
PARP Inhibitors and Myeloid Neoplasms: A Double-Edged Sword

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Chemical Biology of PARP Inhibitors
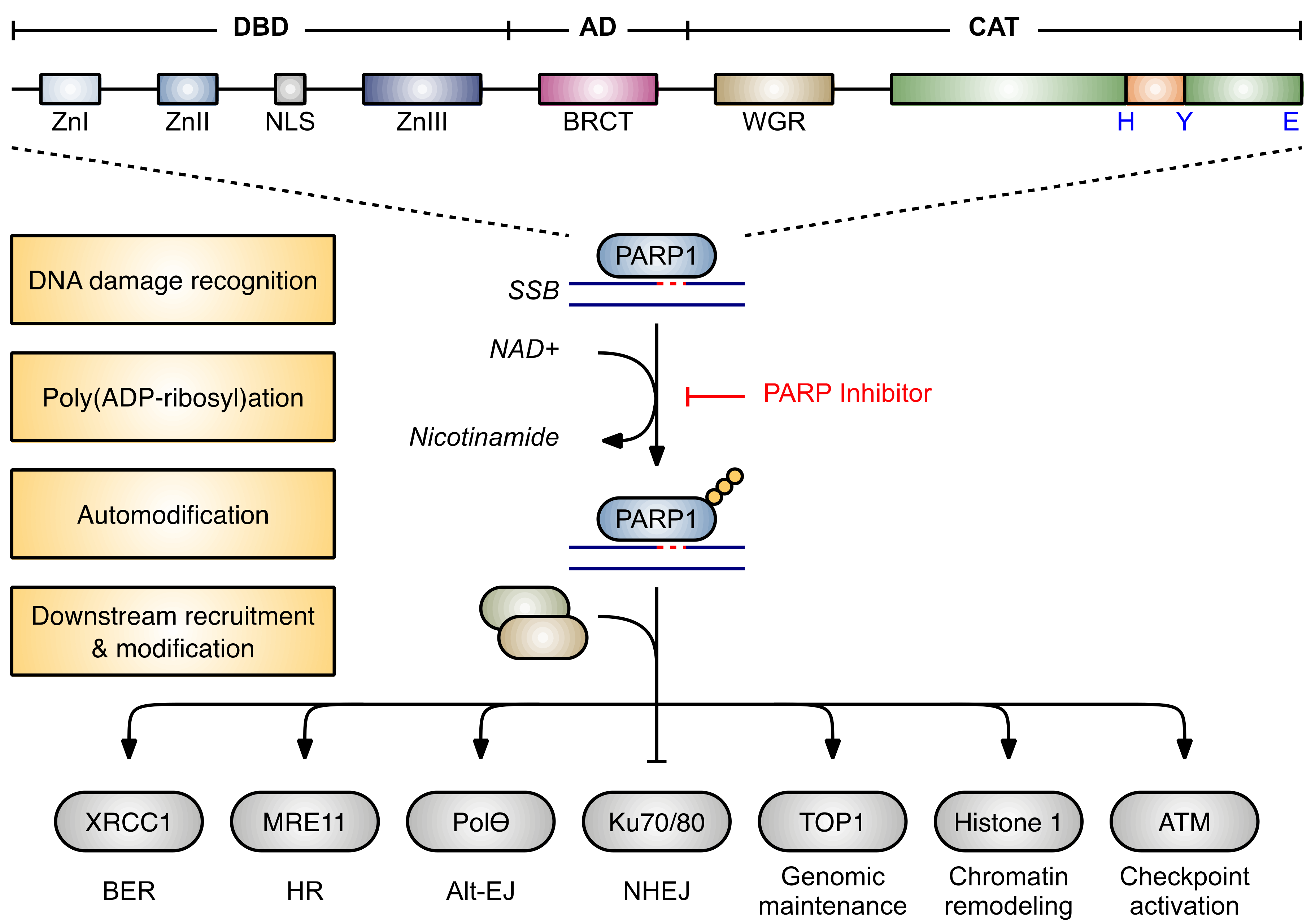
2.1. Structure and Function of ADP-Ribosyltransferases

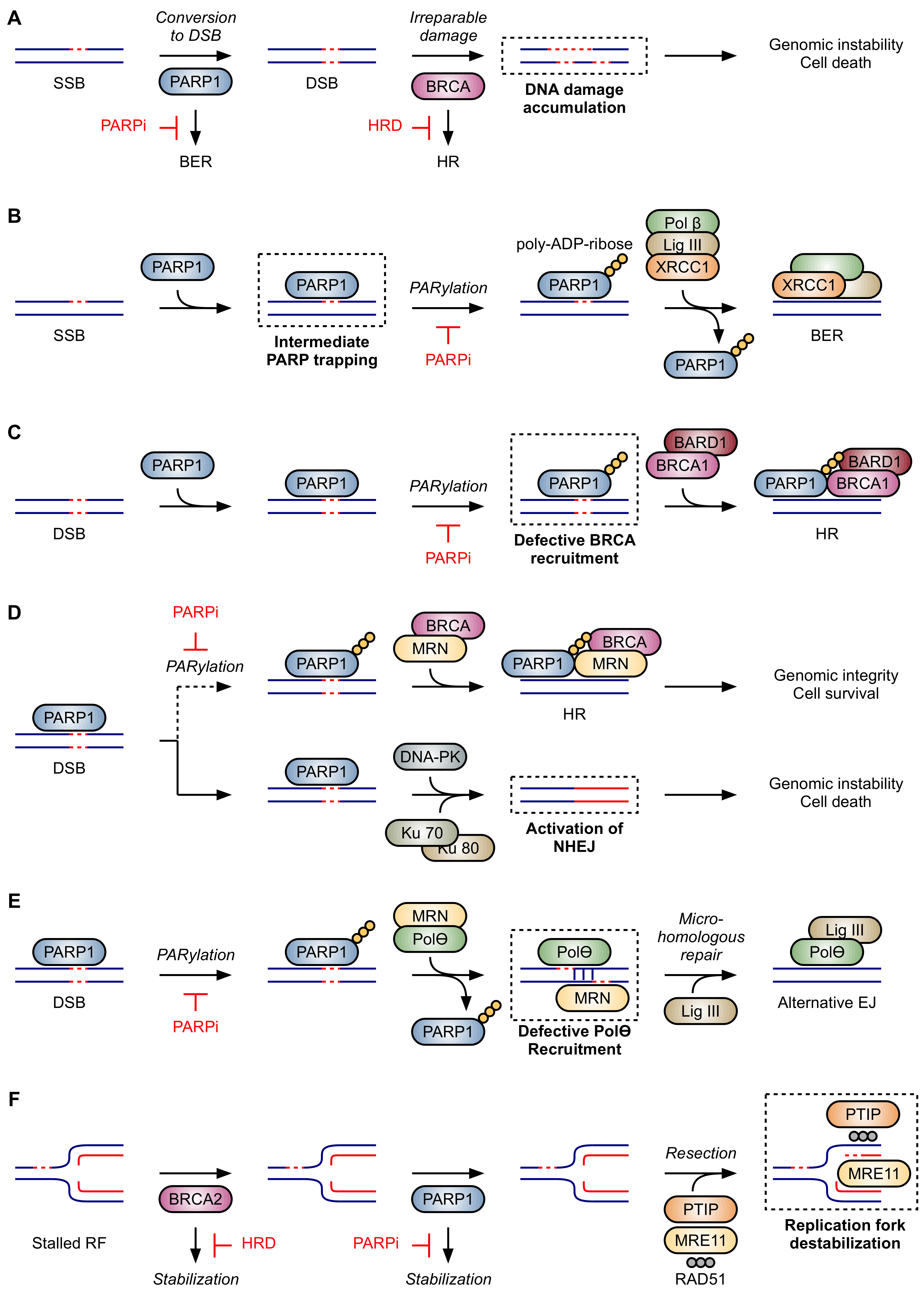
2.2. Proposed Mechanisms of Synthetic Lethality in HRD
2.2.1. Inhibition of Base Excision Repair
2.2.2. PARP Trapping
2.2.3. Impaired Recruitment of BRCA1
2.2.4. Activation of NHEJ
2.2.5. Inhibition of Alt-EJ
2.2.6. Destabilization of Stalled Replication Forks
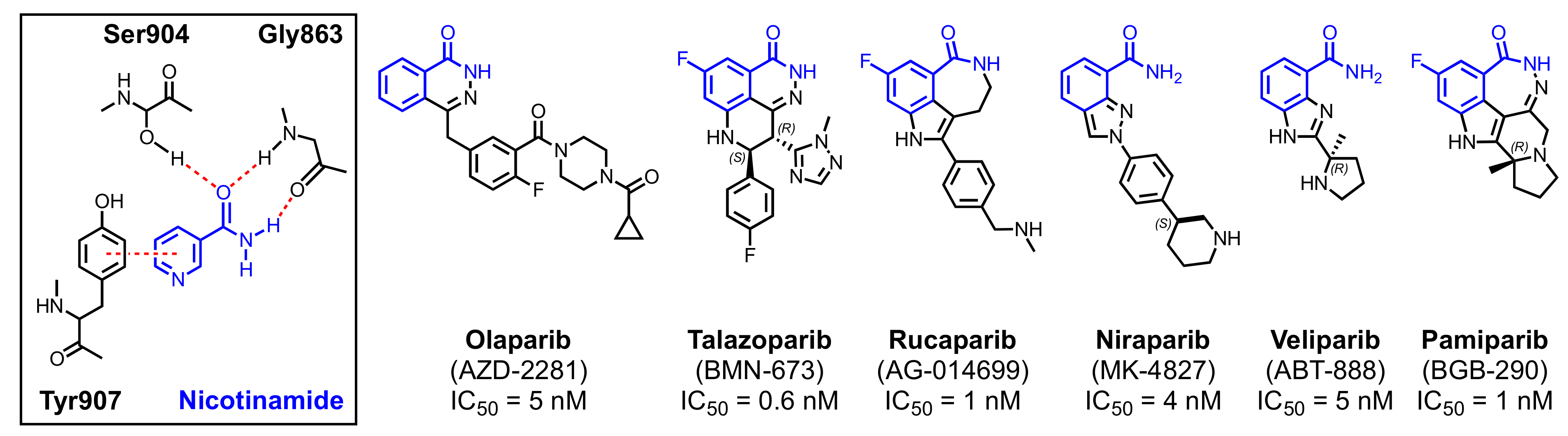
2.3. Clinical PARP Inhibitors
2.3.1. Olaparib
2.3.2. Talazoparib
2.3.3. Rucaparib
2.3.4. Niraparib
2.3.5. Veliparib
2.3.6. Pamiparib
3. PARP Inhibitors for the Treatment of Myeloid Neoplasms
3.1. Rationale for PARP Inhibition in Myeloid Neoplasms
3.2. Pre-Clinical Efficacy in Myeloid Neoplasms

{kind=link}
{kind=link}
{kind=link}
| Disease | Genotype(s) | Phenotype | Results of PARPi Monotherapy | Ref(s) |
|---|---|---|---|---|
| AML | FLT3-ITD mutant | Upregulation of RAD51 via STAT5 activation. Rapid depletion of γH2AX with highly active DSB repair. | Modest anti-leukemic activity seen with PARPi monotherapy in cell lines. Reduction in AML-initiating FLT3-ITD+ cells and clonogenic cells in bone marrow under hypoxic conditions. No significant reduction in leukemic burden or prolongation of survival in primary FLT3-ITD+ AML murine xenografts. | [158,162] |
| AML | IDH1/2 mutant | Increased 2HG inhibits KDM4A/B, ALKBH, ATR, and ATM to induce HRD and DSB persistence. | Primary IDH1/2-mutant AML cells possessed a 2HG-dependent DSB repair defect that conferred sensitivity to PARPi in vitro; sensitivity was reversed with IDH1/2 inhibitors. | [149,150,151] |
| AML | RUNX1- RUNX1T1 (AML1-ETO) positive | Downregulation of DNA repair genes, including BRCA2. High mutation frequency with mutator phenotype. Aberrant TET1 expression and DNA methylation. | Reduced colony-forming potential in RUNX1-RUNX1T1 transformed primary cells and patient-derived cell-lines. Prolonged survival in RUNX1-RUNX1T1 AML xenograft model. DNA damage-induced differentiation of PML-RARα transformed leukemic blasts. | [38,42,148,163] |
| AML | Cohesin (STAG2) mutant | High dependency on DDR pathways. Increased replication fork stalling. | AML (including STAG2-mutant) cell lines were sensitive to PARPi both in vitro and in vivo (xenograft model). Primary STAG2-mutant AML samples exhibited dose-dependent sensitivity to PARPi. PARPi depleted cohesin-mutant clones in a Tet2/Stag2-mutant murine model of MDS/AML. | [153] |
| APL | PML-RARα positive | Reduced MSH6, MLH1, BRCA1, and RAD51 expression. Repression of CHEK1, CHK2, and several BER genes induces a mutator phenotype. | Reduced colony-forming potential in PML-RARα transformed primary cells and patient-derived cell-lines. Suppressed disease onset in an ATRA-resistant APL xenograft model. DNA damage-induced differentiation of PML-RARα transformed leukemic blasts. | [38,39,152,164] |
| CML | BCR-ABL positive | Reduced translation of BRCA1 mRNA. Functional BRCA1 deficiency. HR downregulation and accumulation of DSBs. | Increased DSBs and reduced clonogenic potential of imatinib-refractory CML cell lines and primary samples, including under hypoxic conditions mimicking the bone marrow microenvironment. Eliminated quiescent cells in an inducible mouse model of chronic-phase CML. Reduced leukemic burden up to 10-fold in a BCR-ABL1+ leukemia xenograft model. | [146,147,159] |
| MLL | MLL-AF9 | High burden of oxidative DNA damage. Increased PARP1 expression and acetylation. | MLL-AF9 transformed murine bone marrow cells were only modestly sensitive to PARPi monotherapy. RUNX1-RUNX1T1-positive murine cells were highly sensitive to PARPi. Reduced the number of leukemic stem cells in primary human AML (MLL-AF9+) samples in vitro. PARPi and cytotoxic drugs (doxorubicin and cytarabine) exert additive anti-MLL-AF9 leukemia effects in mice. No significant reduction in leukemic burden was seen in a syngeneic mouse model of MLL-AF9+ leukemia (except when PARP inhibition was combined with cytotoxic drugs). MLL-AF9-transformed cells were resistant to olaparib monotherapy. No significant effect of olaparib on mice transplanted with wild-type MLL-AF9 leukemic cells. Hoxa9-deficient MLL-AF9 cells were highly sensitive to PARPi. | [38,165,166,167] |
| MPN | JAK2 (V617F) MPL (W515L) CALR (del52) positive | Reduced formation of RAD51 foci. Modest down-regulation of BRCA1/2. Accumulation of ROS-induced DSBs. | Modest in vitro sensitivity across several MPN cell lines, though sensitivity of primary MPN samples was variable. Primary MPN cells exhibited reduced colony formation in vitro after PARPi treatment. Veliparib monotherapy did not significantly prolong survival in a murine xenograft model. | [160,168,169] |
3.3. Clinical Efficacy in Myeloid Neoplasms
3.4. Biomarkers of PARPi Sensitivity
3.5. Mechanisms of PARP Inhibitor Resistance
3.6. Challenges and Future Directions in Development of PARP Inhibitors for Myeloid Neoplasms
4. Myeloid Neoplasms Emerging with PARP Inhibitor Therapy
4.1. Recognition of PARP Inhibitor Related Myeloid Neoplasms
- Is there a subset of patients who are at a particularly high risk of developing therapy-related MDS or AML while receiving treatment with a PARPi?
- If so, how can we identify this group of high-risk patients to better stratify the risks and benefits of PARPi therapy?
- Do germline mutations in BRCA1, BRCA2, BARD1, RAD51, TP53, or PALB2—which are commonly encountered in patients with ovarian or breast cancer—confound the picture by increasing the risk of therapy-related MDS and AML?
- Is the risk of therapy-related myeloid neoplasms cumulative with continued PARPi therapy?
- What is the contribution of other DNA-damaging modalities—including conventional chemotherapy and radiation therapy—to the emergence of therapy-related myeloid neoplasms?
4.2. Epidemiology and Characteristics of PARPi-Related Myeloid Neoplasms
4.3. Reconciling the Contradictory Effects of PARP Inhibitors
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tempero, M.A. NCCN guidelines updates: Pancreatic cancer. J. Natl. Compr. Cancer Netw. 2019, 17, 603–605. [Google Scholar]
- Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Barroilhet, L.; Behbakht, K.; Berchuck, A.; Berek, J.S.; Chen, L.M.; Cristea, M.; DeRosa, M. Ovarian cancer, version 1.2019 featured updates to the nccn guidelines. J. Natl. Compr. Cancer Netw. 2019, 17, 896–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradishar, W.J.; Moran, M.S.; Abraham, J.; Aft, R.; Agnese, D.; Allison, K.H.; Blair, S.L.; Burstein, H.J.; Dang, C.; Elias, A.D. NCCN guidelines® insights: Breast cancer, version 4.2021: Featured updates to the NCCN guidelines. J. Natl. Compr. Cancer Netw. 2021, 19, 484–493. [Google Scholar] [CrossRef]
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Behrman, S.W.; Benson, A.B.; Cardin, D.B.; Chiorean, E.G.; Chung, V.; Czito, B.; Del Chiaro, M. Pancreatic adenocarcinoma, version 2.2021, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Barroilhet, L.; Behbakht, K.; Berchuck, A.; Chen, L.-M.; Cristea, M.; DeRosa, M.; Eisenhauer, E.L. Ovarian cancer, version 2.2020, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 191–226. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, E.; Srinivas, S.; Antonarakis, E.S.; Armstrong, A.J.; Bekelman, J.E.; Cheng, H.; D’Amico, A.V.; Davis, B.J.; Desai, N.; Dorff, T. NCCN guidelines insights: Prostate cancer, version 1.2021: Featured updates to the NCCN guidelines. J. Natl. Compr. Cancer Netw. 2021, 19, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef] [Green Version]
- De Lorenzo, S.B.; Patel, A.G.; Hurley, R.M.; Kaufmann, S.H. The elephant and the blind men: Making sense of PARP inhibitors in homologous recombination deficient tumor cells. Front. Oncol. 2013, 3, 228. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Poveda, A.; Floquet, A.; Ledermann, J.A.; Asher, R.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Pignata, S.; Friedlander, M.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 620–631. [Google Scholar] [CrossRef]
- Morice, P.M.; Leary, A.; Dolladille, C.; Chrétien, B.; Poulain, L.; González-Martín, A.; Moore, K.; O’Reilly, E.M.; Ray-Coquard, I.; Alexandre, J. Myelodysplastic syndrome and acute myeloid leukaemia in patients treated with PARP inhibitors: A safety meta-analysis of randomised controlled trials and a retrospective study of the WHO pharmacovigilance database. Lancet Haematol. 2021, 8, e122–e134. [Google Scholar] [CrossRef]
- Amé, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Gagné, J.-P.; Ethier, C.; Defoy, D.; Bourassa, S.; Langelier, M.-F.; Riccio, A.A.; Pascal, J.M.; Moon, K.-M.; Foster, L.J.; Ning, Z. Quantitative site-specific ADP-ribosylation profiling of DNA-dependent PARPs. DNA Repair 2015, 30, 68–79. [Google Scholar] [CrossRef]
- Jungmichel, S.; Rosenthal, F.; Altmeyer, M.; Lukas, J.; Hottiger, M.O.; Nielsen, M.L. Proteome-wide identification of poly (ADP-Ribosyl) ation targets in different genotoxic stress responses. Mol. Cell 2013, 52, 272–285. [Google Scholar] [CrossRef] [Green Version]
- Gagne, J.-P.; Pic, E.; Isabelle, M.; Krietsch, J.; Ethier, C.; Paquet, É.; Kelly, I.; Boutin, M.; Moon, K.-M.; Foster, L.J. Quantitative proteomics profiling of the poly (ADP-ribose)-related response to genotoxic stress. Nucleic Acids Res. 2012, 40, 7788–7805. [Google Scholar] [CrossRef] [Green Version]
- Gibson, B.A.; Zhang, Y.; Jiang, H.; Hussey, K.M.; Shrimp, J.H.; Lin, H.; Schwede, F.; Yu, Y.; Kraus, W.L. Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 2016, 353, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [Green Version]
- Caron, M.C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.P.; Langelier, M.F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D. Survival with olaparib in metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Karp, J.E.; Thomas, B.M.; Greer, J.M.; Sorge, C.; Gore, S.D.; Pratz, K.W.; Smith, B.D.; Flatten, K.S.; Peterson, K.; Schneider, P. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clin. Cancer Res. 2012, 18, 6723–6731. [Google Scholar] [CrossRef] [Green Version]
- Suarez, F.; Mahlaoui, N.; Canioni, D.; Andriamanga, C.; Dubois d’Enghien, C.; Brousse, N.; Jais, J.-P.; Fischer, A.; Hermine, O.; Stoppa-Lyonnet, D. Incidence, presentation, and prognosis of malignancies in ataxia-telangiectasia: A report from the French national registry of primary immune deficiencies. J. Clin. Oncol. 2015, 33, 202–208. [Google Scholar] [CrossRef]
- Esposito, M.T.; Zhao, L.; Fung, T.K.; Rane, J.K.; Wilson, A.; Martin, N.; Gil, J.; Leung, A.Y.; Ashworth, A.; So, C.W. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat. Med. 2015, 21, 1481–1490. [Google Scholar] [CrossRef]
- Alcalay, M.; Meani, N.; Gelmetti, V.; Fantozzi, A.; Fagioli, M.; Orleth, A.; Riganelli, D.; Sebastiani, C.; Cappelli, E.; Casciari, C.; et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J. Clin. Investig. 2003, 112, 1751–1761. [Google Scholar] [CrossRef] [Green Version]
- Faraoni, I.; Compagnone, M.; Lavorgna, S.; Angelini, D.F.; Cencioni, M.T.; Piras, E.; Panetta, P.; Ottone, T.; Dolci, S.; Venditti, A.; et al. BRCA1, PARP1 and γH2AX in acute myeloid leukemia: Role as biomarkers of response to the PARP inhibitor olaparib. Biochim. Biophys. Acta 2015, 1852, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; So, C.W. PARP-inhibitor-induced synthetic lethality for acute myeloid leukemia treatment. Exp. Hematol. 2016, 44, 902–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejci, O.; Wunderlich, M.; Geiger, H.; Chou, F.S.; Schleimer, D.; Jansen, M.; Andreassen, P.R.; Mulloy, J.C. p53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress-induced death. Blood 2008, 111, 2190–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, M.C.; Marconi, G.; Feenstra, J.D.M.; Fonzi, E.; Papayannidis, C.; Ghelli Luserna di Rorá, A.; Padella, A.; Solli, V.; Franchini, E.; Ottaviani, E.; et al. Chromothripsis in acute myeloid leukemia: Biological features and impact on survival. Leukemia 2018, 32, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Gaymes, T.J.; Mufti, G.J.; Rassool, F.V. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the Ku70/86 heterodimer. Cancer Res. 2002, 62, 2791–2797. [Google Scholar]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Krietsch, J.; Rouleau, M.; Pic, É.; Ethier, C.; Dawson, T.M.; Dawson, V.L.; Masson, J.Y.; Poirier, G.G.; Gagné, J.P. Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Mol. Aspects Med. 2013, 34, 1066–1087. [Google Scholar] [CrossRef]
- Messner, S.; Hottiger, M.O. Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol. 2011, 21, 534–542. [Google Scholar] [CrossRef]
- Realini, C.A.; Althaus, F.R. Histone shuttling by poly(ADP-ribosylation). J. Biol. Chem. 1992, 267, 18858–18865. [Google Scholar] [CrossRef]
- Kraus, W.L.; Hottiger, M.O. PARP-1 and gene regulation: Progress and puzzles. Mol. Aspects Med. 2013, 34, 1109–1123. [Google Scholar] [CrossRef]
- Quénet, D.; El Ramy, R.; Schreiber, V.; Dantzer, F. The role of poly(ADP-ribosyl)ation in epigenetic events. Int. J. Biochem. Cell Biol. 2009, 41, 60–65. [Google Scholar] [CrossRef]
- Wang, T.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Chock, P.B.; Yang, D.C. Polyubiquitylation of PARP-1 through ubiquitin K48 is modulated by activated DNA, NAD+, and dipeptides. J. Cell. Biochem. 2008, 104, 318–328. [Google Scholar] [CrossRef]
- Wang, Z.; Michaud, G.A.; Cheng, Z.; Zhang, Y.; Hinds, T.R.; Fan, E.; Cong, F.; Xu, W. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev. 2012, 26, 235–240. [Google Scholar] [CrossRef] [Green Version]
- Aravind, L. The WWE domain: A common interaction module in protein ubiquitination and ADP ribosylation. Trends Biochem. Sci. 2001, 26, 273–275. [Google Scholar] [CrossRef]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef] [Green Version]
- Bai, P.; Cantó, C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Dantzer, F.; Schreiber, V.; Niedergang, C.; Trucco, C.; Flatter, E.; De La Rubia, G.; Oliver, J.; Rolli, V.; Ménissier-de Murcia, J.; de Murcia, G. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie 1999, 81, 69–75. [Google Scholar] [CrossRef]
- De Murcia, J.M.; Niedergang, C.; Trucco, C.; Ricoul, M.; Dutrillaux, B.; Mark, M.; Oliver, F.J.; Masson, M.; Dierich, A.; LeMeur, M.; et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. USA 1997, 94, 7303–7307. [Google Scholar] [CrossRef] [Green Version]
- Masson, M.; Niedergang, C.; Schreiber, V.; Muller, S.; Menissier-de Murcia, J.; de Murcia, G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol. 1998, 18, 3563–3571. [Google Scholar] [CrossRef] [Green Version]
- Trucco, C.; Oliver, F.J.; de Murcia, G.; Ménissier-de Murcia, J. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 1998, 26, 2644–2649. [Google Scholar] [CrossRef]
- Schultz, N.; Lopez, E.; Saleh-Gohari, N.; Helleday, T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003, 31, 4959–4964. [Google Scholar] [CrossRef]
- Langelier, M.F.; Pascal, J.M. PARP-1 mechanism for coupling DNA damage detection to poly(ADP-ribose) synthesis. Curr. Opin. Struct. Biol. 2013, 23, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradwohl, G.; Ménissier de Murcia, J.M.; Molinete, M.; Simonin, F.; Koken, M.; Hoeijmakers, J.H.; de Murcia, G. The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA. Proc. Natl. Acad. Sci. USA 1990, 87, 2990–2994. [Google Scholar] [CrossRef] [Green Version]
- Kulczyk, A.W.; Yang, J.C.; Neuhaus, D. Solution structure and DNA binding of the zinc-finger domain from DNA ligase IIIalpha. J. Mol. Biol. 2004, 341, 723–738. [Google Scholar] [CrossRef]
- Altmeyer, M.; Messner, S.; Hassa, P.O.; Fey, M.; Hottiger, M.O. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009, 37, 3723–3738. [Google Scholar] [CrossRef] [Green Version]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: Structural and functional insights into DNA-dependent PARP-1 activity. J. Biol. Chem. 2011, 286, 10690–10701. [Google Scholar] [CrossRef] [Green Version]
- Pion, E.; Bombarda, E.; Stiegler, P.; Ullmann, G.M.; Mély, Y.; de Murcia, G.; Gérard, D. Poly(ADP-ribose) polymerase-1 dimerizes at a 5’ recessed DNA end in vitro: A fluorescence study. Biochemistry 2003, 42, 12409–12417. [Google Scholar] [CrossRef]
- Pion, E.; Ullmann, G.M.; Amé, J.C.; Gérard, D.; de Murcia, G.; Bombarda, E. DNA-induced dimerization of poly(ADP-ribose) polymerase-1 triggers its activation. Biochemistry 2005, 44, 14670–14681. [Google Scholar] [CrossRef]
- Tao, Z.; Gao, P.; Liu, H.W. Identification of the ADP-ribosylation sites in the PARP-1 automodification domain: Analysis and implications. J. Am. Chem. Soc. 2009, 131, 14258–14260. [Google Scholar] [CrossRef]
- Mendoza-Alvarez, H.; Alvarez-Gonzalez, R. Poly(ADP-ribose) polymerase is a catalytic dimer and the automodification reaction is intermolecular. J. Biol. Chem. 1993, 268, 22575–22580. [Google Scholar] [CrossRef]
- Schreiber, V.; Amé, J.C.; Dollé, P.; Schultz, I.; Rinaldi, B.; Fraulob, V.; Ménissier-de Murcia, J.; de Murcia, G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem. 2002, 277, 23028–23036. [Google Scholar] [CrossRef] [Green Version]
- Prokhorova, E.; Zobel, F.; Smith, R.; Zentout, S.; Gibbs-Seymour, I.; Schutzenhofer, K.; Peters, A.; Groslambert, J.; Zorzini, V.; Agnew, T.; et al. Serine-linked PARP1 auto-modification controls PARP inhibitor response. Nat. Commun. 2021, 12, 4055. [Google Scholar] [CrossRef]
- Suskiewicz, M.J.; Zobel, F.; Ogden, T.E.H.; Fontana, P.; Ariza, A.; Yang, J.C.; Zhu, K.; Bracken, L.; Hawthorne, W.J.; Ahel, D.; et al. HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature 2020, 579, 598–602. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Buch-Larsen, S.C.; Prokhorova, E.; Elsborg, J.D.; Rebak, A.; Zhu, K.; Ahel, D.; Lukas, C.; Ahel, I.; Nielsen, M.L. The regulatory landscape of the human HPF1- and ARH3-dependent ADP-ribosylome. Nat. Commun. 2021, 12, 5893. [Google Scholar] [CrossRef]
- Li, M.; Yu, X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013, 23, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Min, W.; Bruhn, C.; Grigaravicius, P.; Zhou, Z.W.; Li, F.; Krüger, A.; Siddeek, B.; Greulich, K.O.; Popp, O.; Meisezahl, C.; et al. Poly(ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat. Commun. 2013, 4, 2993. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Petit, S.A.; Ficarro, S.B.; Toomire, K.J.; Xie, A.; Lim, E.; Cao, S.A.; Park, E.; Eck, M.J.; Scully, R.; et al. PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination-mediated DNA repair. Cancer Discov. 2014, 4, 1430–1447. [Google Scholar] [CrossRef] [Green Version]
- Otto, H.; Reche, P.A.; Bazan, F.; Dittmar, K.; Haag, F.; Koch-Nolte, F. In silico characterization of the family of PARP-like poly(ADP-ribosyl)transferases (pARTs). BMC Genom. 2005, 6, 139. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.E.; Eisenberg, D. Crystal structure of diphtheria toxin bound to nicotinamide adenine dinucleotide. Biochemistry 1996, 35, 1137–1149. [Google Scholar] [CrossRef]
- Kleine, H.; Poreba, E.; Lesniewicz, K.; Hassa, P.O.; Hottiger, M.O.; Litchfield, D.W.; Shilton, B.H.; Lüscher, B. Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Mol. Cell 2008, 32, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juarez-Salinas, H.; Sims, J.L.; Jacobson, M.K. Poly(ADP-ribose) levels in carcinogen-treated cells. Nature 1979, 282, 740–741. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Robert, I.; Dantzer, F.; Reina-San-Martin, B. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J. Exp. Med. 2009, 206, 1047–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, A.; Siemann, M.; Grabos, M.; Murmann, T.; Pantelias, G.E.; Iliakis, G. Requirement for Parp-1 and DNA ligases 1 or 3 but not of Xrcc1 in chromosomal translocation formation by backup end joining. Nucleic Acids Res. 2014, 42, 6380–6392. [Google Scholar] [CrossRef] [Green Version]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [Green Version]
- Hottiger, M.O.; Hassa, P.O.; Lüscher, B.; Schüler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef]
- Amé, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Höger, T.; Ménissier-de Murcia, J.; de Murcia, G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [Green Version]
- Rulten, S.L.; Fisher, A.E.; Robert, I.; Zuma, M.C.; Rouleau, M.; Ju, L.; Poirier, G.; Reina-San-Martin, B.; Caldecott, K.W. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol. Cell 2011, 41, 33–45. [Google Scholar] [CrossRef]
- Boehler, C.; Gauthier, L.R.; Mortusewicz, O.; Biard, D.S.; Saliou, J.M.; Bresson, A.; Sanglier-Cianferani, S.; Smith, S.; Schreiber, V.; Boussin, F.; et al. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc. Natl. Acad. Sci. USA 2011, 108, 2783–2788. [Google Scholar] [CrossRef] [Green Version]
- Wahlberg, E.; Karlberg, T.; Kouznetsova, E.; Markova, N.; Macchiarulo, A.; Thorsell, A.G.; Pol, E.; Frostell, Å.; Ekblad, T.; Öncü, D.; et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat. Biotechnol. 2012, 30, 283–288. [Google Scholar] [CrossRef]
- Thorsell, A.G.; Ekblad, T.; Karlberg, T.; Löw, M.; Pinto, A.F.; Trésaugues, L.; Moche, M.; Cohen, M.S.; Schüler, H. Structural basis for potency and promiscuity in poly(ADP-ribose) polymerase (PARP) and tankyrase inhibitors. J. Med. Chem. 2017, 60, 1262–1271. [Google Scholar] [CrossRef]
- Haince, J.F.; Kozlov, S.; Dawson, V.L.; Dawson, T.M.; Hendzel, M.J.; Lavin, M.F.; Poirier, G.G. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 2007, 282, 16441–16453. [Google Scholar] [CrossRef] [Green Version]
- Zaremba, T.; Curtin, N.J. PARP inhibitor development for systemic cancer targeting. Anticancer Agents Med. Chem. 2007, 7, 515–523. [Google Scholar] [CrossRef]
- Ratnam, K.; Low, J.A. Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clin. Cancer Res. 2007, 13, 1383–1388. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-ribose) polymerase inhibitors: Recent advances and future development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Saleh-Gohari, N.; Bryant, H.E.; Schultz, N.; Parker, K.M.; Cassel, T.N.; Helleday, T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol. Cell. Biol. 2005, 25, 7158–7169. [Google Scholar] [CrossRef] [Green Version]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Gottipati, P.; Vischioni, B.; Schultz, N.; Solomons, J.; Bryant, H.E.; Djureinovic, T.; Issaeva, N.; Sleeth, K.; Sharma, R.A.; Helleday, T. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010, 70, 5389–5398. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Q.; Auer, B.; Stingl, L.; Berghammer, H.; Haidacher, D.; Schweiger, M.; Wagner, E.F. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995, 9, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Han, E.K.; Anderson, M.; Shi, Y.; Semizarov, D.; Wang, G.; McGonigal, T.; Roberts, L.; Lasko, L.; Palma, J.; et al. Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol. Cancer Res. 2009, 7, 1686–1692. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.G.; Flatten, K.S.; Schneider, P.A.; Dai, N.T.; McDonald, J.S.; Poirier, G.G.; Kaufmann, S.H. Enhanced killing of cancer cells by poly(ADP-ribose) polymerase inhibitors and topoisomerase inhibitors reflects poisoning of both enzymes. J. Biol. Chem. 2012, 287, 4198–4210. [Google Scholar] [CrossRef] [Green Version]
- Gagné, J.P.; Isabelle, M.; Lo, K.S.; Bourassa, S.; Hendzel, M.J.; Dawson, V.L.; Dawson, T.M.; Poirier, G.G. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008, 36, 6959–6976. [Google Scholar] [CrossRef] [Green Version]
- Küpper, J.H.; de Murcia, G.; Bürkle, A. Inhibition of poly(ADP-ribosyl)ation by overexpressing the poly(ADP-ribose) polymerase DNA-binding domain in mammalian cells. J. Biol. Chem. 1990, 265, 18721–18724. [Google Scholar] [CrossRef]
- Molinete, M.; Vermeulen, W.; Bürkle, A.; Ménissier-de Murcia, J.; Küpper, J.H.; Hoeijmakers, J.H.; de Murcia, G. Overproduction of the poly(ADP-ribose) polymerase DNA-binding domain blocks alkylation-induced DNA repair synthesis in mammalian cells. EMBO J. 1993, 12, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.; Fernandez-Capetillo, O.; Kruhlak, M.J.; Pilch, D.R.; Staudt, D.W.; Lee, A.; Bonner, R.F.; Bonner, W.M.; Nussenzweig, A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 2003, 5, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Paddock, M.N.; Bauman, A.T.; Higdon, R.; Kolker, E.; Takeda, S.; Scharenberg, A.M. Competition between PARP-1 and Ku70 control the decision between high-fidelity and mutagenic DNA repair. DNA Repair 2011, 10, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Hochegger, H.; Dejsuphong, D.; Fukushima, T.; Morrison, C.; Sonoda, E.; Schreiber, V.; Zhao, G.Y.; Saberi, A.; Masutani, M.; Adachi, N.; et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 2006, 25, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Yang, K.; Dejsuphong, D.; Hirota, K.; Takeda, S.; D’Andrea, A.D. The USP1/UAF1 complex promotes double-strand break repair through homologous recombination. Mol. Cell. Biol. 2011, 31, 2462–2469. [Google Scholar] [CrossRef] [Green Version]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Farkkila, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.G.; Cortes, U.; Patnaik, S.; Jasin, M.; Wang, Z.Q. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004, 23, 3872–3882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malanga, M.; Althaus, F.R. Poly(ADP-ribose) reactivates stalled DNA topoisomerase I and Induces DNA strand break resealing. J. Biol. Chem. 2004, 279, 5244–5248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruf, A.; Mennissier de Murcia, J.; de Murcia, G.; Schulz, G.E. Structure of the catalytic fragment of poly(AD-ribose) polymerase from chicken. Proc. Natl. Acad. Sci. USA 1996, 93, 7481–7485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruf, A.; de Murcia, G.; Schulz, G.E. Inhibitor and NAD+ binding to poly(ADP-ribose) polymerase as derived from crystal structures and homology modeling. Biochemistry 1998, 37, 3893–3900. [Google Scholar] [CrossRef]
- Lawlor, D.; Martin, P.; Busschots, S.; Thery, J.; O’Leary, J.J.; Hennessy, B.T.; Stordal, B. PARP Inhibitors as P-glyoprotein Substrates. J. Pharm. Sci. 2014, 103, 1913–1920. [Google Scholar] [CrossRef]
- Henneman, L.; van Miltenburg, M.H.; Michalak, E.M.; Braumuller, T.M.; Jaspers, J.E.; Drenth, A.P.; de Korte-Grimmerink, R.; Gogola, E.; Szuhai, K.; Schlicker, A.; et al. Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1-deficient metaplastic breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 8409–8414. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, J.; Roberts, G.; Luger, K. Histone parylation factor 1 contributes to the inhibition of PARP1 by cancer drugs. Nat. Commun. 2021, 12, 736. [Google Scholar] [CrossRef]
- Menear, K.A.; Adcock, C.; Boulter, R.; Cockcroft, X.L.; Copsey, L.; Cranston, A.; Dillon, K.J.; Drzewiecki, J.; Garman, S.; Gomez, S.; et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: A novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J. Med. Chem. 2008, 51, 6581–6591. [Google Scholar] [CrossRef]
- Wang, B.; Chu, D.; Feng, Y.; Shen, Y.; Aoyagi-Scharber, M.; Post, L.E. Discovery and characterization of (8S,9R)-5-Fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-tetrahydro-3H-pyrido [4,3,2-de]phthalazin-3-one (BMN 673, Talazoparib), a novel, highly potent, and orally efficacious poly(ADP-ribose) polymerase-1/2 inhibitor, as an anticancer agent. J. Med. Chem. 2016, 59, 335–357. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Thomas, H.D.; Calabrese, C.R.; Batey, M.A.; Canan, S.; Hostomsky, Z.; Kyle, S.; Maegley, K.A.; Newell, D.R.; Skalitzky, D.; Wang, L.Z.; et al. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol. Cancer Ther. 2007, 6, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Zhou, Y.; Zhao, W.; Jiao, H.; Chen, Y.; Yang, Y.; Li, Z. Identification of novel PARP-1 inhibitors: Drug design, synthesis and biological evaluation. Bioorgan. Med. Chem. Lett. 2015, 25, 4557–4561. [Google Scholar] [CrossRef]
- Jones, P.; Altamura, S.; Boueres, J.; Ferrigno, F.; Fonsi, M.; Giomini, C.; Lamartina, S.; Monteagudo, E.; Ontoria, J.M.; Orsale, M.V.; et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): A novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J. Med. Chem. 2009, 52, 7170–7185. [Google Scholar] [CrossRef]
- Jones, P.; Wilcoxen, K.; Rowley, M.; Toniatti, C. Niraparib: A poly(ADP-ribose) polymerase (PARP) inhibitor for the treatment of tumors with defective homologous recombination. J. Med. Chem. 2015, 58, 3302–3314. [Google Scholar] [CrossRef]
- Donawho, C.K.; Luo, Y.; Luo, Y.; Penning, T.D.; Bauch, J.L.; Bouska, J.J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 2007, 13, 2728–2737. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ren, B.; Liu, Y.; Jiang, B.; Guo, Y.; Wei, M.; Luo, L.; Kuang, X.; Qiu, M.; Lv, L.; et al. Discovery of Pamiparib (BGB-290), a Potent and Selective Poly (ADP-ribose) Polymerase (PARP) Inhibitor in Clinical Development. J. Med. Chem. 2020, 63, 15541–15563. [Google Scholar] [CrossRef]
- Xiong, Y.; Guo, Y.; Liu, Y.; Wang, H.; Gong, W.; Liu, Y.; Wang, X.; Gao, Y.; Yu, F.; Su, D.; et al. Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia 2020, 22, 431–440. [Google Scholar] [CrossRef]
- Markham, A. Pamiparib: First approval. Drugs 2021, 81, 1343–1348. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Esposito, M.T.; So, C.W. DNA damage accumulation and repair defects in acute myeloid leukemia: Implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma 2014, 123, 545–561. [Google Scholar] [CrossRef]
- Faraoni, I.; Giansanti, M.; Voso, M.T.; Lo-Coco, F.; Graziani, G. Targeting ADP-ribosylation by PARP inhibitors in acute myeloid leukaemia and related disorders. Biochem. Pharmacol. 2019, 167, 133–148. [Google Scholar] [CrossRef]
- Santos, M.A.; Faryabi, R.B.; Ergen, A.V.; Day, A.M.; Malhowski, A.; Canela, A.; Onozawa, M.; Lee, J.E.; Callen, E.; Gutierrez-Martinez, P.; et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 2014, 514, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Gaymes, T.J.; Shall, S.; MacPherson, L.J.; Twine, N.A.; Lea, N.C.; Farzaneh, F.; Mufti, G.J. Inhibitors of poly ADP-ribose polymerase (PARP) induce apoptosis of myeloid leukemic cells: Potential for therapy of myeloid leukemia and myelodysplastic syndromes. Haematologica 2009, 94, 638–646. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Uzui, K.; Nishi, R.; Shigemi, H.; Ueda, T. Gemtuzumab ozogamicin and olaparib exert synergistic cytotoxicity in CD33-positive HL-60 myeloid leukemia cells. Anticancer Res. 2014, 34, 5487–5494. [Google Scholar]
- Wang, L.; Cai, W.; Zhang, W.; Chen, X.; Dong, W.; Tang, D.; Zhang, Y.; Ji, C.; Zhang, M. Inhibition of poly(ADP-ribose) polymerase 1 protects against acute myeloid leukemia by suppressing the myeloproliferative leukemia virus oncogene. Oncotarget 2015, 6, 27490–27504. [Google Scholar] [CrossRef] [Green Version]
- Nieborowska-Skorska, M.; Sullivan, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Hoser, G.; Maifrede, S.; Martinez, E.; Di Marcantonio, D.; Bolton-Gillespie, E.; Cramer-Morales, K.; et al. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J. Clin. Investig. 2017, 127, 2392–2406. [Google Scholar] [CrossRef]
- Podszywalow-Bartnicka, P.; Wolczyk, M.; Kusio-Kobialka, M.; Wolanin, K.; Skowronek, K.; Nieborowska-Skorska, M.; Dasgupta, Y.; Skorski, T.; Piwocka, K. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle 2014, 13, 3727–3741. [Google Scholar] [CrossRef] [Green Version]
- Forster, V.J.; Nahari, M.H.; Martinez-Soria, N.; Bradburn, A.K.; Ptasinska, A.; Assi, S.A.; Fordham, S.E.; McNeil, H.; Bonifer, C.; Heidenreich, O.; et al. The leukemia-associated RUNX1/ETO oncoprotein confers a mutator phenotype. Leukemia 2016, 30, 250–253. [Google Scholar] [CrossRef] [Green Version]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, R.J.; Radivoyevitch, T.; Nagata, Y.; Khurshed, M.; Przychodzen, B.; Makishima, H.; Xu, M.; Bleeker, F.E.; Wilmink, J.W.; Carraway, H.E.; et al. IDH1/2 mutations sensitize acute myeloid leukemia to PARP inhibition and this is reversed by IDH1/2-mutant inhibitors. Clin. Cancer Res. 2018, 24, 1705–1715. [Google Scholar] [CrossRef] [Green Version]
- Sule, A.; Van Doorn, J.; Sundaram, R.K.; Ganesa, S.; Vasquez, J.C.; Bindra, R.S. Targeting IDH1/2 mutant cancers with combinations of ATR and PARP inhibitors. NAR Cancer 2021, 3, zcab018. [Google Scholar] [CrossRef] [PubMed]
- Casorelli, I.; Tenedini, E.; Tagliafico, E.; Blasi, M.F.; Giuliani, A.; Crescenzi, M.; Pelosi, E.; Testa, U.; Peschle, C.; Mele, L.; et al. Identification of a molecular signature for leukemic promyelocytes and their normal counterparts: Focus on DNA repair genes. Leukemia 2006, 20, 1978–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tothova, Z.; Valton, A.L.; Gorelov, R.A.; Vallurupalli, M.; Krill-Burger, J.M.; Holmes, A.; Landers, C.C.; Haydu, J.E.; Malolepsza, E.; Hartigan, C.; et al. Cohesin mutations alter DNA damage repair and chromatin structure and create therapeutic vulnerabilities in MDS/AML. JCI Insight 2021, 6, e142149. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Liu, Y.; Chou, F.J.; Yang, C. Genotoxic therapy and resistance mechanism in gliomas. Pharmacol. Ther. 2021, 228, 107922. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Ray Chaudhuri, A.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the cytotoxic effects of PARP inhibitors with DNA demethylating agents—A potential therapy for cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogan, A.A.; Mclaughlin, L.J.; Topper, M.; Muvarak, N.; Stojanovic, L.; Creed, T.M.; Bentzen, S.; Civin, C.I.; Baer, M.R.; Kingsbury, T.J.; et al. DNA demethylating agents generate a brcaness effect in multiple sporadic tumor types: Prediction for sensitivity to PARP inhibitors in AML. Blood 2017, 130, 3347. [Google Scholar] [CrossRef]
- Maifrede, S.; Nieborowska-Skorska, M.; Sullivan-Reed, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Le, B.V.; Solecka, M.; Lian, Z.; Belyaeva, E.A.; Nersesyan, A.; et al. Tyrosine kinase inhibitor-induced defects in DNA repair sensitize FLT3(ITD)-positive leukemia cells to PARP1 inhibitors. Blood 2018, 132, 67–77. [Google Scholar] [CrossRef]
- Podszywalow-Bartnicka, P.; Maifrede, S.; Le, B.V.; Nieborowska-Skorska, M.; Piwocka, K.; Skorski, T. PARP1 inhibitor eliminated imatinib-refractory chronic myeloid leukemia cells in bone marrow microenvironment conditions. Leuk. Lymphoma 2019, 60, 262–264. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Maifrede, S.; Dasgupta, Y.; Sullivan, K.; Flis, S.; Le, B.V.; Solecka, M.; Belyaeva, E.A.; Kubovcakova, L.; Nawrocki, M.; et al. Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms. Blood 2017, 130, 2848–2859. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.W.; Koh, B.D.; Zhang, J.S.; Flatten, K.S.; Schneider, P.A.; Billadeau, D.D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Kaufmann, S.H. Poly(ADP-ribose) polymerase inhibitors sensitize cancer cells to death receptor-mediated apoptosis by enhancing death receptor expression. J. Biol. Chem. 2014, 289, 20543–20558. [Google Scholar] [CrossRef] [Green Version]
- Seedhouse, C.H.; Hunter, H.M.; Lloyd-Lewis, B.; Massip, A.M.; Pallis, M.; Carter, G.I.; Grundy, M.; Shang, S.; Russell, N.H. DNA repair contributes to the drug-resistant phenotype of primary acute myeloid leukaemia cells with FLT3 internal tandem duplications and is reversed by the FLT3 inhibitor PKC412. Leukemia 2006, 20, 2130–2136. [Google Scholar] [CrossRef]
- Bamezai, S.; He, J.; Sahin, D.; Mohr, F.; Ciccarone, F.; Vegi, N.M.; Pulikkottil Jose, A.; Mulaw, M.A.; Caiafa, P.; Döhner, K.; et al. The PARP inhibitor olaparib antagonizes leukemic growth induced by TET1 overexpression in AML1-ETO positive acute myeloid leukemia. Blood 2016, 128, 4063. [Google Scholar] [CrossRef]
- Giansanti, M.; De Gabrieli, A.; Prete, S.P.; Ottone, T.; Divona, M.D.; Karimi, T.; Ciccarone, F.; Voso, M.T.; Graziani, G.; Faraoni, I. Poly(ADP-ribose) polymerase inhibitors for arsenic trioxide-resistant acute promyelocytic leukemia: Synergistic in vitro antitumor effects with hypomethylating agents or high-dose vitamin C. J. Pharmacol. Exp. Ther. 2021, 377, 385–397. [Google Scholar] [CrossRef]
- Maifrede, S.; Martinez, E.; Nieborowska-Skorska, M.; Di Marcantonio, D.; Hulse, M.; Le, B.V.; Zhao, H.; Piwocka, K.; Tempera, I.; Sykes, S.M.; et al. MLL-AF9 leukemias are sensitive to PARP1 inhibitors combined with cytotoxic drugs. Blood Adv. 2017, 1, 1467–1472. [Google Scholar] [CrossRef]
- Zhao, L.; So, C.W.E. PARPi potentiates with current conventional therapy in MLL leukemia. Cell Cycle 2017, 16, 1861–1869. [Google Scholar] [CrossRef] [Green Version]
- Piao, J.; Takai, S.; Kamiya, T.; Inukai, T.; Sugita, K.; Ohyashiki, K.; Delia, D.; Masutani, M.; Mizutani, S.; Takagi, M. Poly (ADP-ribose) polymerase inhibitors selectively induce cytotoxicity in TCF3-HLF-positive leukemic cells. Cancer Lett. 2017, 386, 131–140. [Google Scholar] [CrossRef]
- Pratz, K.W.; Koh, B.D.; Patel, A.G.; Flatten, K.S.; Poh, W.; Herman, J.G.; Dilley, R.; Harrell, M.I.; Smith, B.D.; Karp, J.E.; et al. Poly (ADP-ribose) polymerase inhibitor hypersensitivity in aggressive myeloproliferative neoplasms. Clin. Cancer Res. 2016, 22, 3894–3902. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.R.; Senyuk, V.; Rodriguez, N.S.; Oh, A.L.; Bonetti, E.; Mahmud, D.; Barosi, G.; Mahmud, N.; Rondelli, D. Synergistic cytotoxic effect of busulfan and the PARP inhibitor veliparib in myeloproliferative neoplasms. Biol. Blood Marrow Transplant. 2019, 25, 855–860. [Google Scholar] [CrossRef]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Falzacappa, M.V.; Ronchini, C.; Faretta, M.; Iacobucci, I.; Di Rorà, A.G.; Martinelli, G.; Meyer, L.H.; Debatin, K.M.; Orecchioni, S.; Bertolini, F.; et al. The combination of the PARP inhibitor rucaparib and 5FU is an effective strategy for treating acute leukemias. Mol. Cancer Ther. 2015, 14, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Bowman, K.J.; White, A.; Golding, B.T.; Griffin, R.J.; Curtin, N.J. Potentiation of anti-cancer agent cytotoxicity by the potent poly(ADP-ribose) polymerase inhibitors NU1025 and NU1064. Br. J. Cancer 1998, 78, 1269–1277. [Google Scholar] [CrossRef] [Green Version]
- Orta, M.L.; Höglund, A.; Calderón-Montaño, J.M.; Domínguez, I.; Burgos-Morón, E.; Visnes, T.; Pastor, N.; Ström, C.; López-lázaro, M.; Helleday, T. The PARP inhibitor olaparib disrupts base excision repair of 5-aza-2′-deoxycytidine lesions. Nucleic Acids Res. 2014, 42, 9108–9120. [Google Scholar] [CrossRef] [Green Version]
- Faraoni, I.; Consalvo, M.I.; Aloisio, F.; Fabiani, E.; Giansanti, M.; Di Cristino, F.; Falconi, G.; Tentori, L.; Di Veroli, A.; Curzi, P.; et al. Cytotoxicity and differentiating effect of the poly(ADP-ribose) polymerase inhibitor olaparib in myelodysplastic syndromes. Cancers 2019, 11, 1371. [Google Scholar] [CrossRef] [Green Version]
- Gaymes, T.J.; Padua, R.A.; Pla, M.; Orr, S.; Omidvar, N.; Chomienne, C.; Mufti, G.J.; Rassool, F.V. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: A mechanism for leukemia-specific HDI-dependent apoptosis? Mol. Cancer Res. 2006, 4, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Nagaria, P.K.; Pawar, N.; Adewuyi, A.; Gojo, I.; Meyers, D.J.; Cole, P.A.; Rassool, F.V. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk. Res. 2016, 45, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Dellomo, A.J.; Abbotts, R.; Eberly, C.L.; Karbowski, M.; Baer, M.R.; Kingsbury, T.J.; Rassool, F.V. PARP1 PARylates and stabilizes STAT5 in FLT3-ITD acute myeloid leukemia and other STAT5-activated cancers. Transl. Oncol. 2021, 15, 101283. [Google Scholar] [CrossRef]
- Garcia, T.B.; Snedeker, J.C.; Baturin, D.; Gardner, L.; Fosmire, S.P.; Zhou, C.; Jordan, C.T.; Venkataraman, S.; Vibhakar, R.; Porter, C.C. A small-molecule inhibitor of WEE1, AZD1775, synergizes with olaparib by impairing homologous recombination and enhancing DNA damage and apoptosis in acute leukemia. Mol. Cancer Ther. 2017, 16, 2058–2068. [Google Scholar] [CrossRef] [Green Version]
- Faraoni, I.; Aloisio, F.; De Gabrieli, A.; Consalvo, M.I.; Lavorgna, S.; Voso, M.T.; Lo-Coco, F.; Graziani, G. The poly(ADP-ribose) polymerase inhibitor olaparib induces up-regulation of death receptors in primary acute myeloid leukemia blasts by NF-κB activation. Cancer Lett. 2018, 423, 127–138. [Google Scholar] [CrossRef]
- Mufti, G.; Estey, E.; Popat, R.; Mattison, R.; Menne, T.; Azar, J.; Bloor, A.; Gaymes, T.; Khwaja, A.; Juckett, M. Results of a phase 1 study of BMN 673, a potent and specific PARP-1/2 inhibitor, in patients with advanced hematological malignancies. In Proceedings of the 19th Congress of the European Hematology Association, Milan, Italy, 12–15 June 2014; pp. 33–34. [Google Scholar]
- Gojo, I.; Beumer, J.H.; Pratz, K.W.; McDevitt, M.A.; Baer, M.R.; Blackford, A.L.; Smith, B.D.; Gore, S.D.; Carraway, H.E.; Showel, M.M.; et al. A phase 1 study of the PARP inhibitor veliparib in combination with temozolomide in acute myeloid leukemia. Clin. Cancer Res. 2017, 23, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Pratz, K.W.; Rudek, M.A.; Gojo, I.; Litzow, M.R.; McDevitt, M.A.; Ji, J.; Karnitz, L.M.; Herman, J.G.; Kinders, R.J.; Smith, B.D.; et al. A phase I study of topotecan, carboplatin and the PARP inhibitor veliparib in acute leukemias, aggressive myeloproliferative neoplasms, and chronic myelomonocytic leukemia. Clin. Cancer Res. 2017, 23, 899–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, G.; Yap, C.; Oldreive, C.; Slade, D.; Bishop, R.; Griffiths, M.; Dyer, M.J.S.; Fegan, C.; Oscier, D.; Pettitt, A.; et al. A multi-centre phase I trial of the PARP inhibitor olaparib in patients with relapsed chronic lymphocytic leukaemia, T-prolymphocytic leukaemia or mantle cell lymphoma. Br. J. Haematol. 2018, 182, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Fritz, C.; Portwood, S.M.; Przespolewski, A.; Wang, E.S. PARP goes the weasel! Emerging role of PARP inhibitors in acute leukemias. Blood Rev. 2021, 45, 100696. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; McDonald, S.; Swift, S.; Turner, N.C.; Ashworth, A. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair 2008, 7, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Lord, C.J.; Iorns, E.; Brough, R.; Swift, S.; Elliott, R.; Rayter, S.; Tutt, A.N.; Ashworth, A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008, 27, 1368–1377. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Aghajanian, C.; Konecny, G.E.; O’Malley, D.M.; et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat. Commun. 2021, 12, 2487. [Google Scholar] [CrossRef]
- Hurley, R.M.; McGehee, C.D.; Nesic, K.; Correia, C.; Weiskittel, T.M.; Kelly, R.L.; Venkatachalam, A.; Hou, X.; Pathoulas, N.M.; Meng, X.W.; et al. Characterization of a RAD51C-silenced high grade serous ovarian cancer model during development of PARP inhibitor resistance. NAR Cancer 2021, 3, zcab028. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Aroumougame, A.; Lobrich, M.; Li, Y.; Chen, D.; Chen, J.; Gong, Z. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 2014, 28, 2693–2698. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Xu, P.; Zhou, X.; Diao, Z.; Ouyang, J.; Yan, G.; Chen, B. MiR-181a enhances drug sensitivity of mixed lineage leukemia-rearranged acute myeloid leukemia by increasing poly(ADP-ribose) polymerase1 acetylation. Leuk. Lymphoma 2021, 62, 136–146. [Google Scholar] [CrossRef]
- Fontana, D.; Ramazzotti, D.; Aroldi, A.; Redaelli, S.; Magistroni, V.; Pirola, A.; Niro, A.; Massimino, L.; Mastini, C.; Brambilla, V.; et al. Integrated genomic, functional, and prognostic characterization of atypical chronic myeloid leukemia. Hemasphere 2020, 4, e497. [Google Scholar] [CrossRef]
- Crisà, E.; Nicolosi, M.; Ferri, V.; Favini, C.; Gaidano, G.; Patriarca, A. Atypical chronic myeloid leukemia: Where are we now? Int. J. Mol. Sci. 2020, 21, 6862. [Google Scholar] [CrossRef]
- Fan, J.; Li, L.; Small, D.; Rassool, F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: Implications for genomic instability and therapy. Blood 2010, 116, 5298–5305. [Google Scholar] [CrossRef] [Green Version]
- Plo, I.; Nakatake, M.; Malivert, L.; de Villartay, J.P.; Giraudier, S.; Villeval, J.L.; Wiesmuller, L.; Vainchenker, W. JAK2 stimulates homologous recombination and genetic instability: Potential implication in the heterogeneity of myeloproliferative disorders. Blood 2008, 112, 1402–1412. [Google Scholar] [CrossRef]
- Al-Ejeh, F.; Kumar, R.; Wiegmans, A.; Lakhani, S.R.; Brown, M.P.; Khanna, K.K. Harnessing the complexity of DNA-damage response pathways to improve cancer treatment outcomes. Oncogene 2010, 29, 6085–6098. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and cancer: A review of clinical implications of senescence and senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef]
- Fleury, H.; Malaquin, N.; Tu, V.; Gilbert, S.; Martinez, A.; Olivier, M.A.; Sauriol, A.; Communal, L.; Leclerc-Desaulniers, K.; Carmona, E.; et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat. Commun. 2019, 10, 2556. [Google Scholar] [CrossRef] [Green Version]
- Saliba, A.N.; John, A.J.; Kaufmann, S.H. Resistance to venetoclax and hypomethylating agents in acute myeloid leukemia. Cancer Drug Resist. 2021, 4, 125–142. [Google Scholar] [CrossRef]
- Ciccarone, F.; Valentini, E.; Bacalini, M.G.; Zampieri, M.; Calabrese, R.; Guastafierro, T.; Mariano, G.; Reale, A.; Franceschi, C.; Caiafa, P. Poly(ADP-ribosyl)ation is involved in the epigenetic control of TET1 gene transcription. Oncotarget 2014, 5, 10356–10367. [Google Scholar] [CrossRef] [Green Version]
- Ciccarone, F.; Valentini, E.; Zampieri, M.; Caiafa, P. 5mC-hydroxylase activity is influenced by the PARylation of TET1 enzyme. Oncotarget 2015, 6, 24333–24347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamezai, S.; Demir, D.; Pulikkottil, A.J.; Ciccarone, F.; Fischbein, E.; Sinha, A.; Borga, C.; Te Kronnie, G.; Meyer, L.H.; Mohr, F.; et al. TET1 promotes growth of T-cell acute lymphoblastic leukemia and can be antagonized via PARP inhibition. Leukemia 2021, 35, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Maifrede, S.; Le, B.V.; Nieborowska-Skorska, M.; Golovine, K.; Sullivan-Reed, K.; Dunuwille, W.M.B.; Nacson, J.; Hulse, M.; Keith, K.; Madzo, J.; et al. TET2 and DNMT3A mutations exert divergent effects on DNA repair and sensitivity of leukemia cells to PARP inhibitors. Cancer Res. 2021, 81, 5089–5101. [Google Scholar] [CrossRef] [PubMed]
- Jing, C.B.; Fu, C.; Prutsch, N.; Wang, M.; He, S.; Look, A.T. Synthetic lethal targeting of TET2-mutant hematopoietic stem and progenitor cells (HSPCs) with TOP1-targeted drugs and PARP1 inhibitors. Leukemia 2020, 34, 2992–3006. [Google Scholar] [CrossRef]
- Efficace, F.; Cottone, F.; Oswald, L.B.; Cella, D.; Patriarca, A.; Niscola, P.; Breccia, M.; Platzbecker, U.; Palumbo, G.A.; Caocci, G.; et al. The IPSS-R more accurately captures fatigue severity of newly diagnosed patients with myelodysplastic syndromes compared with the IPSS index. Leukemia 2020, 34, 2451–2459. [Google Scholar] [CrossRef]
- Aoki, D.; Chiyoda, T. PARP inhibitors and quality of life in ovarian cancer. Lancet Oncol. 2018, 19, 1012–1014. [Google Scholar] [CrossRef]
- Higgins, A.; Shah, M.V. Genetic and genomic landscape of secondary and therapy-related acute myeloid leukemia. Genes 2020, 11, 749. [Google Scholar] [CrossRef]
- Shih, A.H.; Chung, S.S.; Dolezal, E.K.; Zhang, S.-J.; Abdel-Wahab, O.I.; Park, C.Y.; Nimer, S.D.; Levine, R.L.; Klimek, V.M. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica 2013, 98, 908. [Google Scholar] [CrossRef] [Green Version]
- Cowell, I.G.; Austin, C.A. Mechanism of generation of therapy related leukemia in response to anti-topoisomerase II agents. Int. J. Environ. Res. Public Health 2012, 9, 2075–2091. [Google Scholar] [CrossRef]
- Itzhar, N.; Dessen, P.; Toujani, S.; Auger, N.; Preudhomme, C.; Richon, C.; Lazar, V.; Saada, V.; Bennaceur, A.; Bourhis, J.H. Chromosomal minimal critical regions in therapy-related leukemia appear different from those of de novo leukemia by high-resolution aCGH. PLoS ONE 2011, 6, e16623. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef] [Green Version]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Cleven, A.H.; Nardi, V.; Ok, C.Y.; Goswami, M.; Dal Cin, P.; Zheng, Z.; Iafrate, A.J.; Abdul Hamid, M.A.; Wang, S.A.; Hasserjian, R.P. High p53 protein expression in therapy-related myeloid neoplasms is associated with adverse karyotype and poor outcome. Mod. Pathol. 2015, 28, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Harrell, M.I.; Norquist, B.M.; Walsh, T.; Brady, M.; Lee, M.; Hershberg, R.; Kalli, K.R.; Lankes, H.; Konnick, E.Q.; et al. Somatic mosaic mutations in PPM1D and TP53 in the blood of women with ovarian carcinoma. JAMA Oncol. 2016, 2, 370–372. [Google Scholar] [CrossRef] [Green Version]
- Bolton, K.L.; Moukarzel, L.A.; Ptashkin, R.; Gao, T.; Patel, M.; Caltabellotta, N.; Braunstein, L.Z.; Aghajanian, C.; Hyman, D.M.; Berger, M.F.; et al. The impact of poly ADP ribose polymerase (PARP) inhibitors on clonal hematopoiesis. J. Clin. Oncol. 2020, 38, 1513. [Google Scholar] [CrossRef]
- Morton, L.M.; Dores, G.M.; Schonfeld, S.J.; Linet, M.S.; Sigel, B.S.; Lam, C.J.; Tucker, M.A.; Curtis, R.E. Association of chemotherapy for solid tumors with development of therapy-related myelodysplastic syndrome or acute myeloid leukemia in the modern era. JAMA Oncol. 2019, 5, 318–325. [Google Scholar] [CrossRef] [Green Version]
- Shenolikar, R.; Durden, E.; Meyer, N.; Lenhart, G.; Moore, K. Incidence of secondary myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) in patients with ovarian or breast cancer in a real-world setting in the United States. Gynecol. Oncol. 2018, 151, 190–195. [Google Scholar] [CrossRef]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [Green Version]
- Korach, J.; Turner, S.; Milenkova, T.; Alecu, I.; McMurtry, E.; Bloomfield, R.; Pujade-Lauraine, E. Incidence of myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) in patients (pts) with a germline (g) BRCA mutation (m) and platinum-sensitive relapsed ovarian cancer (PSR OC) receiving maintenance olaparib in SOLO2: Impact of prior lines of platinum therapy. J. Clin. Oncol. 2018, 36, 5548. [Google Scholar]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Banerjee, S.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; et al. Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1721–1731. [Google Scholar] [CrossRef]
- Tew, W.P.; Lacchetti, C.; Ellis, A.; Maxian, K.; Banerjee, S.; Bookman, M.; Jones, M.B.; Lee, J.M.; Lheureux, S.; Liu, J.F.; et al. PARP inhibitors in the management of ovarian cancer: ASCO guideline. J. Clin. Oncol. 2020, 38, 3468–3493. [Google Scholar] [CrossRef]
- Wethington, S.L.; Wahner-Hendrickson, A.E.; Swisher, E.M.; Kaufmann, S.H.; Karlan, B.Y.; Fader, A.N.; Dowdy, S.C. PARP inhibitor maintenance for primary ovarian cancer—A missed opportunity for precision medicine. Gynecol. Oncol. 2021, 163, 11–13. [Google Scholar] [CrossRef]
- Kayser, S.; Doehner, K.; Krauter, J.; Koehne, C.-H.; Horst, H.A.; Held, G.; von Lilienfeld-Toal, M.; Wilhelm, S.; Kuendgen, A.; Goetze, K. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood 2011, 117, 2137–2145. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.; Khalife-Hachem, S.; Grinda, T.; Kfoury, M.; Garciaz, S.; Pasquier, F.; Vargaftig, J.; Uzunov, M.; Belhabri, A.; Bertoli, S. Letter to the editor: Therapy-related myeloid neoplasms following treatment with PARP inhibitors: New molecular insights. Ann. Oncol. 2021, 32, 1046–1048. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Aghajanian, C.; Lorusso, D.; Colombo, N.; Dean, A.; Weberpals, J.; et al. Preexisting TP53-variant clonal hematopoiesis and risk of secondary myeloid neoplasms in patients with high-grade ovarian cancer treated with rucaparib. JAMA Oncol. 2021, 7, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Kartner, N.; Evernden-Porelle, D.; Bradley, G.; Ling, V. Detection of P-glycoprotein in multidrug-resistant cell lines by monoclonal antibodies. Nature 1985, 316, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Mayer, U.; Wagenaar, E.; Mol, C.A.; van Deemter, L.; Smit, J.J.; van der Valk, M.A.; Voordouw, A.C.; Spits, H.; van Tellingen, O.; et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc. Natl. Acad. Sci. USA 1997, 94, 4028–4033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson-Miller, C.L.; May, R.D.; Tomaszewski, J.; Osborn, B.; Murphy, M.J.; Page, J.G.; Parchment, R.E. Differential toxicity of camptothecin, topotecan and 9-aminocamptothecin to human, canine, and murine myeloid progenitors (CFU-GM) in vitro. Cancer Chemother. Pharmacol. 1997, 39, 467–472. [Google Scholar] [CrossRef]


| Class | Agent(s) | PARPi(s) | Mechanism(s) | Results of Combination Therapy | Ref(s) |
|---|---|---|---|---|---|
| Alkylating agents | Temozolomide Busulfan | Olaparib Veliparib | Temozolomide induced abasic sites and resultant SSBs. Busulfan stalled replication forks through DNA strand crosslinking. Combination with olaparib, but not veliparib, significantly increased PARP trapping. | PARPi showed synergy with temozolomide (CI < 0.3) and busulfan (CI 0.40–0.55) in vitro. With temozolomide, olaparib was >100-fold more potent than veliparib due to enhanced PARP trapping with olaparib. Busulfan plus veliparib was associated with activation of the ATR-Chk1 pathway and G2/M arrest in MPN cell lines, and modestly prolonged survival in a murine xenograft model of MPN-AML. | [169,170] |
| Conventional chemotherapy | Doxorubicin Daunorubicin Cytarabine 5-Fluorouracil | Olaparib Talazoparib Rucaparib | Increased abundance and phosphorylation of H2AX and CHK1. Accumulation of oxidative DNA damage. Suppression of ATM. | Increased PARPi sensitivity in vitro with accumulation of DNA damage, replication arrest, and apoptosis. Synergistic cytotoxicity against primary IDH1/2-mutant AML cells associated with ATM suppression. Rucaparib cooperates with 5-FU to accumulate DSBs in vitro and significantly enhance cytotoxicity in a syngeneic murine model of AML. Olaparib potentiates anti-leukemogenic activity of conventional chemotherapy in MLL. | [150,165,166,171] |
| Topoisomerase poisons | Camptothecin Etoposide | Olaparib Veliparib | Camptothecin-induced DNA lesions induce replication fork stalling, which depend in part on PARP1 for restart. | PARPi treatment was synergistic with camptothecin (CI < 0.3) in vitro; no increase in PARP/DNA complexes was detected using an insensitive assay, but genetic studies suggest a key role for PARP trapping. No synergy was seen with PARPi plus etoposide. | [104,170,172] |
| DNMT inhibitors | Decitabine Azacitidine | Olaparib Talazoparib | Downregulation of RAD51, BRCA1/2, FEN1, and FANCD2 leads to HRD. Trapped both DNMT and PARP1 at sites of DNA damage. Repair of decitabine-induced DNA lesions is mediated by BER and requires XRCC1, recruitment of which is impaired by PARPi. | Decitabine plus olaparib was synthetically lethal in a large panel of AML cell lines, with synergy driven by PARPi-mediated inhibition of XRCC1 recruitment. DNMTi treatment induced HRD in most primary AML samples, and combination therapy (decitabine + talazoparib) significantly reduced subsequent colony formation. Combination therapy reduced leukemic burden and prolonged survival in murine AML xenografts. Synergistic antiproliferative effects against ATO-sensitive and ATO-resistant APL cell lines, with PARPi and demethylating agents (azacitidine, decitabine, and ascorbate). Synergistic cytotoxic and differentiating effects on primary MDS cells grown on culture. | [143,156,157,164,173,174] |
| HDAC inhibitors | Entinostat Trichostatin A Apcidin | PJ34 EB47 KU-0058948 Talazoparib | Induced DNA damage, phosphorylation of H2AX and ATM, and ultimately apoptosis. Promoted PARP trapping and impaired NHEJ via differential acetylation of Ku70/80. | HDAC inhibition enhanced PARP trapping, and co-treatment with a PARPi significantly increased apoptosis in AML cell lines. Synergistic cytotoxicity was seen with MS275 + PARPi combination therapy in select AML cell lines. | [143,175,176] |
| JAK2 inhibitors | Ruxolitinib | Olaparib Talazoparib | Impaired BRCA-mediated HR and DNA-PK-mediated NHEJ, thereby increasing sensitivity to PARP inhibition. | Ruxolitinib enhanced PARPi sensitivity in both MPN cell lines and primary samples with synergistic cytotoxicity in vitro. The combination of ruxolitinib, hydroxyurea, and talazoparib provided significantly greater cytoreduction than mono-/doublet therapy in both a murine MPN model and primary MPN xenograft model. | [160] |
| BCR-ABL inhibitors | Imatinib | Talazoparib | Downregulation of RAD51 and LIG4 to impair HR and NHEJ, respectively | Induction of DSBs and reduced clonogenic potential of imatinib-refractory CML cell lines and primary samples. Reduction in LSC-enriched quiescent cells in an inducible mouse model of chronic-phase CML. Extended disease latency in both primary and secondary recipient mice in a primary xenograft model. Synergistic 40-fold reduction in disease burden in a BCR-ABL1+ leukemia xenograft model. | [146,159] |
| FLT3-ITD inhibitors | Quizartinib | Olaparib Talazoparib Veliparib | Downregulation of BRCA1/2, PALB2, RAD51, and LIG4 impairs HR and NHEJ to induce HRD. Combination therapy caused accumulation of lethal DSBs. PARPi destabilize STAT5 to reduce aberrant FLT3-ITD signaling | Combination therapy exhibited synergistic activity against proliferating and quiescent leukemic stem/progenitor cells, eliminating both from primary AML samples. Combination therapy reduced leukemic burden in primary AML xenograft mice and prolonged survival in secondary recipients. PARPi and TKI combination therapy exhibited synergistic cytotoxicity in both TKI-sensitive and TKI-resistant AML cell lines. | [158,177] |
| WEE1 inhibitors | AZD1775 | Olaparib | Inhibition of WEE1 impairs HR by indirectly inhibiting BRCA2. Combination therapy resulted in elevated γH2AX, accumulation of DNA damage, and induction of apoptosis. | Mild synergy between WEE1 and PARP inhibition was seen in cell lines harboring FLT3-ITD, while FLT3 wild-type cells were relatively insensitive, independent of TP53 status. Significantly prolonged survival in a murine model of FLT3-ITD+ AML and reduced colony formation in primary AML samples. | [178] |
| TRAIL | rTRAIL | Olaparib Veliparib | PARPi upregulate TNFRSF6 and TNFRSF10B expression via potentiation of the Sp1 transcription factor and NF-kB, increasing sensitivity to TRAIL. | Both olaparib and veliparib enhanced the sensitivity of myeloid cell lines to TRAIL in vitro. Though olaparib had no consistent activity alone, it sensitized most primary AML isolates to TRAIL and reduced colony formation. | [161,179] |
| Antibody drug conjugates | Gemtuzumab ozogamicin | Olaparib | Calicheamicin induces both SSBs and DSBs, invoking PARP activation. | The IC50 value for GO was reduced from 24 to 13 ng/mL when combined with olaparib; the CI was 0.86, indicating synergistic cytotoxicity. | [144] |
| Trial | Year | Intervention(s) | Phase | Disease(s) a | N | CRR | ORR | OS b | Ref |
|---|---|---|---|---|---|---|---|---|---|
| NCT01399840 | 2014 | Talazoparib | I | AML/MDS CLL/MCL | 25 8 | 0% 0% | 0% 0% | N/A | [180] |
| NCT01139970 | 2017 | Veliparib + Temozolomide | I | AML | 48 | 17% | 33% | 5.3 | [181] |
| NCT00588991 | 2017 | Veliparib + Topotecan ± Carboplatin | I | AML, MPN, CMML | 99 | 14% | 33% | 15.3 c | [182] |
| ISRCTN34386131 | 2017 | Olaparib | I | CLL, MCL, T-PLL | 15 | 0% | 0% | 4.3 | [183] |
| Trial | Phase | Intervention(s) | Population(s) a | Status |
|---|---|---|---|---|
| NCT03289910 | II | Topotecan + Carboplatin ± Veliparib | AML, MDS, MPN, CMML | Active (not recruiting) |
| NCT02878785 | I/II | Talazoparib + Decitabine | AML (phase I) AML, untreated (phase II) | Active (not recruiting) |
| NCT03953898 | II | Olaparib | IDH1/2-mutant AML/MDS | Recruiting |
| NCT03974217 | I | Talazoparib | Cohesin-mutant AML/MDS | Recruiting |
| Authors | N | PARPi | Myeloid Neoplasm | SOT | Karyotype | NGS | SOT Status at Diagnosis | Median OS |
|---|---|---|---|---|---|---|---|---|
| Martin et al. [238] | 20 | Olaparib (94%) Rucaparib (6%) | AML (45%) MDS (55%) | Ovarian | 95% complex | DDR pathway mutations in 83% | 55% in CR | 4.3 months |
| Kwan et al. [239] | 22 | Rucaparib | AML 41% MDS 59% * | Ovarian | 53% complex; 80% with chrom. 5 or 7 alteration | NR | NR | NR |
| Morice et al. [19] | 178 | Olaparib (75%) Niraparib (18%) Rucaparib (6%) Talazoparib (1%) Veliparib (1%) | AML (44%) MDS (56%) | Ovarian (85%) Prostate (7%) Breast (5%) Pancreatic (2%) | NR | NR | Response (85%) Progression (15%) | NR (45% had died on follow-up) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Csizmar, C.M.; Saliba, A.N.; Swisher, E.M.; Kaufmann, S.H. PARP Inhibitors and Myeloid Neoplasms: A Double-Edged Sword. Cancers 2021, 13, 6385. https://doi.org/10.3390/cancers13246385
Csizmar CM, Saliba AN, Swisher EM, Kaufmann SH. PARP Inhibitors and Myeloid Neoplasms: A Double-Edged Sword. Cancers. 2021; 13(24):6385. https://doi.org/10.3390/cancers13246385
Chicago/Turabian StyleCsizmar, Clifford M., Antoine N. Saliba, Elizabeth M. Swisher, and Scott H. Kaufmann. 2021. "PARP Inhibitors and Myeloid Neoplasms: A Double-Edged Sword" Cancers 13, no. 24: 6385. https://doi.org/10.3390/cancers13246385
APA StyleCsizmar, C. M., Saliba, A. N., Swisher, E. M., & Kaufmann, S. H. (2021). PARP Inhibitors and Myeloid Neoplasms: A Double-Edged Sword. Cancers, 13(24), 6385. https://doi.org/10.3390/cancers13246385