
Impact of a Faulty Germinal Center Reaction on the Pathogenesis of Primary Diffuse Large B Cell Lymphoma of the Central Nervous System

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Historic Background
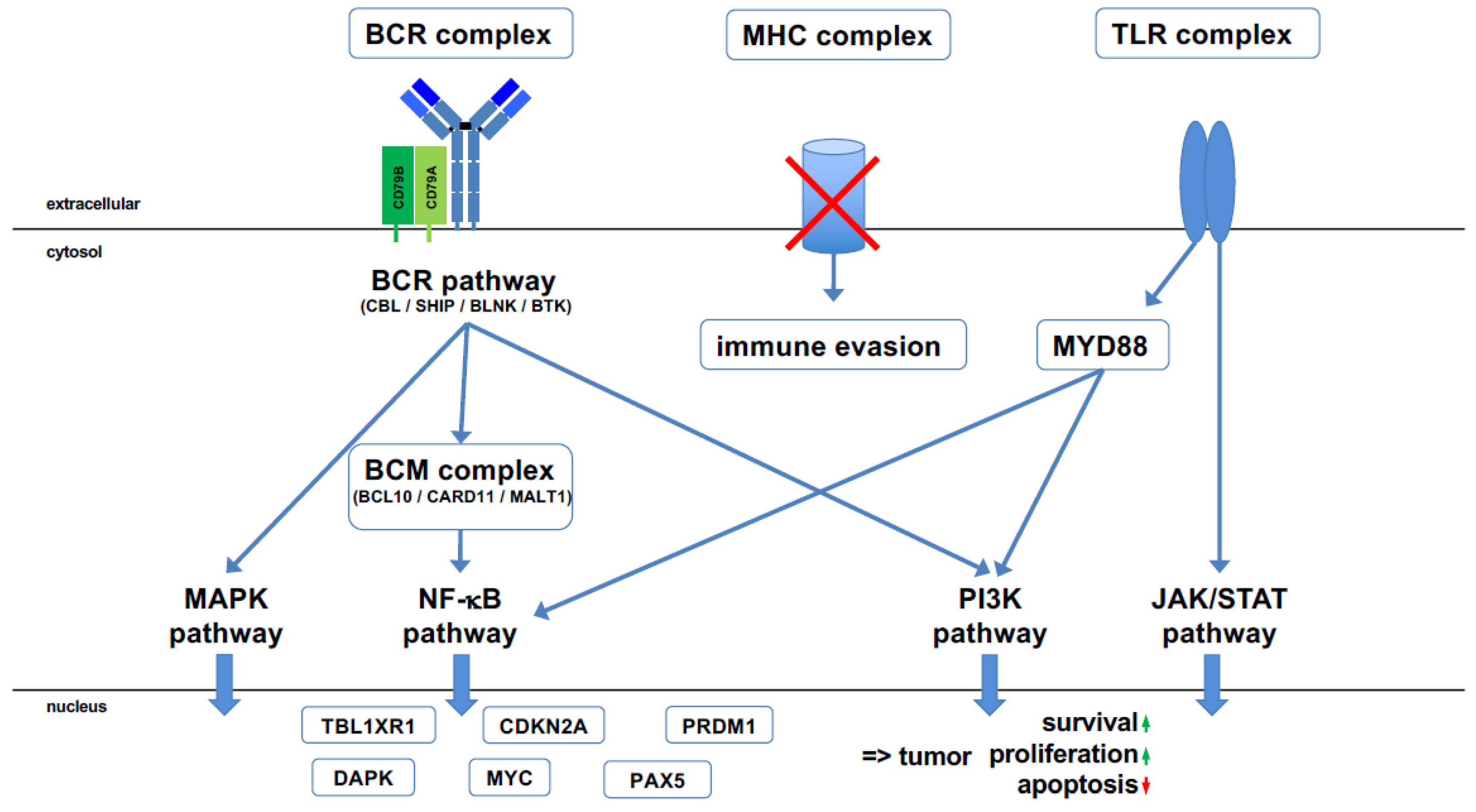
2. Molecular Pathogenesis of PCNSL
- (1)
- What is the cellular origin of these tumor cells?
- (2)
- What are the pathogenetically relevant genetic, epigenetic, transcriptional, and proteomic alterations of these tumor cells?
- (3)
- What are the reasons for the tropism and exclusive manifestation of this tumor in the CNS?
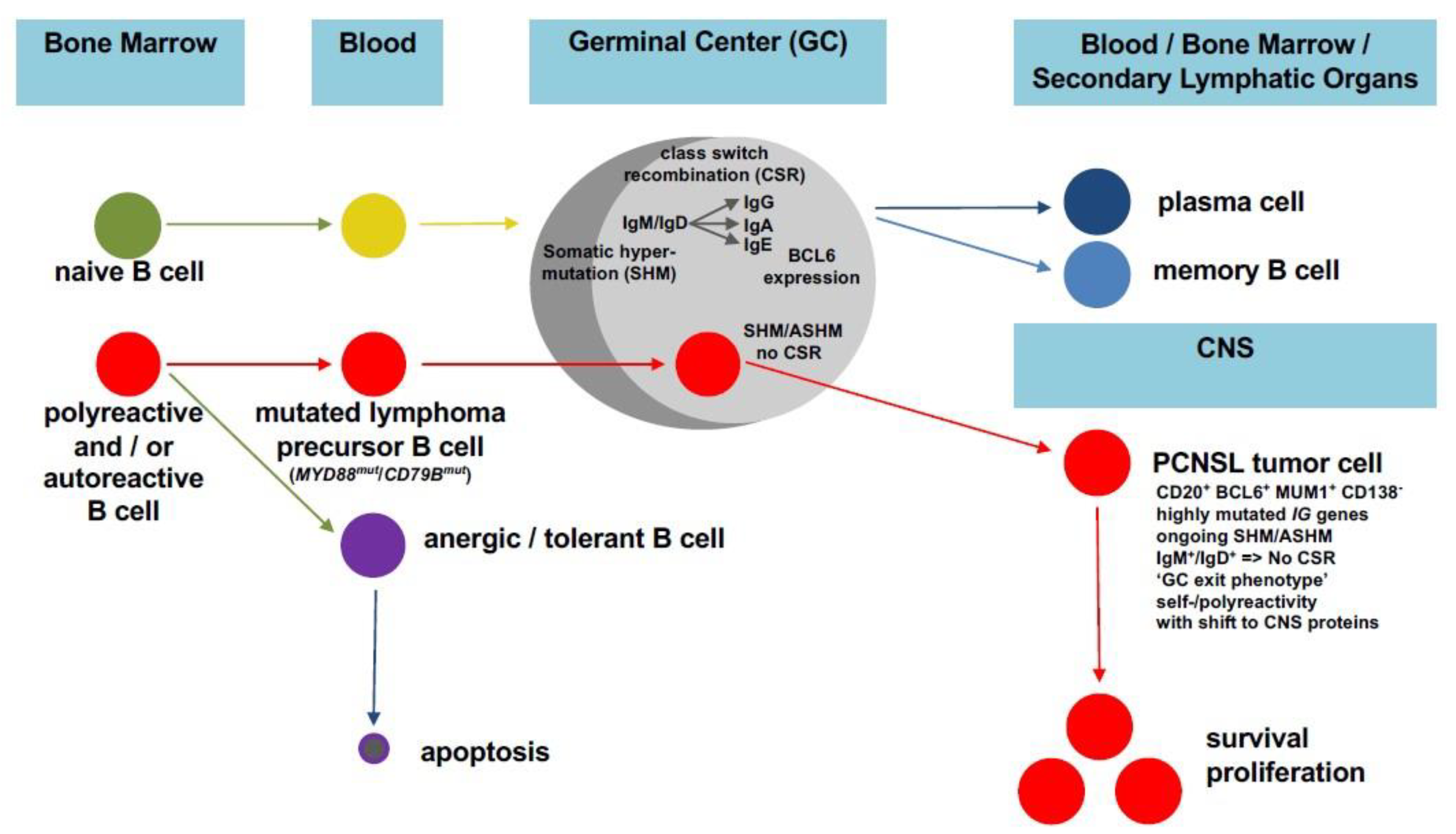
3. A Faulty GC Reaction as Key Process for PCNSL Pathogenesis
4. Tumor Cell Adaptation to the CNS Microenvironment in PCNSL
5. Role of Preclinical Animal Models in Studying PCNSL Pathogenesis
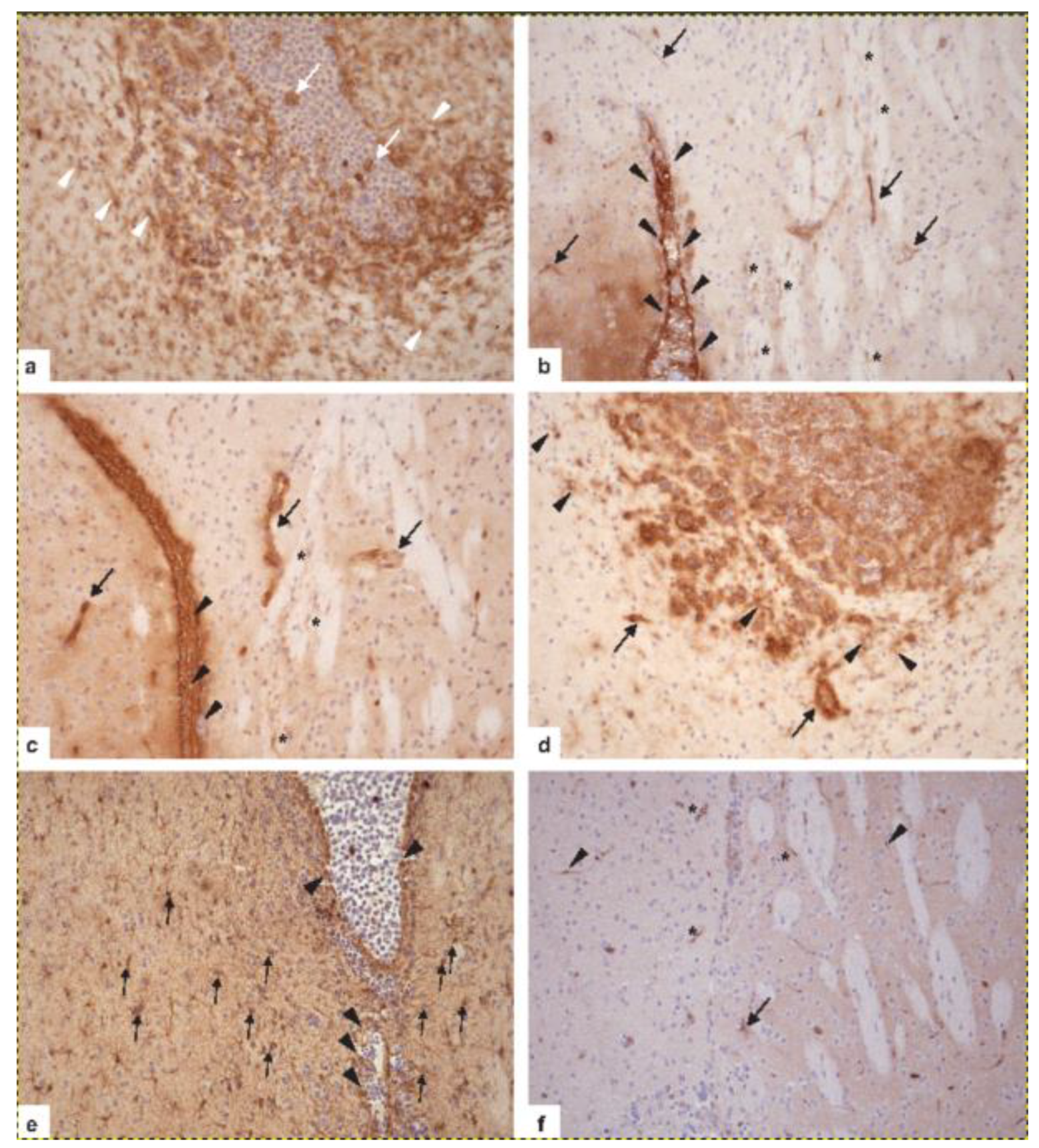
6. Neuropathological Diagnostics of PCNSL
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deckert, M.; Paulus, W.; Kluin, P.; Ferry, J. Lymphomas. In WHO Classification of Tumours of the Central Nervous System, Revised 4th ed.; Louis, D.N., Ohgaki, H., Wiestler, O.D., Cavenee, W.K., Bosman, F.T., Jaffe, E.S., Lakhani, S.R., Ohgaki, H., Eds.; World Health Organization Classification of Tumours; IARC: Lyon, France, 2016; pp. 272–277. [Google Scholar]
- Kluin, P.; Deckert, M.; Ferry, J.A. Primary diffuse large B-cell Lymphoma of the CNS. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Bosman, F.T., Jaffe, E.S., Lakhani, S.R., et al., Eds.; World Health Organization Classification of Tumours; IARC: Lyon, France, 2017; Volume 2, pp. 300–302. [Google Scholar]
- Bailey, P. Intercranial Sarcomatous Tumors of Leptomeningeal Origin. Arch. Surg. 1929, 18, 1359–1402. [Google Scholar] [CrossRef]
- Lennert, K.; Feller, A.C. The Kiel Classification. In Histopathology of Non-Hodgkin’s Lymphomas: Based on the Updated Kiel Classification; Springer: Berlin/Heidelberg, Germany, 1992; pp. 13–52. [Google Scholar]
- Gandhi, M.K.; Hoang, T.; Law, S.C.; Brosda, S.; O’Rourke, K.; Tobin, J.W.D.; Vari, F.; Murigneux, V.; Fink, L.; Gunawardana, J.; et al. EBV-associated primary CNS lymphoma occurring after immunosuppression is a distinct immunobiological entity. Blood 2021, 137, 1468–1477. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Küppers, R.; Schlüter, D.; Spieker, T.; Van Roost, D.; Schaller, C.; Reifenberger, G.; Wiestler, O.D.; Deckert-Schlüter, M. Primary central nervous system lymphomas are derived from germinal-center B cells and show a preferential usage of the V4–34 gene segment. Am. J. Pathol. 1999, 155, 2077–2086. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Siebert, R.; Deckert, M. Primary lymphoma of the central nervous system: Just DLBCL or not? Blood 2009, 113, 7–10. [Google Scholar] [CrossRef]
- Deckert, M.; Montesinos-Rongen, M.; Brunn, A.; Siebert, R. Systems biology of primary CNS lymphoma: From genetic aberrations to modeling in mice. Acta Neuropathol. 2014, 127, 175–188. [Google Scholar] [CrossRef]
- Löw, S.; Han, C.H.; Batchelor, T.T. Primary central nervous system lymphoma. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418793562. [Google Scholar] [CrossRef] [Green Version]
- Mondello, P.; Mian, M.; Bertoni, F. Primary central nervous system lymphoma: Novel precision therapies. Crit. Rev. Oncol. 2019, 141, 139–145. [Google Scholar] [CrossRef]
- Thompsett, A.R.; Ellison, D.W.; Stevenson, F.K.; Zhu, D. V(H) gene sequences from primary central nervous system lymphomas indicate derivation from highly mutated germinal center B cells with ongoing mutational activity. Blood 1999, 94, 1738–1746. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Purschke, F.; Küppers, R.; Deckert, M. Immunoglobulin Repertoire of Primary Lymphomas of the Central Nervous System. J. Neuropathol. Exp. Neurol. 2014, 73, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-Rongen, M.; Van Roost, D.; Schaller, C.; Wiestler, O.D.; Deckert, M. Primary diffuse large B-cell lymphomas of the central nervous system are targeted by aberrant somatic hypermutation. Blood 2004, 103, 1869–1875. [Google Scholar] [CrossRef] [Green Version]
- Vater, I.; Montesinos-Rongen, M.; Schlesner, M.; Haake, A.; Purschke, F.; Sprute, R.; Mettenmeyer, N.; Nazzal, I.; Nagel, I.; Gutwein, J.; et al. The mutational pattern of primary lymphoma of the central nervous system determined by whole-exome sequencing. Leukemia 2015, 29, 677–685. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Schmitz, R.; Courts, C.; Stenzel, W.; Bechtel, D.; Niedobitek, G.; Blümcke, I.; Reifenberger, G.; von Deimling, A.; Jungnickel, B.; et al. Absence of immuno-globulin class switch in primary lymphomas of the central nervous system. Am. J. Pathol. 2005, 166, 1773–1779. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-Rongen, M.; Zühlke-Jenisch, R.; Gesk, S.; Martin-Subero, J.I.; Schaller, C.; Van Roost, D.; Wiestler, O.D.; Deckert, M.; Siebert, R. Interphase cytogenetic analysis of lymphoma-associated chromosomal breakpoints in primary diffuse large B-cell lymphomas of the central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-Rongen, M.; Akasaka, T.; Zühlke-Jenisch, R.; Schaller, C.; Van Roost, D.; Wiestler, O.D.; Siebert, R.; Deckert, M. Molecular Characterization of BCL6 Breakpoints in Primary Diffuse Large B-cell Lymphomas of the Central Nervous System Identifies GAPD as Novel Translocation Partner. Brain Pathol. 2006, 13, 534–538. [Google Scholar] [CrossRef]
- Schwindt, H.; Akasaka, T.; Zühlke-Jenisch, R.; Hans, V.; Schaller, C.; Klapper, W.; Dyer, M.J.; Siebert, R.; Deckert, M. Chromosomal translocations fusing the BCL6 gene to different partner loci are recurrent in primary central nervous system lymphoma and may be as-sociated with aberrant somatic hypermutation or defective class switch recombination. J. Neuropathol. Exp. Neurol. 2006, 65, 776–782. [Google Scholar] [CrossRef] [Green Version]
- Cady, F.M.; O’Neill, B.P.; Law, M.E.; Decker, P.A.; Kurtz, D.; Giannini, C.; Porter, A.B.; Kurtin, P.J.; Johnston, P.B.; Dogan, A.; et al. Del(6)(q22) and BCL6 Rearrangements in Primary CNS Lymphoma Are Indicators of an Aggressive Clinical Course. J. Clin. Oncol. 2008, 26, 4814–4819. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-Rongen, M.; Godlewska, E.; Brunn, A.; Wiestler, O.D.; Siebert, R.; Deckert, M. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol. 2011, 122, 791–792. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Brunn, A.; Tuchscherer, A.; Borchmann, P.; Schorb, E.; Kasenda, B.; Altmüller, J.; Illerhaus, G.; Ruge, M.I.; Maarouf, M.; et al. Analysis of Driver Mutational Hot Spots in Blood-Derived Cell-Free DNA of Patients with Primary Central Nervous System Lymphoma Obtained before Intracerebral Biopsy. J. Mol. Diagn. 2020, 22, 1300–1307. [Google Scholar] [CrossRef]
- Chapuy, B.; Roemer, M.G.M.; Stewart, C.; Tan, Y.; Abo, R.P.; Zhang, L.; Dunford, A.J.; Meredith, D.M.; Thorner, A.R.; Jordanova, E.S.; et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood 2016, 127, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-Rongen, M.; Schäfer, E.; Siebert, R.; Deckert, M. Genes regulating the B cell receptor pathway are recurrently mutated in primary central nervous system lymphoma. Acta Neuropathol. 2012, 124, 905–906. [Google Scholar] [CrossRef]
- Courts, C.; Montesinos-Rongen, M.; Brunn, A.; Bug, S.; Siemer, D.; Hans, V.; Blümcke, I.; Klapper, W.; Schaller, C.; Wiestler, O.D.; et al. Recurrent Inactivation of the PRDM1 Gene in Primary Central Nervous System Lymphoma. J. Neuropathol. Exp. Neurol. 2008, 67, 720–727. [Google Scholar] [CrossRef]
- Gonzalez-Aguilar, A.; Idbaih, A.; Boisselier, B.; Habbita, N.; Rossetto, M.; Laurenge, A.; Bruno, A.; Jouvet, A.; Polivka, M.; Adam, C.; et al. Recurrent Mutations of MYD88 and TBL1XR1 in Primary Central Nervous System Lymphomas. Clin. Cancer Res. 2012, 18, 5203–5211. [Google Scholar] [CrossRef] [Green Version]
- Booman, M.; Szuhai, K.; Rosenwald, A.; Hartmann, E.; Kluin-Nelemans, J.C.; De Jong, D.; Schuuring, E.; Kluin, P.M. Genomic alterations and gene expression in primary diffuse large B-cell lymphomas of immune-privileged sites: The importance of apoptosis and immunomodulatory pathways. J. Pathol. 2008, 216, 209–217. [Google Scholar] [CrossRef]
- Schwindt, H.; Vater, I.; Kreuz, M.; Montesinos-Rongen, M.; Brunn, A.; Richter, J.; Gesk, S.; Ammerpohl, O.; Wiestler, O.D.; Hasenclever, D.; et al. Chromosomal imbalances and partial uniparental disomies in primary central nervous system lymphoma. Leukemia 2009, 23, 1875–1884. [Google Scholar] [CrossRef] [Green Version]
- Cobbers, J.M.; Wolter, M.; Reifenberger, J.; Ring, G.U.; Jessen, F.; An, H.X.; Niederacher, D.; Schmidt, E.E.; Ichimura, K.; Floeth, F.; et al. Frequent inactivation of CDKN2A and rare mutation of TP53 in PCNSL. Brain Pathol. 1998, 8, 263–276. [Google Scholar] [CrossRef]
- Nayyar, N.; White, M.D.; Gill, C.M.; Lastrapes, M.; Bertalan, M.; Kaplan, A.; D’Andrea, M.R.; Bihun, I.; Kaneb, A.; Dietrich, J.; et al. MYD88 L265P mutation and CDKN2A loss are early mutational events in primary central nervous system diffuse large B-cell lymphomas. Blood Adv. 2019, 3, 375–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montesinos-Rongen, M.; Brunn, A.; Bentink, S.; Basso, K.; Lim, W.K.; Klapper, W.; Schaller, C.; Reifenberger, G.; Rubenstein, J.; Wiestler, O.D.; et al. Gene expression profiling suggests primary central nervous system lymphomas to be derived from a late germinal center B cell. Leukemia 2007, 22, 400–405. [Google Scholar] [CrossRef]
- Courts, C.; Montesinos-Rongen, M.; Martin-Subero, J.I.; Brunn, A.; Siemer, D.; Zuhlke-Jenisch, R.; Pels, H.; Jürgens, A.; Schlegel, U.; Schmidt-Wolf, I.G.H.; et al. Transcriptional Profiling of the Nuclear Factor-κB Pathway Identifies a Subgroup of Primary Lymphoma of the Central Nervous System with Low BCL10 Expression. J. Neuropathol. Exp. Neurol. 2007, 66, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-Rongen, M.; Schmitz, R.; Brunn, A.; Gesk, S.; Richter, J.; Hong, K.; Wiestler, O.D.; Siebert, R.; Küppers, R.; Deckert, M. Mutations of CARD11 but not TNFAIP3 may activate the NF-κB pathway in primary CNS lymphoma. Acta Neuropathol. 2010, 120, 529–535. [Google Scholar] [CrossRef]
- Riemersma, S.A.; Jordanova, E.S.; Schop, R.F.; Philippo, K.; Looijenga, L.H.; Schuuring, E.; Kluin, P.M. Extensive genetic alterations of the HLA region, including homozygous de-letions of HLA class II genes in B-cell lymphomas arising in immune-privileged sites. Blood 2000, 96, 3569–3577. [Google Scholar] [CrossRef]
- Weber, T.; Weber, R.; Kaulich, K.; Actor, B.; Meyer-Puttlitz, B.; Lampel, S.; Büschges, R.; Weigel, R.; Deckert-Schlüter, M.; Schmiedek, P.; et al. Characteristic Chromosomal Imbalances in Primary Central Nervous System Lymphomas of the Diffuse Large B-Cell Type. Brain Pathol. 2006, 10, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Jordanova, E.S.; Riemersma, S.A.; Philippo, K.; Giphart-Gassler, M.; Schuuring, E.; Kluin, P.M. Hemizygous deletions in the HLA region account for loss of heterozygosity in the ma-jority of diffuse large B-cell lymphomas of the testis and the central nervous system. Genes Chromosomes Cancer 2002, 35, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Jordanova, E.S.; Riemersma, S.A.; Philippo, K.; Schuuring, E.; Kluin, P.M. β2-microglobulin aberrations in diffuse large B-cell lymphoma of the testis and the central nervous system. Int. J. Cancer 2003, 103, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Ammerpohl, O.; Martin-Subero, J.I.; Montesinos-Rongen, M.; Bibikova, M.; Wickham-Garcia, E.; Wiestler, O.D.; Deckert, M.; Siebert, R. Array-based DNA methylation profiling of primary lymphomas of the central nervous system. BMC Cancer 2009, 9, 455. [Google Scholar] [CrossRef] [Green Version]
- Bruno, A.; Boisselier, B.; Labreche, K.; Marie, Y.; Polivka, M.; Jouvet, A.; Adam, C.; Figa-rella-Branger, D.; Miquel, C.; Eimer, S.; et al. Mutational analysis of primary central nerv-ous system lymphoma. Oncotarget 2014, 5, 5065–5075. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, K.; Kawazu, M.; Kojima, S.; Ueno, T.; Sai, E.; Soda, M.; Ueda, H.; Yasuda, T.; Yamaguchi, H.; Lee, J.; et al. Genomic characterization of primary central nervous system lymphoma. Acta Neuropathol. 2016, 131, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Tateishi, K.; Niwa, T.; Matsushita, Y.; Tamura, K.; Kinoshita, M.; Tanaka, K.; Fukushima, S.; Takami, H.; Arita, H.; et al. Recurrent mutations ofCD79BandMYD88are the hallmark of primary central nervous system lymphomas. Neuropathol. Appl. Neurobiol. 2016, 42, 279–290. [Google Scholar] [CrossRef]
- Fontanilles, M.; Marguet, F.; Bohers, É.; Viailly, P.J.; Dubois, S.; Bertrand, P.; Camus, V.; Mareschal, S.; Ruminy, P.; Maingonnat, C.; et al. Non-invasive detection of somatic mutations using next-generation sequencing in primary central nervous system lymphoma. Oncotarget 2017, 8, 48157–48168. [Google Scholar] [CrossRef] [Green Version]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef]
- Hübschmann, D.; Kleinheinz, K.; Wagener, R.; Bernhart, S.H.; López, C.; Toprak, U.H.; Sungalee, S.; Ishaque, N.; Kretzmer, H.; Kreuz, M.; et al. Mutational mechanisms shaping the coding and noncoding genome of germinal center derived B-cell lymphomas. Leukemia 2021, 35, 2002–2016. [Google Scholar] [CrossRef] [PubMed]
- Grommes, C.; Tang, S.S.; Wolfe, J.; Kaley, T.J.; Daras, M.; Pentsova, E.I.; Piotrowski, A.F.; Stone, J.; Lin, A.; Nolan, C.P.; et al. Phase 1b trial of an ibrutinib-based combination therapy in recurrent/refractory CNS lymphoma. Blood 2019, 133, 436–445. [Google Scholar] [CrossRef]
- Soussain, C.; Choquet, S.; Blonski, M.; Leclercq, D.; Houillier, C.; Rezai, K.; Bijou, F.; Houot, R.; Boyle, E.; Gressin, R.; et al. Ibrutinib monotherapy for relapse or refractory primary CNS lymphoma and primary vitreoretinal lymphoma: Final analysis of the phase II ‘proof-of-concept’ iLOC study by the Lymphoma study association (LYSA) and the French oculo-cerebral lymphoma (LOC) network. Eur. J. Cancer 2019, 117, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Wirsching, H.-G.; Weller, M.; Balabanov, S.; Roth, P. Targeted Therapies and Immune Checkpoint Inhibitors in Primary CNS Lymphoma. Cancers 2021, 13, 3073. [Google Scholar] [CrossRef]
- MacLennan, I.C.M.; Gulbranson-Judge, A.; Toellner, K.-M.; Casamayor-Palleja, M.; Sze, D.M.-Y.; Chan, E.Y.-T.; Luther, S.A.; Orbea, H.A. The changing preference of T and B cells for partners as T-dependent antibody responses develop. Immunol. Rev. 1997, 156, 53–66. [Google Scholar] [CrossRef]
- Rajewsky, K. Clonal selection and learning in the antibody system. Nat. Cell Biol. 1996, 381, 751–758. [Google Scholar] [CrossRef]
- Küppers, R.; Dalla-Favera, R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene 2001, 20, 5580–5594. [Google Scholar] [CrossRef] [Green Version]
- Küppers, R.; Klein, U.; Hansmann, M.-L.; Rajewsky, K. Cellular Origin of Human B-Cell Lymphomas. N. Engl. J. Med. 1999, 341, 1520–1529. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Purschke, F.G.; Brunn, A.; May, C.; Nordhoff, E.; Marcus, K.; Deckert, M. Primary Central Nervous System (CNS) Lymphoma B Cell Receptors Recognize CNS Proteins. J. Immunol. 2015, 195, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Spies, E.; Fichtner, M.; Müller, F.; Krasemann, S.; Illerhaus, G.; Glatzel, M.; Binder, M.; Trepel, M. Comment on “Primary Central Nervous System (CNS) Lymphoma B Cell Receptors Recognize CNS Proteins”. J. Immunol. 2015, 195, 4549–4550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurner, L.; Preuss, K.-D.; Bewarder, M.; Kemele, M.; Fadle, N.; Regitz, E.; Altmeyer, S.; Schormann, C.; Poeschel, V.; Ziepert, M.; et al. Hyper-N-glycosylated SAMD14 and neurabin-I as driver autoantigens of primary central nervous system lymphoma. Blood 2018, 132, 2744–2753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montesinos-Rongen, M.; Terrao, M.; May, C.; Marcus, K.; Blümcke, I.; Hellmich, M.; Küp-pers, R.; Brunn, A.; Deckert, M. The process of somatic hypermutation increases polyreac-tivity for central nervous system antigens in primary central nervous system lymphoma. Haematologica 2021, 106, 708–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, M.; Przekopowitz, M.; Taudien, S.; Lollies, A.; Ronge, V.; Drees, B.; Lindemann, M.; Hillen, U.; Engler, H.; Singer, B.B.; et al. Functional capacities of human IgM memory B cells in early inflammatory responses and secondary germinal center reactions. Proc. Natl. Acad. Sci. USA 2015, 112, E546–E555. [Google Scholar] [CrossRef] [Green Version]
- Seifert, M.; Küppers, R. Human memory B cells. Leukemia 2016, 30, 2283–2292. [Google Scholar] [CrossRef]
- Braggio, E.; Van Wier, S.; Ojha, J.; McPhail, E.; Asmann, Y.W.; Egan, J.; Ayres-Silva, J.; Schiff, D.; Lopes, M.B.; Decker, P.A.; et al. Genome-Wide Analysis Uncovers Novel Recurrent Alterations in Primary Central Nervous System Lymphomas. Clin. Cancer Res. 2015, 21, 3986–3994. [Google Scholar] [CrossRef] [Green Version]
- Siebert, R.; Rosenwald, A.; Staudt, L.M.; Morris, S.W. Molecular features of B-cell lymphoma. Curr. Opin. Oncol. 2001, 13, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Victora, G.; Ying, C.Y.; Phan, R.T.; Saito, M.; Nussenzweig, M.C.; Dalla-Favera, R. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat. Immunol. 2012, 13, 1083–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venturutti, L.; Melnick, A.M. The dangers of déjà vu: Memory B cells as the cells of origin of ABC-DLBCLs. Blood 2020, 136, 2263–2274. [Google Scholar] [CrossRef]
- Venturutti, L.; Teater, M.; Zhai, A.; Chadburn, A.; Babiker, L.; Kim, D.; Béguelin, W.; Lee, T.C.; Kim, Y.; Chin, C.R.; et al. TBL1XR1 Mutations Drive Extranodal Lymphoma by Inducing a Pro-tumorigenic Memory Fate. Cell 2020, 182, 297–316.e27. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of Ectopic B-cell Follicles with Germinal Centers in the Meninges of Patients with Secondary Progressive Multiple Sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.; Jellinger, K. Comparison of integrin adhesion molecules expressed by primary brain lymphomas and nodal lymphomas. Acta Neuropathol. 1993, 86, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Brunn, A.; Montesinos-Rongen, M.; Strack, A.; Reifenberger, G.; Mawrin, C.; Schaller, C.; Deckert, M. Expression pattern and cellular sources of chemokines in primary central nervous system lymphoma. Acta Neuropathol. 2007, 114, 271–276. [Google Scholar] [CrossRef]
- Fangazio, M.; Ladewig, E.; Gomez, K.; Garcia-Ibanez, L.; Kumar, R.; Teruya-Feldstein, J.; Rossi, D.; Filip, I.; Pan-Hammarström, Q.; Inghirami, G.; et al. Genetic mechanisms of HLA-I loss and immune escape in diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2104504118. [Google Scholar] [CrossRef] [PubMed]
- Riemersma, S.A.; Oudejans, J.J.; Vonk, M.J.; Dreef, E.J.; Prins, F.A.; Jansen, P.M.; Vermeer, M.H.; Blok, P.; Kibbelaar, R.E.; Muris, J.J.; et al. High numbers of tumour-infiltrating activated cytotoxic T lymphocytes, and frequent loss of HLA class I and II expression, are features of aggressive B cell lymphomas of the brain and testis. J. Pathol. 2005, 206, 328–336. [Google Scholar] [CrossRef]
- Ponzoni, M.; Berger, F.; Chassagne-Clement, C.; Tinguely, M.; Jouvet, A.; Ferreri, A.J.M.; Dell’Oro, S.; Terreni, M.R.; Doglioni, C.; Weis, J.; et al. Reactive perivascular T-cell infiltrate predicts survival in primary central nervous system B-cell lymphomas. Br. J. Haematol. 2007, 138, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Bruck, W.; Nervensystems, N.L.U.L.L.D.; Brunn, A.; Klapper, W.; Kuhlmann, T.; Metz, I.; Paulus, W.; Deckert, M. Differenzialdiagnose lymphoider Infiltrate im Zentralnervensystem. Der Pathol. 2013, 34, 186–197. [Google Scholar] [CrossRef]
- Deckert, M.; Brunn, A.; Montesinos-Rongen, M.; Terreni, M.R.; Ponzoni, M. Primary lymphoma of the central nervous system-a diagnostic challenge. Hematol. Oncol. 2013, 32, 57–67. [Google Scholar] [CrossRef]
- Waldera-Lupa, D.M.; Etemad-Parishanzadeh, O.; Brocksieper, M.; Kirchgaessler, N.; Seidel, S.; Kowalski, T.; Montesinos-Rongen, M.; Deckert, M.; Schlegel, U.; Stühler, K. Proteomic changes in cerebrospinal fluid from primary central nervous system lymphoma patients are associated with protein ectodomain shedding. Oncotarget 2017, 8, 110118–110132. [Google Scholar] [CrossRef] [Green Version]
- Salzburg, J.; Burkhardt, B.; Zimmermann, M.; Wachowski, O.; Woessmann, W.; Oschlies, I.; Klapper, W.; Wacker, H.-H.; Ludwig, W.-D.; Niggli, F.; et al. Prevalence, Clinical Pattern, and Outcome of CNS Involvement in Childhood and Adolescent Non-Hodgkin’s Lymphoma Differ by Non-Hodgkin’s Lymphoma Subtype: A Berlin-Frankfurt-Münster Group Report. J. Clin. Oncol. 2007, 25, 3915–3922. [Google Scholar] [CrossRef] [PubMed]
- Donnou, S.; Galand, C.; Daussy, C.; Crozet, L.; Fridman, W.H.; Sautès-Fridman, C.; Fisson, S. Immune adaptive microenvironment profiles in intracerebral and intrasplenic lymphomas share common characteristics. Clin. Exp. Immunol. 2011, 165, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Montesinos-Rongen, M.; Sanchez-Ruiz, M.; Brunn, A.; Hong, K.; Bens, S.; Perales, S.R.; Cigudosa, J.C.; Siebert, R.; Deckert, M. Mechanisms of Intracerebral Lymphoma Growth Delineated in a Syngeneic Mouse Model of Central Nervous System Lymphoma. J. Neuropathol. Exp. Neurol. 2013, 72, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Brunn, A.G.; Nagel, I.; Montesinos-Rongen, M.; Klapper, W.; Vater, I.; Paulus, W.; Hans, V.; Blümcke, I.; Weis, J.; Siebert, R.; et al. Frequent triple-hit expression of MYC, BCL2, and BCL6 in primary lymphoma of the central nervous system and absence of a favorable MYClowBCL2low subgroup may underlie the inferior prognosis as compared to systemic diffuse large B cell lymphomas. Acta Neuropathol. 2013, 126, 603–605. [Google Scholar] [CrossRef]
- Dierlamm, J.; Murga Penas, E.M.; Bentink, S.; Wessendorf, S.; Berger, H.; Hummel, M.; Klapper, W.; Lenze, D.; Rosenwald, A.; Haralambieva, E.; et al. Gain of chromosome re-gion 18q21 including the MALT1 gene is associated with the activated B-cell-like gene ex-pression subtype and increased BCL2 gene dosage and protein expression in diffuse large B-cell lymphoma. Haematologica 2008, 93, 688–696. [Google Scholar] [CrossRef]
- Horn, H.; Ziepert, M.; Becher, C.; Barth, T.F.E.; Bernd, H.-W.; Feller, A.C.; Klapper, W.; Hummel, M.; Stein, H.; Hansmann, M.-L.; et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013, 121, 2253–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grommes, C.; DeAngelis, L. Primary CNS Lymphoma. J. Clin. Oncol. 2017, 35, 2410–2418. [Google Scholar] [CrossRef]
- Liu, Y.; Hernandez, A.M.; Shibata, D.; Cortopassi, G.A. BCL2 translocation frequency rises with age in humans. Proc. Natl. Acad. Sci. USA 1994, 91, 8910–8914. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alterations | Frequencies [%] (Number of Cases) | Techniques | References | |
|---|---|---|---|---|
| Average mutation frequencies of variable regions of IGHV genes | 13 (10) | IG PCR; Sanger sequencing | [6] | |
| 18 (5) | IG PCR; Sanger sequencing | [11] | ||
| 10 (50) | IG PCR; Sanger sequencing | [12] | ||
| Ongoing process of SHM | 100 (3/3) | IG PCR; cloning; Sanger sequencing | [6] | |
| 60 (3/5) | IG PCR; cloning; Sanger sequencing | [11] | ||
| Aberrant SHM targeting genes with oncogenic potential | 90 (9/10) | PCR; Sanger sequencing | [13] | |
| 100 (9/9) | WES | [14] | ||
| Impaired CSR with Sµdel | IgM/IgD IgG Sµdel | 100 (11/11) 0 (0/11) 64 (7/11) | IG PCR, IGHC-RT-PCR, LD-PCR; Sanger sequencing, Southern blot | [15] |
| Recurrent translocations affecting the BCL6 gene | 23 (3/13) | FISH; LDI-PCR; Sanger sequencing | [16,17] | |
| 38 (14/37) | FISH; LDI-PCR; Sanger sequencing | [18] | ||
| 17 (13/75) | FISH | [19] | ||
| Recurrent translocations affecting the IG loci | 38 (5/13) | FISH | [16] | |
| 13 (10/75) | FISH | [19] | ||
| MYD88 L265P mutations | 36 (5/14) | PCR; Sanger sequencing | [20] | |
| 85 (23/27) | NGS | [21] | ||
| 86 (12/14) | WES; RNA-Seq | [22] | ||
| CD79B Y196X mutations | 8 (2/25) | PCR; Sanger sequencing | [23] | |
| 59 (16/27) | NGS | [21] | ||
| 64 (9/14) | WES; RNA-Seq | [22] | ||
| Other alterations of the B cell receptor pathway | INPP5D CBL BLNK | 20 (5/25) 4 (1/25) 4 (1/25) | PCR; Sanger sequencing | [23] |
| Mutations of PRDM1 | 19 (4/21) | PCR; Sanger sequencing | [24] | |
| Mutations of TBL1XR1 | 14 (4/29) | WES; PCR; Sanger sequencing | [25] | |
| 36 (5/14) | WES; RNA-Seq | [22] | ||
| 18q21 gains (BCL2 and MALT1) | 38 (5/13) | FISH | [16] | |
| 22 (2/9) | Array CGH | [26] | ||
| Chromosome 12 gains | 44 (4/9) | Array CGH | [26] | |
| 26 (5/19) | SNP array | [27] | ||
| 9p13 gains (PAX5) | 21 (4/19) | SNP array | [27] | |
| 7q31 gains | 21 (4/19) | SNP array | [27] | |
| 10q23 losses (PTEN) | 21 (4/19) | SNP array | [27] | |
| 9p21 losses (CDKN2A) | 50 (10/20) | FD-PCR | [28] | |
| 32 (6/19) | SNP array | [27] | ||
| 44 (16/36) | WES | [29] | ||
| 8q12 losses (TOX) | 32 (6/19) | SNP array | [27] | |
| 6p21 losses (MHC locus) | 56 (5/9) | Array CGH | [26] | |
| 74 (14/19) | SNP array | [27] | ||
| 79 (23/29) | WES; PCR; Sanger sequencing | [25] | ||
| 6q21 losses (PRDM1) | 56 (5/9) | Array CGH | [26] | |
| 52 (10/19) | SNP array | [27] | ||
| Cell-of-origin | ABC non-cl GCB | 24 43 33 | RNA array | [30] |
| Summary: Germinal center (GC) B cell-derived tumor cells with a GC exit phenotype; PIM1, MYD88, and CD79B mutations; activated NF-kB and BCR pathways; frequently deleted CDKN2A and MHC loci with a high expression of BCL2 and MYC, currently summarized as group MCD/C5/MYD88. | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montesinos-Rongen, M.; Brunn, A.; Sanchez-Ruiz, M.; Küppers, R.; Siebert, R.; Deckert, M. Impact of a Faulty Germinal Center Reaction on the Pathogenesis of Primary Diffuse Large B Cell Lymphoma of the Central Nervous System. Cancers 2021, 13, 6334. https://doi.org/10.3390/cancers13246334
Montesinos-Rongen M, Brunn A, Sanchez-Ruiz M, Küppers R, Siebert R, Deckert M. Impact of a Faulty Germinal Center Reaction on the Pathogenesis of Primary Diffuse Large B Cell Lymphoma of the Central Nervous System. Cancers. 2021; 13(24):6334. https://doi.org/10.3390/cancers13246334
Chicago/Turabian StyleMontesinos-Rongen, Manuel, Anna Brunn, Monica Sanchez-Ruiz, Ralf Küppers, Reiner Siebert, and Martina Deckert. 2021. "Impact of a Faulty Germinal Center Reaction on the Pathogenesis of Primary Diffuse Large B Cell Lymphoma of the Central Nervous System" Cancers 13, no. 24: 6334. https://doi.org/10.3390/cancers13246334
APA StyleMontesinos-Rongen, M., Brunn, A., Sanchez-Ruiz, M., Küppers, R., Siebert, R., & Deckert, M. (2021). Impact of a Faulty Germinal Center Reaction on the Pathogenesis of Primary Diffuse Large B Cell Lymphoma of the Central Nervous System. Cancers, 13(24), 6334. https://doi.org/10.3390/cancers13246334