
Therapeutic Implications of TGFβ in Cancer Treatment: A Systematic Review


Abstract
:Simple Summary
Abstract
1. Introduction
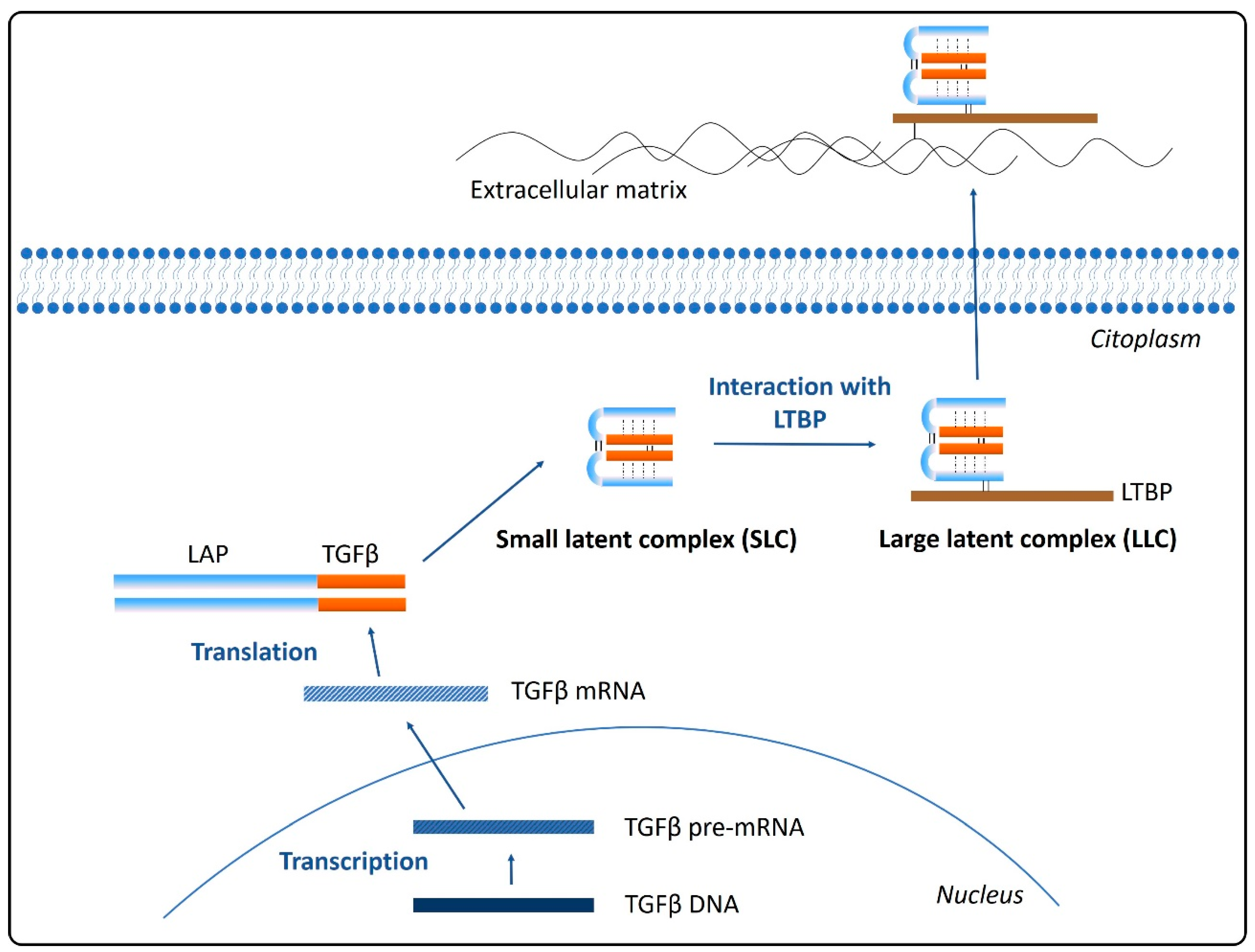
2. Synthesis and Secretion of TGFβ
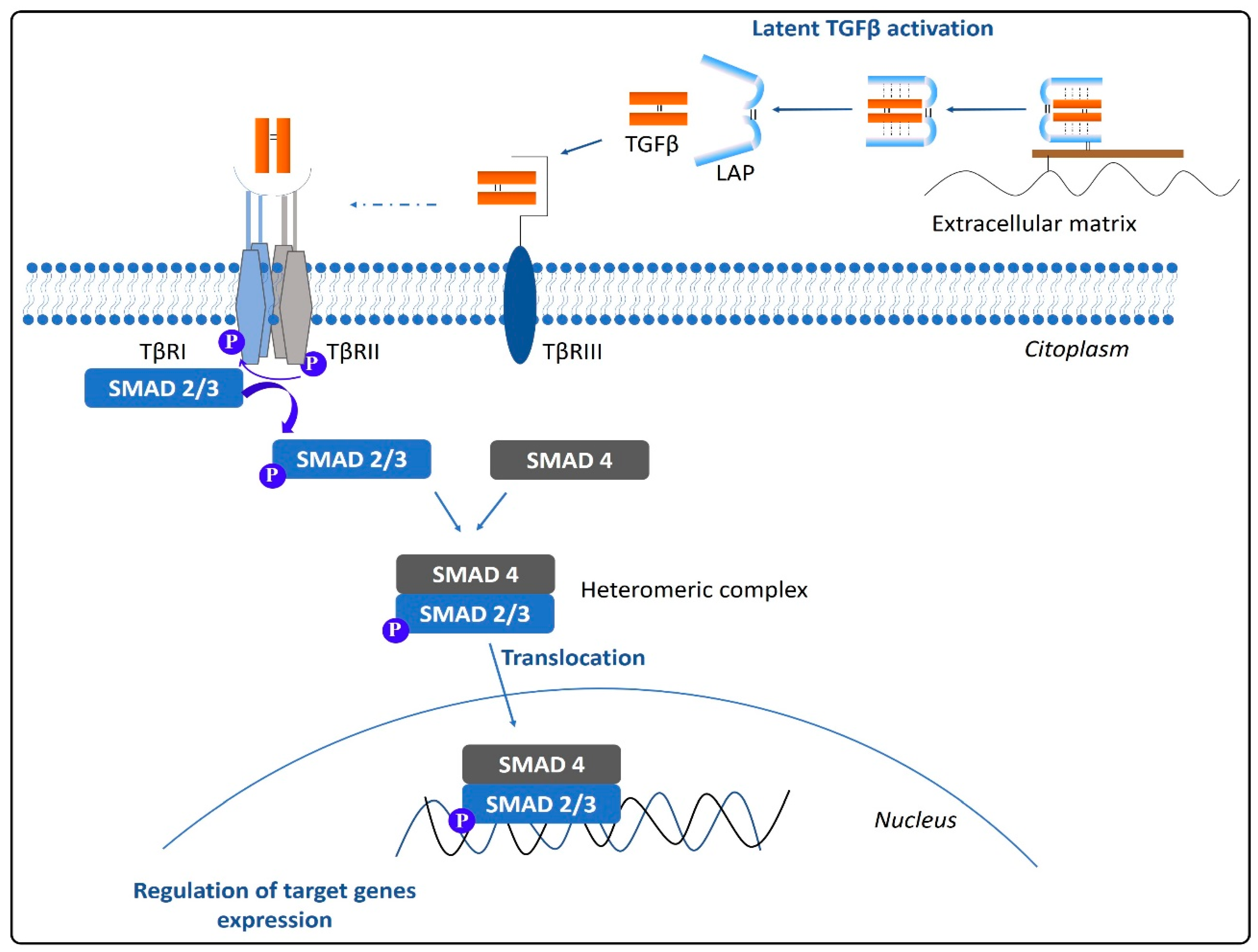
3. TGFβ Activation and Signaling
4. Results
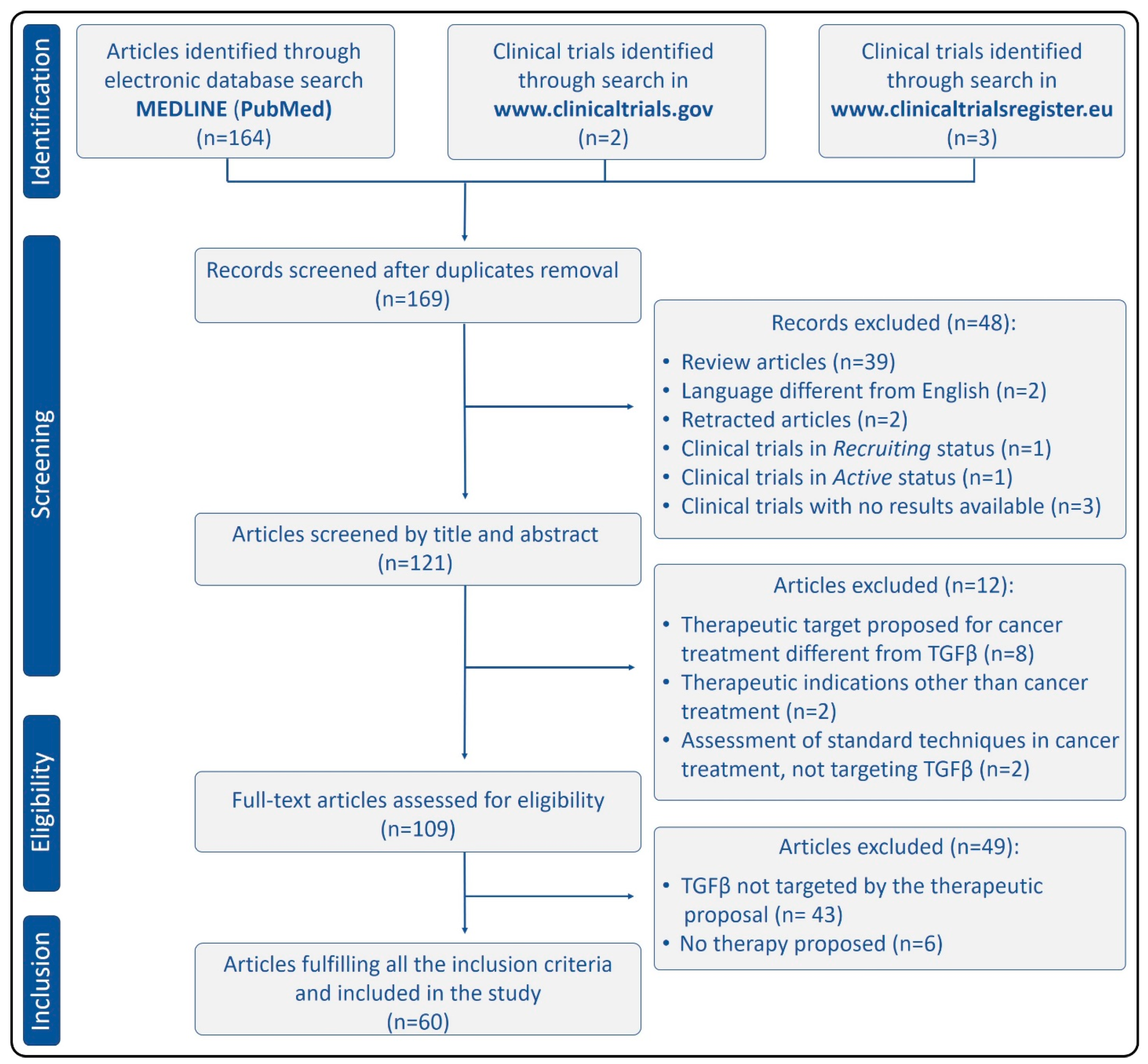
4.1. Search Results
4.2. Review of Eligible Studies
4.2.1. MicroRNAs
4.2.2. Small Interfering RNA
4.2.3. Long Non-Coding RNA Activated by TGFβ (lnc-ATB)
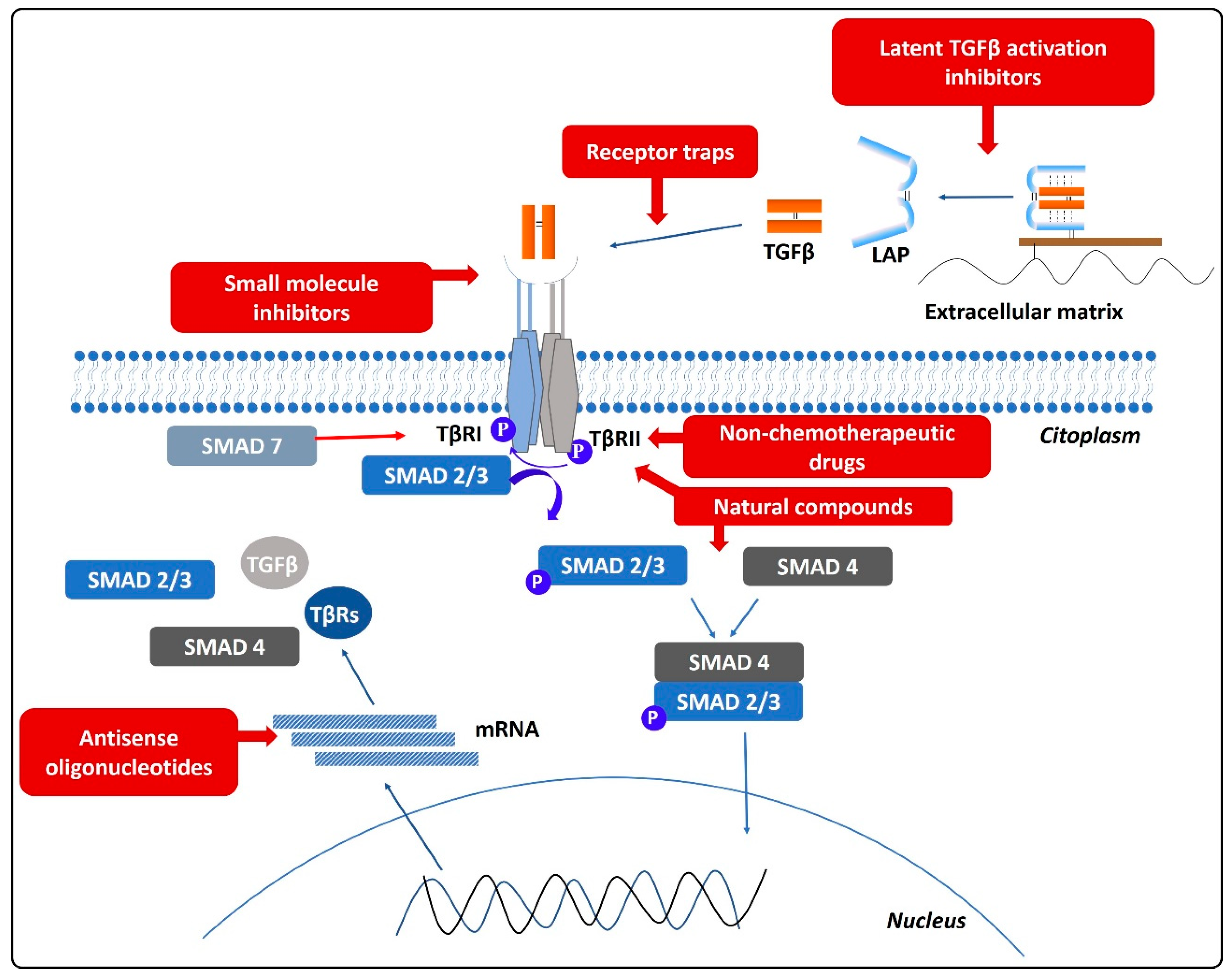
4.2.4. Small Molecule Inhibitors (SMI)
4.2.5. Receptor Traps
4.2.6. Strategies Targeting Latent TGFβ Activation
4.2.7. Natural Compounds
4.2.8. Drugs Approved for Other Diseases
4.2.9. Strategies Targeting Molecules with an Effect on TGFβ Regulation
5. Discussion
6. Methods
6.1. Study Design
6.2. Search Strategy
6.3. Exclusion/Inclusion Criteria
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, A.; Shuler, C.F.; Gulka, A.O.D.; Hanai, J.I. TGF-β signaling and the epithelial-mesenchymal transition during palatal fusion. Int. J. Mol. Sci. 2018, 19, 3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Živicová, V.; Lacina, L.; Mateu, R.; Smetana, K.; Kavková, R.; Krejcí, E.D.; Grim, M.; Kvasilová, A.; Borský, J.; Strnad, H.; et al. Analysis of dermal fibroblasts isolated from neonatal and child cleft lip and adult skin: Developmental implications on reconstructive surgery. Int. J. Mol. Med. 2017, 40, 1323–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFβ. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, K.; Suzuki, H.I. TGF-β signaling in cellular senescence and aging-related pathology. Int. J. Mol. Sci. 2019, 20, 5002. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.H.; Landström, M.; Moustakas, A. Mechanism of TGF-β signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Roberts, A.B. Tumor suppressor activity of the TGF-β pathway in human cancers. Cytokine Growth Factor Rev. 1996, 7, 93–102. [Google Scholar] [CrossRef]
- Levy, L.; Hill, C.S. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006, 17, 41–58. [Google Scholar] [CrossRef]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. Smad7 induces tumorigenicity by blocking TGF-β-induced growth inhibition and apoptosis. Exp. Cell Res. 2005, 307, 231–246. [Google Scholar] [CrossRef]
- Grady, W.M.; Willis, J.E.; Trobridge, P.; Romero-Gallo, J.; Munoz, N.; Olechnowicz, J.; Ferguson, K.; Gautam, S.; Markowitz, S.D. Proliferation and Cdk4 expression in microsatellite unstable colon cancers with TGFBR2 mutations. Int. J. Cancer 2006, 118, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Toonkel, R.L.; Borczuk, A.C.; Powell, C.A. TGF-β signaling pathway in lung adenocarcinoma invasion. J. Thorac. Oncol. 2010, 5, 153–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lücke, C.D.; Philpott, A.; Metcalfe, J.C.; Thompson, A.M.; Hughes-Davies, L.; Kemp, P.R.; Hesketh, R. Inhibiting mutations in the transforming growth factor β type 2 receptor in recurrent human breast cancer. Cancer Res. 2001, 61, 482–485. [Google Scholar] [PubMed]
- Parsons, R.; Myeroff, L.; Liu, B.; Willson, J.; Markowitz, S.; Kinzler, K.; Vogelstein, B. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res. 1994, 55, 5548–5550. [Google Scholar]
- Wang, D.; Kanuma, T.; Mizunuma, H.; Takama, F.; Ibuki, Y.; Wake, N.; Mogi, A.; Shitara, Y.; Takenoshita, S. Analysis of specific gene mutations in the transforming growth factor-β signal transduction pathway in human ovarian cancer. Cancer Res. 2000, 60, 4507–4512. [Google Scholar]
- Chen, T.; Carter, D.; Garrigue-Antar, L.; Reiss, M. Transforming growth factor β type I receptor kinase mutant associated with metastatic breast cancer. Cancer Res. 1998, 58, 4805–4810. [Google Scholar]
- Goggins, M.; Shekher, M.; Turnacioglu, K.; Yeo, C.J.; Hruban, R.H.; Kern, S.E. Genetic alterations of the transforming growth factor receptor β genes in pancreatic and biliary adenocarcinomas. Cancer Res. 1998, 58, 5329–5332. [Google Scholar]
- Goudie, D.R.; D’Alessandro, M.; Merriman, B.; Lee, H.; Szeverényi, I.; Avery, S.; O’Connor, B.D.; Nelson, S.F.; Coats, S.E.; Stewart, A.; et al. Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat. Genet. 2011, 43, 365–371. [Google Scholar] [CrossRef]
- Singh, K.K.; Rommel, K.; Mishra, A.; Karck, M.; Haverich, A.; Schmidtke, J.; Arslan-Kirchner, M. TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Hum. Mutat. 2006, 27, 770–777. [Google Scholar] [CrossRef]
- Wang, C.; Liu, P.; Wu, H.; Cui, P.; Li, Y.; Liu, Y. MicroRNA-323-3p inhibits cell invasion and metastasis in pancreatic ductal adenocarcinoma via direct suppression of SMAD2 and SMAD3. Oncotarget 2016, 7, 14912–14924. [Google Scholar] [CrossRef] [Green Version]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierie, B.; Moses, H.L. Gain or loss of TGFβ signaling in mammary carcinoma cells can promote metastasis. Cell Cycle 2009, 8, 3319–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierie, B.; Moses, H.L. Transforming growth factor beta (TGF-β) and inflammation in cancer. Cytokine Growth Factor Rev. 2011, 21, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Persepct. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [Green Version]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.D.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin αvβ6 binds and activates latent TGFβ1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Budi, E. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, 1–58. [Google Scholar] [CrossRef] [Green Version]
- Vilchis-Landeros, M.; Juárez, P.; López-Casillas, F. Receptores y funciones del TGF-beta, una citocina crucial en la cicatrización. Gac. Med. Mex. 2003, 139, 126–143. [Google Scholar]
- Robertson, I.B.; Rifkin, D.B. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–27. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Sankar, S.; Mahooti-Brooks, N.; Centrella, M.; McCarthy, T.L.; Madri, J.A. Expression of Transforming Growth Factor Type III Receptor in Vascular Endothelial Cells Increases Their Responsiveness to Transforming Growth Factor β2. J. Biol. Chem. 1995, 270, 13567–13572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Zhang, Y.; Feng, X.H. Smads: Transcriptional activators of TGF-β responses. Cell 1998, 95, 737–740. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Davis-Dusenbery, B.N.; Hata, A. Smad-mediated miRNA processing. RNA Biol. 2011, 8, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014, 106, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Kusanagi, K.; Inoue, H. Divergence and convergence of TGF-β/BMP signaling. J. Cell. Physiol. 2001, 187, 265–276. [Google Scholar] [CrossRef]
- Ning, J.; Zhao, Y.; Ye, Y.; Yu, J. Opposing roles and potential antagonistic mechanism between TGF-β and BMP pathways: Implications for cancer progression. EBioMedicine 2019, 41, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Haque, S.; Morris, J.C. Transforming growth factor-β: A therapeutic target for cancer. Hum. Vaccines Immunother. 2017, 13, 1741–1750. [Google Scholar] [CrossRef]
- Oft, M.; Peli, J.; Rudaz, C.; Schwarz, H.; Beug, H.; Reichmann, E. TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996, 10, 2462–2477. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R.; Brown, A.; Papkoff, J.; Scambler, P.; Shackleford, G.; McMahon, A.; Moon, R.; Varmus, H. A New Nomenclature for int-1 and Related Genes: The Wnt Gene Family. J. Can. Chiropr. Assoc. 1991, 35, 179. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. BMJ 2009, 339, 332–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giner, M.; Montoya, M.; Vázquez, M.; Miranda, C.; Miranda, M.; Perez-Cano, R. What are microRNAs? Potential biomarkers and therapeutic targets in osteoporosis. Rev. Osteoporos. Metab. Miner. 2016, 8, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.; Wang, X.; Lin, Q.; Zhang, J.; Ren, W.; Xu, G. Transforming growth factor-β/miR-143-3p/cystatin B axis is a therapeutic target in human ovarian cancer. Int. J. Oncol. 2019, 55, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Wahid, F.; Shehzad, A.; Khan, T.; Young, Y. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Biochim. Biophys. Acta 2010, 1803, 1231–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.; Hu, C.; Yang, H.; Cao, L.; An, J. Transforming growth factor-β-induced miR-143 expression in regulation of non-small cell lung cancer cell viability and invasion capacity in vitro and in vivo. Int. J. Oncol. 2014, 45, 1977–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, L.; Xue, Y.; Wang, J.; Fu, Y.; Wang, X.; Xiao, G.; Hao, M.; Sun, X.; Wang, Y.; Fu, G.; et al. The feedback loop between miR-124 and TGF-β pathway plays a significant role in non-small cell lung cancer metastasis. Carcinogenesis 2016, 37, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.; Zhang, H.; Sun, W. MicroRNA-155 promotes gastric cancer growth and invasion by negatively regulating transforming growth factor-β receptor 2. Cancer Sci. 2018, 109, 618–628. [Google Scholar] [CrossRef] [Green Version]
- Velázquez, K.T.; Enos, R.T.; Mcclellan, J.L.; Cranford, T.L.; Chatzistamou, I.; Singh, U.P.; Nagarkatti, M.; Nagarkatti, P.S.; Fan, D.; Murphy, E.A. MicroRNA-155 deletion promotes tumorigenesis in the azoxymethane-dextran sulfate sodium model of colon cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G347–G358. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Li, Y.; Jiang, F.; Wang, X.; Zhang, J.; Shen, J.; Yang, X. Inhibition of transforming growth factor beta ⁄ SMAD signal by MiR-155 is involved in arsenic trioxide-induced anti-angiogenesis in prostate cancer. Cancer Sci. 2014, 105, 1541–1549. [Google Scholar] [CrossRef]
- Hu, S.; Chen, H.; Zhang, Y.; Wang, C.; Liu, K.; Wang, H.; Luo, J. MicroRNA-520c inhibits glioma cell migration and invasion by the suppression of transforming growth factor-β receptor type 2. Oncol. Rep. 2017, 37, 1691–1697. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.; Zhang, H.; Duan, J.; Liu, R.; Deng, T.; Bai, M.; Huang, D.; Li, H.; Ning, T.; Zhang, L.; et al. MiR-17-5p regulates cell proliferation and migration by targeting transforming growth factor-β receptor 2 in gastric cancer. Oncotarget 2016, 7, 33286–33296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Xu, Y.; Zhao, J.; Liu, Q.; Feng, W.; Fan, J.; Wang, P. miR-367 promotes epithelial-to-mesenchymal transition and invasion of pancreatic ductal adenocarcinoma cells by targeting the Smad7-TGF-b signalling pathway. Br. J. Cancer 2015, 112, 1367–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Shin, H.; Lee, Y.S.; Lee, Y.C. miR-106b modulates cancer stem cell characteristics through TGF-b/Smad signaling in CD44-positive gastric cancer cells. Lab. Investig. 2014, 94, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Zhu, J.; Wu, G.; Cao, L.; Tan, Z.; Zhang, S.; Jiang, L.; Wu, J. Antagonizing miR-455-3p inhibits chemoresistance and aggressiveness in esophageal squamous cell carcinoma. Mol. Cancer 2017, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Zhang, H.; Bai, X.; Liu, X. Suppressive role of miR-592 in breast cancer by repressing TGF-β 2. Oncol. Lett. 2017, 38, 3447–3454. [Google Scholar] [CrossRef]
- Niu, G.; Li, B.; Sun, L.; An, C. MicroRNA-153 Inhibits Osteosarcoma Cells Proliferation and Invasion by Targeting. PLoS ONE 2015, 20, e0119225. [Google Scholar] [CrossRef]
- Wang, Q.; Li, X.; Zhu, Y.; Yang, P. MicroRNA-16 suppresses epithelial-mesenchymal transition—Related gene expression in human glioma. Mol. Med. Rep. 2014, 10, 3310–3314. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhong, Y.; Chen, J.; Lin, X.; Lin, Z.; Wang, N.; Lin, S. Radiation Enhances the Epithelial– Mesenchymal Transition of A549 Cells via miR3591-5p/USP33/PPM1A. Cell Physiol. Biochem. 2018, 50, 721–733. [Google Scholar] [CrossRef]
- Chae, D.-K.; Ban, E.; Yoo, Y.S.; Kim, E.E.; Baik, J.-H.; Song, E.J. miR-27a regulates the TGF-β signaling pathway by targeting SMAD2 and SMAD4 in lung cancer. Mol. Carcinog. 2017, 56, 1992–1998. [Google Scholar] [CrossRef]
- Han, Y.; Jia, C.; Cong, X.; Yu, F.; Cai, H.; Fang, S.; Cai, L.; Yang, H.; Sun, Y.; Li, D.; et al. Increased expression of TGFβR2 Is associated with the clinical outcome of non-small cell lung cancer patients treated with chemotherapy. PLoS ONE 2015, 10, e0134682. [Google Scholar] [CrossRef] [Green Version]
- Vigorito, E.; Kohlhaas, S.; Lu, D.; Leyland, R. miR-155: An ancient regulator of the immune system. Immunol. Rev. 2013, 253, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zeng, Z.; Fan, S.; Wang, J.; Yang, J.; Zhou, Y.; Li, X.; Huang, D.; Liang, F.; Wu, M.; et al. Evaluation of the prognostic value of TGF-β superfamily type I receptor and TGF-β type II receptor expression in nasopharyngeal carcinoma using high-throughput tissue microarrays. J. Mol. Histol. 2012, 43, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sun, P.; Wang, C.; Sun, T. Downregulation of microRNA-155 accelerates cell growth and invasion by targeting c-myc in human gastric carcinoma cells. Oncol. Rep. 2014, 32, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Huffaker, T.B.; Hu, R.; Runtsch, M.C.; Bake, E.; Chen, X.; Zhao, J.; Round, J.L.; Baltimore, D.; O’Connell, R.M. Epistasis between MicroRNAs 155 and 146a during T Cell-Mediated Antitumor Immunity. Cell Rep. 2012, 2, 1697–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yu, F.; Jia, X.; Iwanowycz, S.; Wang, Y.; Huang, S.; Ai, W.; Fan, D. MicroRNA-155 deficiency enhances the recruitment and functions of myeloid-derived suppressor cells in tumor microenvironment and promotes solid tumor growth. Int. J. Cancer 2015, 136, E602–E613. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, B.; Niu, L.; Ge, L. MiR-592 functions as a tumor suppressor in human non-small cell lung cancer by targeting SOX9. Oncol. Rep. 2017, 37, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.Y.; Zhao, J.Y.; Li, B.L.; Gao, K.; Song, Y.; Liu, M.Y.; Yang, X.J.; Xue, Y.; Wen, A.D.; Shi, L. miR-592/WSB1/HIF-1α axis inhibits glycolytic metabolism to decrease hepatocellular carcinoma growth. Oncotarget 2016, 7, 35257–35269. [Google Scholar] [CrossRef]
- Lv, Z.; Rao, P.; Li, W. MiR-592 represses FOXO3 expression and promotes the proliferation of prostate cancer cells. Int. J. Clin. Exp. Med. 2015, 8, 15246–15253. [Google Scholar]
- Fu, Q.; Du, Y.; Yang, C.; Zhang, D.; Zhang, N.; Liu, X.; Cho, W.C.; Yang, Y. An oncogenic role of miR-592 in tumorigenesis of human colorectal cancer by targeting Forkhead Box O3A (FoxO3A). Expert Opin. Ther. Targets 2016, 20, 771–782. [Google Scholar] [CrossRef]
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 15, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Nakamura-López, Y.; Esparza-Aguilar, M.; Garrido-Olvera, L.; Palomar-Olguín, V.M.; Gallardo-Pérez, J.C. Aplicaciones terapéuticas del ARN de interferencia. Bioquimia 2009, 34, 26–36. [Google Scholar]
- Li, J.; Liu, H.A.O.; Yu, J.; Yu, H. Chemoresistance to doxorubicin induces epithelial-mesenchymal transition via upregulation of transforming growth factor β signaling in HCT116 colon cancer cells. Mol. Med. Rep. 2015, 12, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.; Han, Z.; Oh, G.; Joo, Y.; Choi, H.J.; Song, J.J. Down-Regulation of TGF-β Expression Sensitizes the Resistance of Hepatocellular Carcinoma Cells to Sorafenib. Yonsei Med. J. 2017, 58, 899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhang, B.; Wu, H.; Cai, J.; Sui, X.; Wang, Y.; Li, H.; Qiu, Y.; Wang, T.; Chen, Z.; et al. CD51 correlates with the TGF-beta pathway and is a functional marker for colorectal cancer stem cells. Oncogene 2016, 36, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, S.R.; Marguerat, S.; Bähler, J. Exploring long non-coding RNAs through sequencing. Semin. Cell Dev. Biol. 2012, 23, 200–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, B.K.; Mueller, A.C.; Dutta, A. Long non-coding RNAs as emerging regulators of differentiation, development, and disease. Transcription 2014, 5, e944014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.J.; Leng, R.X.; Fan, Y.G.; Pan, H.F.; Ye, D.Q. Translation of noncoding RNAs: Focus on lncRNAs, pri-miRNAs, and circRNAs. Exp. Cell Res. 2017, 361, 1–8. [Google Scholar] [CrossRef]
- Sanchez Calle, A.; Kawamura, Y.; Yamamoto, Y.; Takeshita, F.; Ochiya, T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018, 109, 2093–2100. [Google Scholar] [CrossRef]
- López-Urrutia, E.; Bustamante Montes, L.P.; Ladrón de Guevara Cervantes, D.; Pérez-Plasencia, C.; Campos-Parra, A.D. Crosstalk Between Long Non-coding RNAs, Micro-RNAs and mRNAs: Deciphering Molecular Mechanisms of Master Regulators in Cancer. Front. Oncol. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Jia, S.; Wang, Y.; Kang, Y.; Zhang, W. Down-regulation of lncRNA-ATB inhibits epithelial–mesenchymal transition of breast cancer cells by increasing miR-141-3p expression. Biochem. Cell Biol. 2019, 97, 193–200. [Google Scholar] [CrossRef]
- Cao, W.; Peng, T.; Zhou, Y. Long noncoding RNA activated by transforming growth factor-β promotes cancer development and is a prognostic marker in cervical cancer. J. Cancer Res. Ther. 2017, 13, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Liu, Y.; Jiang, L.; Zeng, Y.; Tang, W. High expression of long non-coding RNA lncRNA-ATB is correlated with metastases and promotes cell migration and invasion in renal cell carcinoma. Jpn. J. Clin. Oncol. 2016, 46, 378–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, X.; Xu, W. Long Noncoding RNA ATB Promotes Proliferation, Migration, and Invasion in Bladder Cancer by Suppressing MicroRNA-126. Oncol. Res. 2018, 26, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Yasuchika, K.; Ishii, T.; Katayama, H.; Yoshitoshi, E.Y.; Ogiso, S.; Minami, T.; Miyauchi, Y.; Kojima, H.; Yamaoka, R.; et al. Identification of keratin 19-positive cancer stem cells associating human hepatocellular carcinoma using CYFRA 21-1. Cancer Med. 2017, 6, 2531–2540. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, R.; Cao, Y.; Hoffmeier, K.; Krezdorn, N.; Jost, L.; Meisel, A.R.; Jüngling, R.; Dituri, F.; Mancarella, S.; Rotter, B.; et al. Precision medicine for hepatocelluar carcinoma using molecular pattern diagnostics: Results from a preclinical pilot study. Cell Death Dis. 2017, 8, e2867. [Google Scholar] [CrossRef] [Green Version]
- Yi, E.; Park, S.; Jung, S.; Jang, W.; Kim, Y. Mitochondrial dysfunction induces EMT through the TGF-β/Smad/Snail signaling pathway in Hep3B hepatocellular carcinoma cells. Int. J. Oncol. 2015, 47, 1845–1853. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Sun, D.; Ren, F.; Pang, S.; Xu, S. CUTL1 induces epithelial-mesenchymal transition in non-small cell lung cancer. Oncol. Rep. 2017, 37, 3068–3074. [Google Scholar] [CrossRef]
- Park, S.A.; Kim, M.J.; Park, S.Y.; Kim, J.S.; Lee, S.J.; Woo, H.A.; Kim, D.K.; Nam, J.S.; Sheen, Y.Y. EW-7197 inhibits hepatic, renal, and pulmonary fibrosis by blocking TGF-β/Smad and ROS signaling. Cell. Mol. Life Sci. 2015, 72, 2023–2039. [Google Scholar] [CrossRef]
- Bouquet, F.; Pal, A.; Pilones, K.A.; Demaria, S.; Hann, B.; Akhurst, R.J.; Babb, J.S.; Lonning, S.M.; DeWyngaert, J.K.; Formenti, S.C.; et al. TGFβ1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin. Cancer Res. 2011, 17, 6754–6765. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Kleber, S.; Röhrich, M.; Timke, C.; Han, N.; Tuettenberg, J.; Martin-Villalba, A.; Debus, J.; Peschke, P.; Wirkner, U.; et al. Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011, 71, 7155–7167. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Al-odaini, A.A.; Wang, Y.; Korah, J.; Xiao, L.; Ali, S.; Lebrun, J.J. KiSS1 gene as a novel mediator of TGFβ-mediated cell invasion in triple negative breast cancer. Cell. Signal. 2018, 42, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Yi, J.Y.; Kim, M.; Song, K.; Kang, S. IM-412 inhibits the invasion of human breast carcinoma cells by blocking FGFR-mediated signaling. Oncol. Rep. 2015, 34, 2731–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellweg, R.; Mooneyham, A.; Chang, Z.; Shetty, M.; Emmings, E.; Iizuka, Y.; Clark, C.; Starr, T.; Abrahante, J.H.; Schütz, F.; et al. RNA Sequencing of Carboplatin- and Paclitaxel-Resistant Endometrial Cancer Cells Reveals New Stratification Markers and Molecular Targets for Cancer Treatment. Horm. Cancer 2018, 9, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.W.; Homanics, G.E.; Lazo, J.S. Targeted Deletion of the Metastasis-Associated Phosphatase Ptp4a3 (PRL-3) Suppresses Murine Colon Cancer. PLoS ONE 2013, 8, e58300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radke, I.; Götte, M.; Kersting, C.; Mattsson, B.; Kiesel, L.; Wülfing, P. Expression and prognostic impact of the protein tyrosine phosphatases PRL-1, PRL-2, and PRL-3 in breast cancer. Br. J. Cancer 2006, 95, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Ooki, A.; Yamashita, K.; Kikuchi, S.; Sakuramoto, S.; Katada, N.; Waraya, M.; Kawamata, H.; Nishimiya, H.; Nakamura, K.; Watanabe, M. Therapeutic potential of PRL-3 targeting and clinical significance of PRL-3 genomic amplification in gastric cancer. BMC Cancer 2011, 11, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.H.; Zhang, Z.Y. Regulatory Mechanisms and Novel Therapeutic Targeting Strategies for Protein Tyrosine Phosphatases. Chem. Rev. 2018, 118, 1069–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wu, X.; Tian, Y. Crosstalk of AP4 and TGF β receptor signaling in NSCLC. Tumor Biol. 2015, 36, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Chihara, Y.; Shimoda, M.; Hori, A.; Ohara, A. A small-molecule inhibitor of SMAD3 attenuates resistance to anti-HER2 drugs in HER2-positive breast cancer cells. Breast Cancer Res. Treat. 2017, 166, 55–68. [Google Scholar] [CrossRef]
- Abera, M.B.; Kazanietz, M.G. Protein Kinase C α Mediates Erlotinib Resistance in Lung Cancer Cells. Mol. Pharmacol. 2015, 87, 832–841. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.J.; Lee, J.W.; Chung, S.H.; Jang, S.Y.; Choi, J. Synthesis and anti-tumor activity of imidazopyrazines as TAK1 inhibitors. Eur. J. Med. Chem. 2019, 163, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Bo, L.; Cui, H.; Fang, Z.; Qun, T.; Xia, C. Inactivation of transforming growth factor-β-activted kinase 1 promotes taxol efficacy in ovarian cancer cells. Biomed. Pharmacother. 2016, 84, 917–924. [Google Scholar] [CrossRef]
- Qin, T.; Barron, L.; Xia, L.; Huang, H.; Villarreal, M.M.; Zwaagstra, J.; Collins, C.; Yang, J.; Zwieb, C.; Kodali, R.; et al. A novel highly potent trivalent TGF-β receptor trap inhibits early-stage tumorigenesis and tumor cell invasion in murine Pten-deficient prostate glands. Oncotarget 2016, 7, 86087–86102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zou, X.; Xia, W.; Gao, H.; Li, Z.; Liu, N.; Xu, Z.; Gao, C.; He, Z.; Niu, W.; et al. Integrin αvβ6 plays a bi-directional regulation role between colon cancer cells and cancer-associated fibroblasts. Biosci. Rep. 2018, 38, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, E.R.; Bibby, L.I.; Slack, R.J. Characterisation of a novel, high affinity and selective αvβ6 integrin RGD-mimetic radioligand. Biochem. Pharmacol. 2016, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, Y.; Liu, X.; Zhang, N.; Mao, G.; Zeng, Q.; Yin, M.; Song, D.; Deng, H. Resibufogenin suppresses transforming growth factor-β-activated kinase 1-mediated nuclear factor-κ B activity through protein kinase C-dependent inhibition of glycogen synthase kinase 3. Cancer Sci. 2018, 109, 3611–3622. [Google Scholar] [CrossRef]
- Tang, X.; Shi, L.; Xie, N.; Liu, Z.; Qian, M.; Meng, F.; Xu, Q.; Zhou, M.; Cao, X.; Zhu, W.; et al. SIRT7 antagonizes TGF-β signaling and inhibits breast cancer metastasis. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Da, C.; Liu, Y.; Zhan, Y.; Liu, K.A.I.; Wang, R. Nobiletin inhibits epithelial-mesenchymal transition of human non-small cell lung cancer cells by antagonizing the TGF-β1/Smad3 signaling pathway. Oncol. Rep. 2016, 35, 2767–2774. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Li, Y.; Mu, J.; Hu, C.; Zhou, M.; Wang, X.; Si, L.; Ning, S.; Li, Z. Glabridin Inhibits Cancer Stem Cell-Like Properties of Human Breast Cancer Cells: An Epigenetic Regulation of miR-148a/SMAD2 signaling. Mol. Carcinog. 2016, 55, 929–940. [Google Scholar] [CrossRef]
- Bokhari, A.; Syed, V. Inhibition of Transforming Growth Factor-β (TGF-β) Signaling by Scutellaria baicalensis and Fritillaria cirrhosa Extracts in Endometrial Cancer. J. Cell Biochem. 2015, 116, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Shi, R.; Chang, L.; Tang, K.; Chen, K.; Yu, G.; Tian, Y.; Guo, Y.; He, W.; Song, X.; et al. Huachansu suppresses human bladder cancer cell growth through the Fas/Fasl and TNF-α/TNFR1 pathway in vitro and in vivo. J. Exp. Clin. Cancer Res. 2015, 34, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.H.; Zhu, X.Y.; Shi, W.D.; Liu, L.M. Huachansu injection inhibits metastasis of pancreatic cancer in mice model of human tumor xenograft. BMC Complement. Altern. Med. 2014, 14, 483. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Garrett, C.R.; Shen, Y.; Liu, L.; Yang, P.; Huo, Y.; Zhao, Q.; Spelman, A.R.; Ng, C.S.; Chang, D.Z.; et al. Prospective randomised evaluation of traditional Chinese medicine combined with chemotherapy: A randomised phase II study of wild toad extract plus gemcitabine in patients with advanced pancreatic adenocarcinomas. Br. J. Cancer 2012, 107, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Daman, Z.; Faghihi, H.; Montazeri, H. Salinomycin Nanoparticles Interfere with Tumor Cell Growth and the Tumor Microenvironment in an Orthotopic Model of Pancreatic Cancer. Drug Dev. Ind. Pharm. 2018, 44, 1434–1442. [Google Scholar] [CrossRef]
- Lee, J.; Wang, T.; Liu, C.; Chien, M.; Chen, M.; Hsu, Y.; Leung, C.; Cheng, S. Dipeptidyl Peptidase IV as a Prognostic Marker and Therapeutic Target in Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2017, 102, 2930–2940. [Google Scholar] [CrossRef]
- Shang, L.; Jia, S.; Jiang, H.; Wang, H. Simvastatin downregulates expression of TGF-β RII and inhibits proliferation of A549 cells via ERK. Tumor Biol. 2015, 36, 4819–4824. [Google Scholar] [CrossRef]
- Tian, X.; Guan, W.; Zhang, L.; Sun, W.; Zhou, D.; Lin, Q.; Ren, W.; Nadeem, L.; Xu, G. Physical interaction of STAT1 isoforms with TGF-β receptors leads to functional crosstalk between two signaling pathways in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 2018, 37, 103. [Google Scholar] [CrossRef]
- Li, S.; Zhang, W.; Wu, C.; Gao, H.; Yu, J.; Wang, X. HOXC10 promotes proliferation and invasion and induces immunosuppressive gene expression in glioma. FEBS J. 2018, 285, 2278–2291. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Wu, D.; Tang, R.; Li, X.; Chen, R.; Xue, S.; Zhang, C.; Sun, X. Silencing of HMGA2 promotes apoptosis and inhibits migration and invasion of prostate cancer cells. J. Biosci. 2016, 41, 229–236. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, W. Down-regulation of tripartite motif protein 59 inhibits proliferation, migration and invasion in breast cancer cells. Biomed. Pharmacother. 2017, 89, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Chen, L.; Luo, N.; Yang, W. Inhibition of TMEM45A suppresses proliferation, induces cell cycle arrest and reduces cell invasion in human ovarian cancer cells. Oncol. Lett. 2015, 33, 3124–3130. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Kim, W.; Lee, J.; Park, J.; Cho, J.K.; Pang, K.; Lee, J.; Kim, D.; Park, S.; Yang, K.; et al. Cytoplasmic DRAK1 overexpressed in head and neck cancers inhibits TGF-β 1 tumor suppressor activity by binding to Smad3 to interrupt its complex formation with Smad4. Oncogene 2014, 34, 5037–5045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, W.-L.; Xu, J.-F.; Hu, J. Regulation of Oral Squamous Cell Carcinoma Proliferation Through Crosstalk Between SMAD7 and CYLD. Cell. Physiol. Biochem. 2016, 38, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Sun, Y. Bone Morphogenetic Protein-7 Inhibits EMT-Associated Genes in Breast Cancer. Cell. Physiol. Biochem. 2015, 37, 1271–1278. [Google Scholar] [CrossRef] [Green Version]
- Tricas, J.M.L. Las posibilidades terapéuticas del ARN de interferencia. Therapeutic possibilities of interference RNA. Farm. Hosp. 2012, 36, 115–117. [Google Scholar] [CrossRef]
- Brioschi, M.; Banfi, C. The application of gene silencing in proteomics: From laboratory to clinic. Expert Rev. Proteom. 2018, 15, 717–732. [Google Scholar] [CrossRef]
- Beg, M.S.; Borad, M.; Sachdev, J.; Hong, D.S.; Smith, S.; Bader, A.; Stoudemire, J.; Kim, S.; Brenner, A. Abstract CT327: Multicenter phase I study of MRX34, a first-in-class microRNA miR-34 mimic liposomal injection. Cancer Res. 2014, 74 (Suppl. 19), CT327. [Google Scholar] [CrossRef]
- Kubowicz, P.; Zelaszczyk, D.; Pekala, E. RNAi in Clinical Studies. Curr. Med. Chem. 2013, 20, 1801–1816. [Google Scholar] [CrossRef]
- BusinessWire. Mirna Therapeutics Halts Phase 1 Clinical Study of MRX34. Available online: https://www.businesswire.com/news/home/20160920006814/en/Mirna-Therapeutics-Halts-Phase-1-Clinical-Study (accessed on 2 December 2020).
- Giaccone, G.; Bazhenova, L.A.; Nemunaitis, J.; Tan, M.; Juhász, E.; Ramlau, R.; Van Den Heuvel, M.M.; Lal, R.; Kloecker, G.H.; Eaton, K.D.; et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur. J. Cancer 2015, 51, 2321–2329. [Google Scholar] [CrossRef] [Green Version]
- Oettle, H.; Seufferlein, T.; Luger, T.; Schmid, R.M.; von Wichert, G.; Endlicher, E.; Garbe, C.; Kaehler, K.K.; Enk, A.; Schneider, A.; et al. Final results of a phase I/II study in patients with pancreatic cancer, malignant melanoma, and colorectal carcinoma with trabedersen. J. Clin. Oncol. 2012, 30, 4034. [Google Scholar] [CrossRef]
- Uckun, F.M.; Qazi, S.; Hwang, L.; Trieu, V.N. Recurrent or refractory high-grade gliomas treated by convection-enhanced delivery of a TGFΒ2-targeting RNA therapeutic: A post-hoc analysis with long-term follow-up. Cancers 2019, 11, 1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faivre, S.; Santoro, A.; Kelley, R.K.; Gane, E.; Costentin, C.E.; Gueorguieva, I.; Smith, C.; Cleverly, A.; Lahn, M.M.; Raymond, E.; et al. Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int. 2019, 39, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (Ly2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef] [Green Version]
- Fiercebiotech. Eli Lilly Cuts 3 Cancer Drugs Amid Q4 Clear-Out. Available online: https://www.fiercebiotech.com/biotech/eli-lilly-cuts-three-cancer-drugs-amid-q4-clear-out (accessed on 15 December 2020).
- Llopiz, D.; Dotor, J.; Casares, N.; Bezunartea, J.; Díaz-Valdés, N.; Ruiz, M.; Aranda, F.; Berraondo, P.; Prieto, J.; Lasarte, J.J.; et al. Peptide inhibitors of transforming growth factor-β enhance the efficacy of antitumor immunotherapy. Int. J. Cancer 2009, 125, 2614–2623. [Google Scholar] [CrossRef]
- Gallo-Oller, G.; Vollmann-Zwerenz, A.; Meléndez, B.; Rey, J.A.; Hau, P.; Dotor, J.; Castresana, J.S. P144, a Transforming Growth Factor beta inhibitor peptide, generates antitumoral effects and modifies SMAD7 and SKI levels in human glioblastoma cell lines. Cancer Lett. 2016, 381, 67–75. [Google Scholar] [CrossRef]
- Katz, L.H.; Li, Y.; Chen, J.-S.; Muñoz, N.M.; Majumdar, A.; Chen, J.; Mishra, L. Targeting TGF-β signaling in cancer. Expert Opin. Ther. Targets 2013, 17, 743–760. [Google Scholar] [CrossRef] [Green Version]
- Aarsen, L.A.K.V.; Leone, D.R.; Ho, S.; Dolinski, B.M.; Mccoon, P.E.; Lepage, D.J.; Kelly, R.; Heaney, G.; Rayhorn, P.; Reid, C.; et al. Antibody-Mediated Blockade of Integrin αvβ6 Inhibits Tumor Progression In vivo by a Transforming Growth Factor-β–Regulated Mechanism. Cancer Res. 2008, 68, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.D.; Chen, B.C.; Kao, S.T.; Liu, C.J.; Yeh, C.C. Genistein inhibits tumor invasion by suppressing multiple signal transduction pathways in human hepatocellular carcinoma cells. BMC Complement. Altern. Med. 2014, 14, 26. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.W.; Xin, L.Y.; Xu, X.; Ji, X.; Fan, L.H. Epithelial-to-mesenchymal transition markers to predict response of Berberine in suppressing lung cancer invasion and metastasis. J. Transl. Med. 2014, 12, 22. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Ye, W.; Wu, J.; Meng, X.; Liu, R.Y.; Ying, X.; Zhou, Y.; Wang, H.; Pan, C.; Huang, W. Overexpression of Sirt7 exhibits oncogenic property and serves as a prognostic factor in colorectal cancer. Clin. Cancer Res. 2014, 20, 3434–3445. [Google Scholar] [CrossRef] [Green Version]
- Malik, S.; Villanova, L.; Tanaka, S.; Aonuma, M.; Roy, N.; Berber, E.; Pollack, J.R.; Michishita-Kioi, E.; Chua, K.F. SIRT7 inactivation reverses metastatic phenotypes in epithelial and mesenchymal tumors. Sci. Rep. 2015, 5, 9841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aljada, A.; Saleh, A.M.; Alkathiri, M.; Shamsa, H.B.; Al-Bawab, A.; Nasr, A. Altered sirtuin 7 expression is associated with early stage breast cancer. Breast Cancer Basic Clin. Res. 2015, 9, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.V.; Ding, Z.; Ho, P.P.; Luo, J.; Agrawal, A.N.; Srinagesh, H.; Axtell, R.; Zhang, H.; Platten, M.; Wyss-coray, T.; et al. Angiotensin II sustains brain inflammation in mice via TGF-β. J. Clin. Investig. 2010, 120, 2782–2794. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Latham, C.W.; Zander, D.S.; Margolin, S.B.; Visner, G.A. Pirfenidone Inhibits Obliterative Airway Disease in Mouse Tracheal Allografts. J. Hear. Lung Transplant. 2005, 24, 1577–1585. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Biswas, S.; Freeman, M.L.; Arteaga, C.L.; Biswas, S.; Guix, M.; Rinehart, C.; Dugger, T.C.; Chytil, A.; Moses, H.L.; Freeman, M.L.; et al. Inhibition of TGF-β with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J. Clin. Investig. 2007, 117, 1305–1313. [Google Scholar] [CrossRef] [Green Version]
- Kodet, O.; Dvořánková, B.; Bendlová, B.; Sýkorová, V.; Krajsová, I.; Štork, J.; Kučera, J.; Szabo, P.; Strnad, H.; Kolář, M.; et al. Microenvironment-driven resistance to B-Raf inhibition in a melanoma patient is accompanied by broad changes of gene methylation and expression in distal fibroblasts. Int. J. Mol. Med. 2018, 41, 2687–2703. [Google Scholar] [CrossRef] [Green Version]
- Fedorenko, I.V.; Smalley, K.S.M. The complexity of microenvironment-mediated drug resistance. Genes Cancer 2015, 6, 367–368. [Google Scholar] [CrossRef]
- Li, H.; Fan, X.; Houghton, J.M. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtor, N.; Betge, J.; Ebert, M.P. CRISPR / Cas9 for cancer research and therapy. Semin. Cancer Biol. 2019, 55, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2014, 13, 788–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, A.J.; Chang, Z.L.; Lorenzini, M.H.; Zah, E.; Chen, Y.Y. TGFβ– responsive CAR-T cells promote anti-tumor immune function. Bioeng. Transl. Med. 2018, 3, 75–86. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Target | miRNA Level in Pathological State | Cancer Type | Reference |
|---|---|---|---|---|
| miR-143-3p | Cystatin | Decreased | Ovarian cancer | Guan et al. [43] |
| miR-143 | SMAD3 | Decreased | NSCLC | Cheng et al. [45] |
| miR-124 | SMAD4 | Decreased | NSCLC | Zu et al. [46] |
| miR-155 | TβRII | Increased | Gastric cancer | Qu et al. [47] |
| miR-155 | SMAD2/3 | Increased | Colon cancer | Velázquez et al. [48] |
| miR-155 | SMAD2/3 | Decreased | Prostate cancer | Ji et al. [49] |
| miR-520c | TβRII | Decreased | Glioma | Hu et al. [50] |
| miR-17-5p | TβRII | Increased | Gastric cancer | Qu et al. [51] |
| miR-323-3p | SMAD2/3 | Decreased | PDAC | Wang et al. [20] |
| miR-367 | SMAD7 | Increased | PDAC | Zhu et al. [52] |
| miR-106b | SMAD7 | Increased | Gastric cancer | Yu et al. [53] |
| miR-455-3p | p-SMAD2 | Increased | ESCC | Liu et al. [54] |
| miR-592 | TGFβ2 | Decreased | Breast cancer | Hou et al. [55] |
| miR-153 | TGFβ2, p-SMAD2/3 | Decreased | Osteosarcoma | Niu et al. [56] |
| miR-16 | TGFβ | Decreased | Glioma | Wang et al. [57] |
| miR-3591-5p | TGFβ, SMAD2/3 | Increased | Lung cancer | Lu et al. [58] |
| miR-27a | SMAD2/4 | Increased | Lung cancer | Chae et al. [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Gil, V. Therapeutic Implications of TGFβ in Cancer Treatment: A Systematic Review. Cancers 2021, 13, 379. https://doi.org/10.3390/cancers13030379
Gómez-Gil V. Therapeutic Implications of TGFβ in Cancer Treatment: A Systematic Review. Cancers. 2021; 13(3):379. https://doi.org/10.3390/cancers13030379
Chicago/Turabian StyleGómez-Gil, Verónica. 2021. "Therapeutic Implications of TGFβ in Cancer Treatment: A Systematic Review" Cancers 13, no. 3: 379. https://doi.org/10.3390/cancers13030379
APA StyleGómez-Gil, V. (2021). Therapeutic Implications of TGFβ in Cancer Treatment: A Systematic Review. Cancers, 13(3), 379. https://doi.org/10.3390/cancers13030379