell 2014, 25, 697–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Chénard-Poirier, M.; Roda, D.; de Miguel, M.; Harris, S.J.; Candilejo, I.M.; Sriskandarajah, P.; Xu, W.; Scaranti, M.; Constantinidou, A.; et al. Intermittent schedules of the oral RAF–MEK inhibitor CH5126766/VS-6766 in patients with RAS/RAF-mutant solid tumours and multiple myeloma: A single-centre, open-label, phase 1 dose-escalation and basket dose-expansion study. Lancet Oncol. 2020, 21, 1478–1488. [Google Scholar] [CrossRef]
- Sanclemente, M.; Francoz, S.; Esteban-Burgos, L.; Bousquet-Mur, E.; Djurec, M.; Lopez-Casas, P.P.; Hidalgo, M.; Guerra, C.; Drosten, M.; Musteanu, M.; et al. c-RAF Ablation Induces Regression of Advanced Kras/Trp53 Mutant Lung Adenocarcinomas by a Mechanism Independent of MAPK Signaling. Cancer Cell 2018, 33, 217–228.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coma, S.; Chowdhury, S.; Pachter, J.A. Abstract 1263: Dual RAF/MEK Inhibitor VS-6766 Enhances Antitumor Efficacy of KRAS-G12C Inhibitors through A Vertical Pathway Inhibition Strategy. In Proceedings of the American Association for Cancer Research Annual Meeting 2021; AACR: Philadelphia, PA, USA, 2021. [Google Scholar]
- Shinde, R.; Terbuch, A.; Little, M.; Caldwell, R.; Kurup, R.; Riisnaes, R.; Crespo, M.; Ruddle, R.; Gurel, B.; Stewart, A.; et al. Abstract CT143: Phase I Study of the Combination of A RAF-MEK Inhibitor CH5126766 and FAK Inhibitor Defactinib in an Intermittent Dosing Schedule with Expansions in KRAS Mutant Cancers. In Proceedings of the Annual Meeting of the American Association for Cancer Research 2020; AACR: Philadelphia, PA, USA, 2020. [Google Scholar]
- Krebs, M.G.; Shinde, R.; Rahman, R.A.; Grochot, R.; Little, M.; King, J.; Kitchin, J.; Parmar, M.; Turner, A.; Mahmud, M.; et al. Abstract CT019: A Phase I Trial of the Combination of the Dual RAF-MEK Inhibitor VS-6766 and the FAK Inhibitor Defactinib: Evaluation of Efficacy in KRAS Mutated NSCLC. In Proceedings of the American Association for Cancer Research Annual Meeting 2021; AACR: Philadelphia, PA, USA, 2021. [Google Scholar]
- Riely, G.J.; Brahmer, J.R.; Planchard, D.; Crinò, L.; Doebele, R.C.; Mas Lopez, L.A.; Gettinger, S.N.; Schumann, C.; Li, X.; Atkins, B.; et al. A randomized discontinuation phase II trial of ridaforolimus in non-small cell lung cancer (NSCLC) patients with KRAS mutations. J. Clin. Oncol. 2012, 40, 7531. [Google Scholar] [CrossRef]
- Vansteenkiste, J.F.; Canon, J.L.; De Braud, F.; Grossi, F.; De Pas, T.; Gray, J.E.; Su, W.C.; Felip, E.; Yoshioka, H.; Gridelli, C.; et al. Safety and Efficacy of Buparlisib (BKM120) in Patients with PI3K Pathway-Activated Non-Small Cell Lung Cancer: Results from the Phase II BASALT-1 Study. J. Thorac. Oncol. 2015, 10, 1319–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.I.; Koczywas, M.; Ulahannan, S.; Janne, P.; Pacheco, J.; Burris, H.; McCoach, C.; Wang, J.S.; Gordon, M.; Haura, E.; et al. A12 The SHP2 Inhibitor RMC-4630 in Patients with KRAS-Mutant Non-Small Cell Lung Cancer: Preliminary Evaluation of a First-in-Man Phase 1 Clinical Trial. J. Thorac. Oncol. 2020, 10, 1319–1327. [Google Scholar] [CrossRef]
- Bery, N.; Miller, A.; Rabbitts, T. A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat Commun. 2020, 11, 3233. [Google Scholar] [CrossRef]
- Shin, W.; Lee, S.K.; Hwang, J.H.; Park, J.C.; Cho, Y.H.; Ro, E.J.; Song, Y.; Seo, H.R.; Choi, K.Y. Identification of Ras-degrading small molecules that inhibit the transformation of colorectal cancer cells independent of β-catenin signaling. Exp. Mol. Med. 2018, 50, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, A.J.; Hahn, W.C. Synthetic lethal vulnerabilities in kras-mutant cancers. Cold Spring Harb. Perspect. Med. 2018, 8, a031518. [Google Scholar] [CrossRef] [PubMed]
- Litvak, A.M.; Drilon, A.E.; Rekhtman, N.; Pietanza, M.C.; Chaft, J.E.; Woo, K.; Paik, P.K.; Kris, M.G.; Riely, G.J. Phase II trial of bortezomib in KRAS G12D mutant lung cancers. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef]
- Goldman, J.W.; Shi, P.; Reck, M.; Paz-Ares, L.; Koustenis, A.; Hurt, K.C. Treatment Rationale and Study Design for the JUNIPER Study: A Randomized Phase III Study of Abemaciclib with Best Supportive Care Versus Erlotinib with Best Supportive Care in Patients with Stage IV Non-Small-Cell Lung Cancer with a Detectable KRAS Mutati. Clin. Lung Cancer 2016, 17, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Goldman, J.W.; Mazieres, J.; Barlesi, F.; Dragnev, K.H.; Koczywas, M.; Göskel, T.; Cortot, A.B.; Girard, N.; Wesseler, C.; Bischoff, H.; et al. A Randomized Phase III Study of Abemaciclib Versus Erlotinib in Patients with Stage IV Non-small Cell Lung Cancer With a Detectable KRAS Mutation Who Failed Prior Platinum-Based Therapy: JUNIPER. Front Oncol. 2020, 10, 578756. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Thennavan, A.; Dolgalev, I.; Chen, T.; Li, J.; Marzio, A.; Poirier, J.T.; Peng, D.; Bulatovic, M.; Mukhopadhyay, S.; et al. ULK1 inhibition overcomes compromised antigen presentation and restores antitumor immunity in LKB1 mutant lung cancer. Nat Cancer. 2021, 2, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J.; et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc. Natl. Acad. Sci. USA 2019, 2019, 2551–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Burke, J.P.; Phan, J.; Burns, M.C.; Olejniczak, E.T.; Waterson, A.G.; Lee, T.; Rossanese, O.W.; Fesik, S.W. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew. Chemie. Int. Ed. 2012, 51, 6140–61403. [Google Scholar] [CrossRef] [Green Version]
- Maurer, T.; Garrenton, L.S.; Oh, A.; Pitts, K.; Anderson, D.J.; Skelton, N.J.; Fauber, B.P.; Pan, B.; Malek, S.; Stokoe, D.; et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, P.D.; Guimarães, C.F.; Cardoso, A.P.; Mendonça, S.; Costa, Â.M.; Oliveira, M.J.; Velho, S. KRAS oncogenic signaling extends beyond cancer cells to orchestrate the microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; Von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-tumor Immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Steinbuck, M.; DeMuth, P.; Seenappa, L. 723 Lymph node-targeted AMP-vaccine enables tumor-directed mKRAS-specific immune responses with potent polyfunctional and cytolytic activity. J. Immunother. Cancer 2020. [Google Scholar] [CrossRef]
- Liu, W.; Yin, Y.; Wang, J.; Shi, B.; Zhang, L.; Qian, D.; Li, C.; Zhang, H.; Wang, S.; Zhu, J.; et al. Kras mutations increase telomerase activity and targeting telomerase is a promising therapeutic strategy for Kras-mutant NSCLC. Oncotarget 2017, 8, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, A.A.; Kolevska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. 2015, 26, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 14, 1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; et al. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]



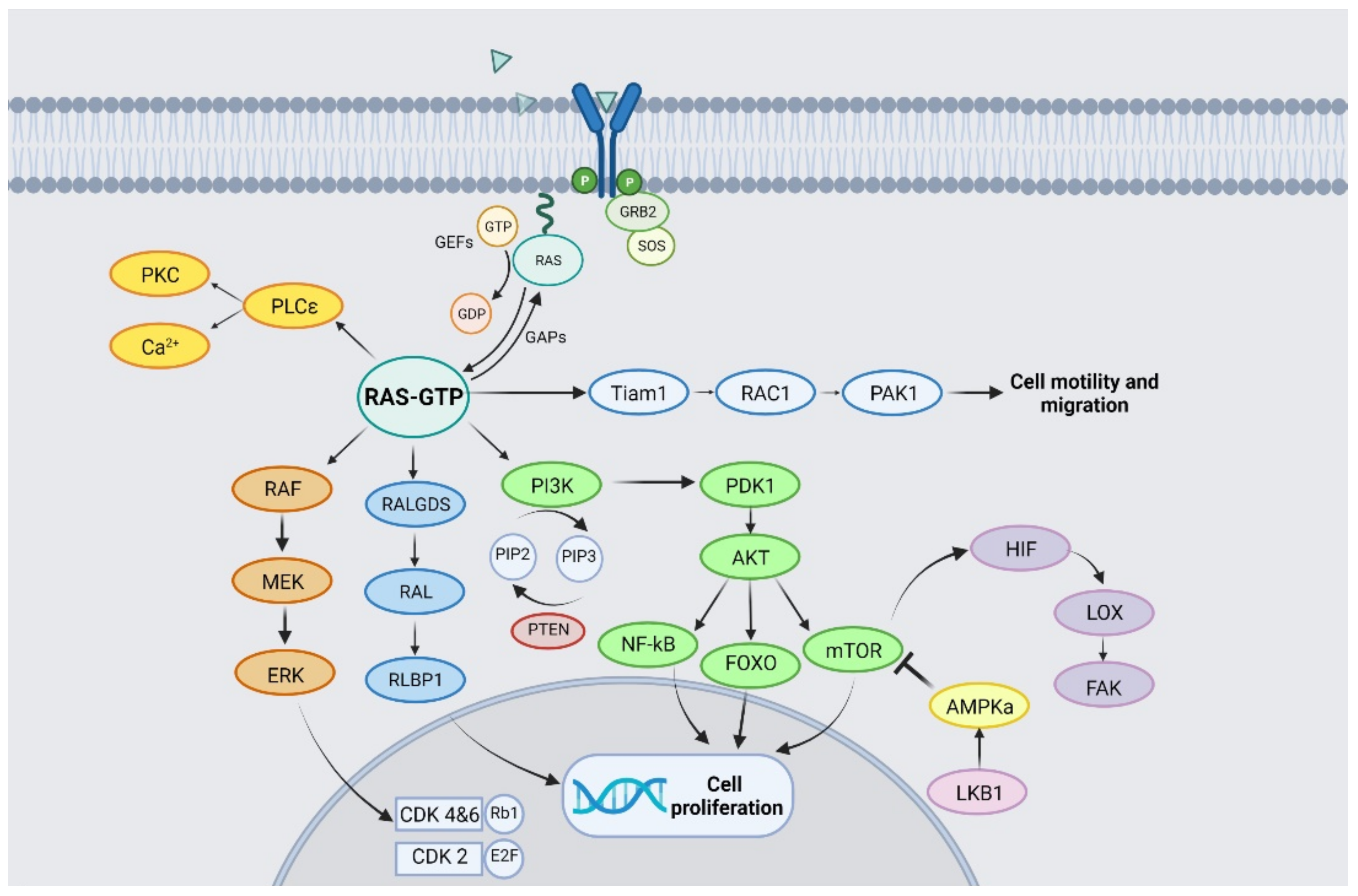{kind=link}
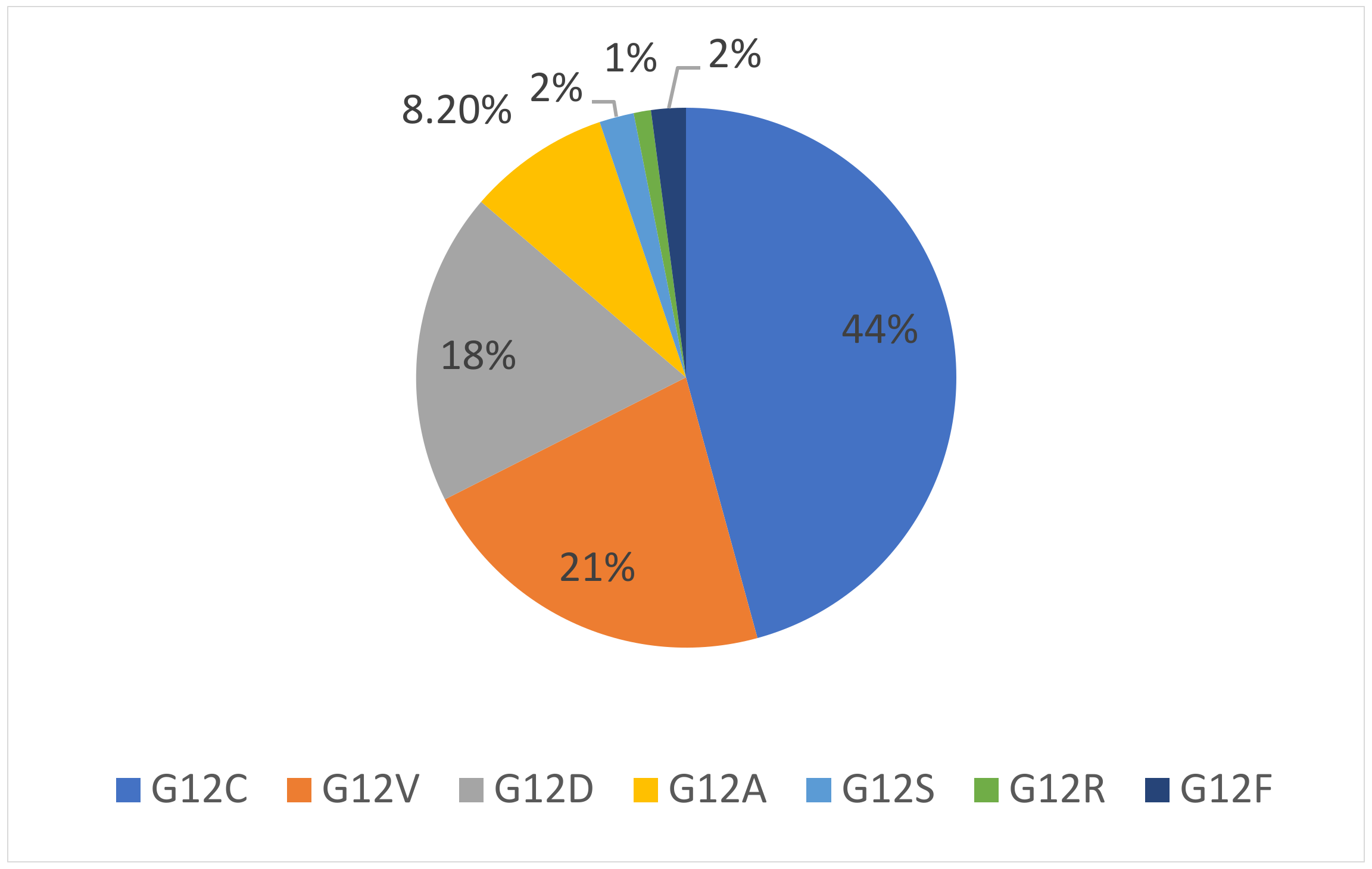{kind=link}
| Mutation Subtype | Codon 12 Transition or Transvertion | Aminoacid Substitution | Mutation Frequency in Lung Adenocarcinoma |
|---|---|---|---|
| G12C | G>T transversion resulting in GGT to TGT | Glycine to cysteine | 44% |
| G12V | G>T transversion resulting in GGT to GTT | Glycine to valine | 18–21% |
| G12D | G>A transition resulting in GGT to GAT | Glycine to aspartic acid | 11–18% |
| G12A | G>C transvertion resulting in GGT to GCT | Glycine to alanine | 8.2% |
| G12S | G>A transition resulting in GGT to AGT | Glycine to serine | 2% |
| G12R | G>C transversion resulting in GGT to CGT | Glycine to arginine | 1% |
| G12F | GG>TT transversion resulting in GGT to TTT | Glycine to phenylalanine | 2% |
| Co-Mutated Genes | Gene- Location | Frequency | Main Features | Ref |
|---|---|---|---|---|
| TP53 | 17p13.1 | 39% | Shorter latency and greater metastatic tendency; high expression of PD-L1 and TILs in TME; good response to immunotherapy. | [23,31] |
| STK11 | 19p13.3 | 20% | Greater tumour growth rate, increased tendency to turn into squamous histology; low expression of PD-L1 levels and reduced TILs in TME; reduced response to chemo-immunotherapy. | [21,23,25,32] |
| KEAP1 | 19p13.2 | 13% | Increased tumour progression rate; low response to platinum-based chemotherapy and immunotherapy. | [23,25,33] |
| ATM | 11q22.3 | 11.9% | Incomplete ATM loss is associated to carcinogenesis in case of p53 deficiency. | [23] |
| CDKN2A and CDKN2B | 9p21.39p21.3 | 20% 12% | Associated to mucinous histology with a lower TTF-1 expression. | [23,25,34] |
| Clinical Trial | Drug(s) Name | Target | Phase | Population and Tumor Characteristics | Estimated or Actual Enrollment | Status |
|---|---|---|---|---|---|---|
| NCT03875820 (FRAME) | VS-6766 plus defactinib | MEK/RAF, FAK, | 1 | Solid advanced tumours, including KRAS mutant NSCLC | 80 | Recruiting |
| NCT04620330 (RAMP-202) | VS-6766 plus defactinib | MEK/RAF, FAK | 1b/2 | Advanced, pre-treated, G12V or other KRAS mutant NSCLC | 100 | Recruiting |
| NCT02407509 | VS-6766 plus everolimus | MEK/RAF, mTOR | 1 | Solid advanced tumours, including KRAS mutant NSCLC, and multiple myeloma | 94 | Recruiting |
| NCT03681483 | VS-6766 | MEK/RAF | 1 | Advanced KRAS mutant lung cancer | 15 | Active, not recruiting |
| NCT03704688 | Ponatinib + trametinib | Bcr-Abl, MEK/MAPK/ERK | 1b/2 | Previously treated KRAS mutant advanced NSCLC | 12 | Active, not recruiting |
| NCT02964689 | Binimetinib + pemetrexed and cisplatin, followed by maintenance with binimetinib + pemetrexed | MEK | 1 | Advanced KRAS mutant NSCLC | 18 | Active, not recruiting |
| NCT03990077 | HL-085 + docetaxel | MEK | 1 | Advanced pre-treated KRAS mutant NSCLC | 27 | Not yet recruiting |
| NCT01912625 | Trametinib + chemoradiation | MEK | 1 | Stage III NSCLC that cannot be removed by surgery | 16 | Active, not recruiting |
| NCT02079740 | Trametinib + navitoclax | MEK, BCL-XL | 1b/2 | Solid advanced tumours with KRAS mutation, including NSCLC | 130 | Recruiting |
| NCT04735068 | Binimetinib + hydroxychloroquine | MAPK, lysosome | 2 | Advanced KRAS mutant NSCLC | 29 | Not yet recruiting |
| NCT04566393 | Expanded Access to ulixertinib (BVD-523) | MAPK pathway | - | Advanced solid tumours (including NSCLC) with MAPK pathway alterations, including KRAS mutations | - | Expanded Access Avalaible |
| NCT02022982 | Palbociclib + PD-0325901 | CDK4/6, MEK | 1b/2 | Solid cancers with KRAS mutations, including NSCLC | 139 | Active, not recruiting |
| NCT03170206 | Palbociclib (PD-0332991) and binimetinib (MEK162) | CDK4/6, MEK | 1b/2 | Advanced KRAS mutant NSCLC | 72 | Recruiting |
| NCT02974725 | LXH254 + LTT462 or trametinib or ribociclib | RAF, ERK/MEK/CDK4/6 | 1b | Advanced solid tumours, including KRAS mutant Non-Small Cell Lung Cancer | 331 | Recruiting |
| NCT03299088 | Pembrolizumab + trametinib | PD-1, MEK | 1 | Advanced KRAS mutant NSCLC | 15 | Active, not recruiting |
| NCT03225664 (BATTLE-2) | Pembrolizumab + trametinib | PD-1, MEK | 1b/2 | Advanced, previously treated NSCLC | 37 | Active, not recruiting |
| NCT01859026 | Binimetinib (MEK162) + erlotinib | MEK, EGFR | 1/1b | Advanced NSCLC harbouring KRAS or EGFR mutation | 43 | Active, not recruiting |
| NCT03520842 | Regorafenib + methotrexate | Multiple kinases | 2 | Recurrent or metastatic KRAS mutated NSCLC | 18 | Recruiting |
| NCT04000529 | TNO155 + spartalizumab or ribociclib | SHP2, PD-1, CDK4/6 | 1 | Solid advanced tumours, including KRAS mutant NSCLC | 126 | Recruiting |
| NCT04916236 (SHERPA) | RMC-4630 + LY3214996 | SHP2, ERK | 1 | KRAS mutant cancers, including NSCLC | 55 | Not yet recruiting |
| NCT03114319 | TNO155 | SHP2 | 1 | Advanced solid tumours, including KRAS G12-mutant NSCLC | 255 | Recruiting |
| NCT03989115 | RMC-4630 + cobimetinib | SHP2, MEK | 1b/2 | Advanced solid tumours, including KRAS G12-mutant NSCLC | 168 | Recruiting |
| NCT03808558 | TVB-2640 | FAS/FASN | 2 | Advanced KRAS mutant NSCLC | 12 | Recruiting |
| NCT03965845 | Telaglenastat (CB-839) + palbociclib | Glutaminase, CDK4/6 | 1b/2 | Solid advanced tumours, including pre-treated, KRAS mutant NSCLC | 85 | Recruiting |
| NCT04263090 | Rigosertib + nivolumab | PI3K/PLK, PD-1 | 1 | Advanced, pre-treated, KRAS mutant NSCLC | 30 | Recruiting |
| NCT03693326 | PDR001 | PD-1 | 2 | Non-small Cell Lung Cancer harbouring mutations including KRAS | 70 | Recruiting |
| NCT04470674 | Carboplatin-pemetrexed +/− durvalumab | PD-L1 | 2 | Advanced, naïve, KRAS mutant and PD-L1 high (≥50%) NSCLC | 50 | Recruiting |
| NCT03777124 | SHR-1210 + apatinib | PD-1, VEGFR2 | 2 | KRAS mutant stage IV non-squamous NSCLC | 230 | Not yet recruiting |
| NCT04853017 (AMPLIFY-201) | ELI-002 (lymph node-targeted therapeutic vaccine) | - | 1/2 | KRAS/NRAS mutant (G12D or G12R) solid tumours, including NSCLC | 159 | Recruiting |
| NCT03095612 | Selinexor (KPT-330) | XPO1 | 1b/2 | Pre-treated advanced KRAS mutant lung cancer | 59 | Recruiting |
| NCT03819387 | NBF-006 (siRNA-based lipid nanoparticle) | - | 1 | Solid advanced tumours including KRAS-Mutant NSCLC | 44 | Recruiting |
| NCT03948763 | mRNA-5671/V941 +/− pembrolizumab | - | 1 | Solid advanced tumours, including KRAS mutant NSCLC | 100 | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacobs, F.; Cani, M.; Malapelle, U.; Novello, S.; Napoli, V.M.; Bironzo, P. Targeting KRAS in NSCLC: Old Failures and New Options for “Non-G12c” Patients. Cancers 2021, 13, 6332. https://doi.org/10.3390/cancers13246332
Jacobs F, Cani M, Malapelle U, Novello S, Napoli VM, Bironzo P. Targeting KRAS in NSCLC: Old Failures and New Options for “Non-G12c” Patients. Cancers. 2021; 13(24):6332. https://doi.org/10.3390/cancers13246332
Chicago/Turabian StyleJacobs, Francesca, Massimiliano Cani, Umberto Malapelle, Silvia Novello, Valerio Maria Napoli, and Paolo Bironzo. 2021. "Targeting KRAS in NSCLC: Old Failures and New Options for “Non-G12c” Patients" Cancers 13, no. 24: 6332. https://doi.org/10.3390/cancers13246332
APA StyleJacobs, F., Cani, M., Malapelle, U., Novello, S., Napoli, V. M., & Bironzo, P. (2021). Targeting KRAS in NSCLC: Old Failures and New Options for “Non-G12c” Patients. Cancers, 13(24), 6332. https://doi.org/10.3390/cancers13246332