
CD137+ T-Cells: Protagonists of the Immunotherapy Revolution


Abstract
:Simple Summary
Abstract
1. Introduction
2. CD137: The Receptor
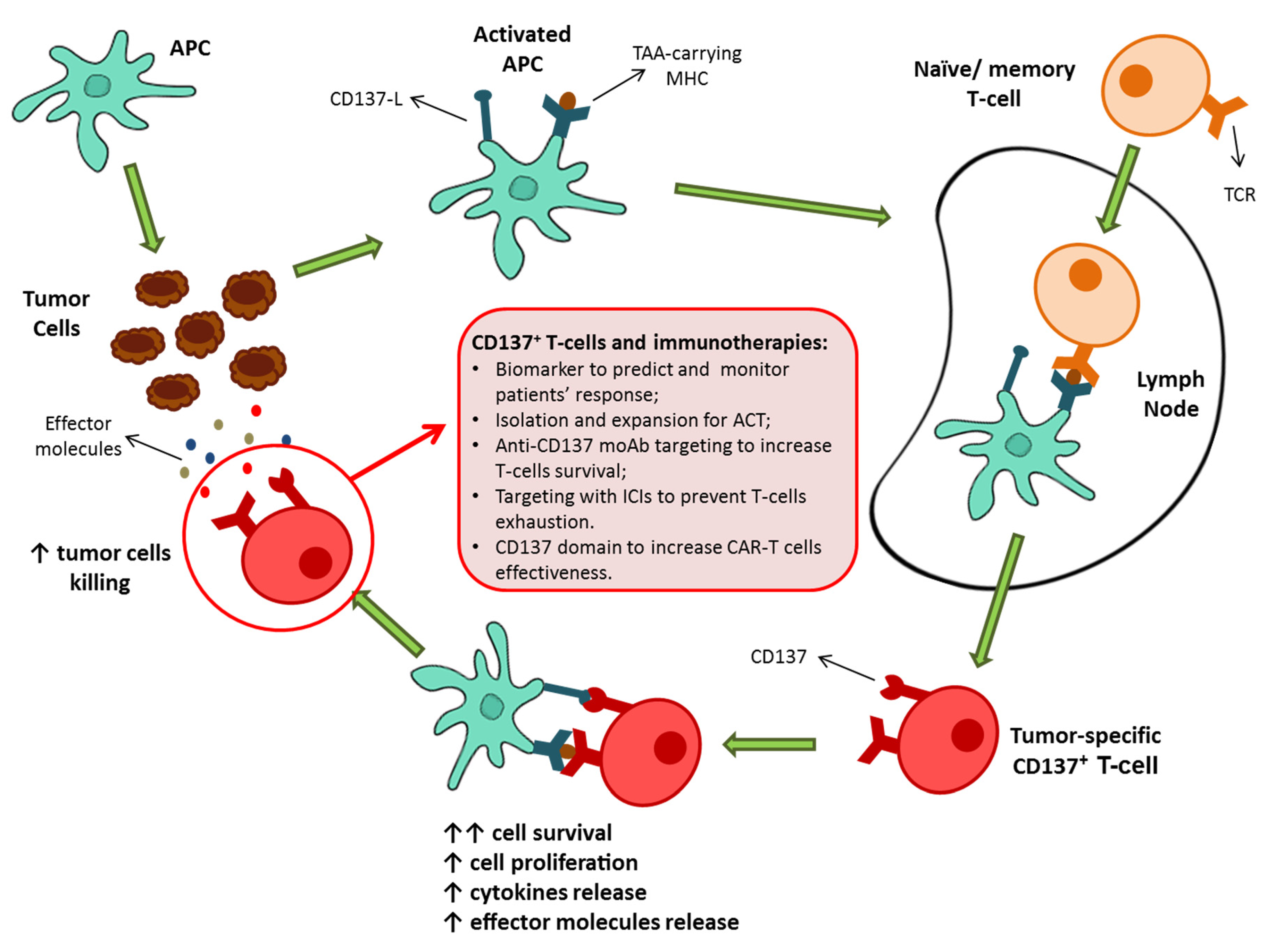
3. CD137+ T-Cells: The Natural Tumor-Specific Population
4. CD137+ T-Cell-Based Therapies as Possible Protagonists for Future Immunotherapies
5. CD137 in Current Clinical Practice: CD137-Targeting Antibodies and CD137 CAR-T Cells
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schalper, K.A.; Brown, J.; Carvajal-Hausdorf, D.; McLaughlin, J.; Velcheti, V.; Syrigos, K.N.; Herbst, R.S.; Rimm, D.L. Objective Measurement and Clinical Significance of TILs in Non–Small Cell Lung Cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boxberg, M.; Leising, L.; Steiger, K.; Jesinghaus, M.; Alkhamas, A.; Mielke, M.; Pfarr, N.; Götz, C.; Wolff, K.D.; Weichert, W.; et al. Composition and Clinical Impact of the Immunologic Tumor Microenvironment in Oral Squamous Cell Carcinoma. J. Immunol. 2019, 202, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagès, F.; Galon, J.; Dieu-Nosjean, M.-C.; Tartour, E.; Sautès-Fridman, C.; Fridman, W.-H. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2009, 29, 1093–1102. [Google Scholar] [CrossRef] [Green Version]
- Erdag, G.; Schaefer, J.T.; Smolkin, M.E.; Deacon, D.H.; Shea, S.M.; Dengel, L.T.; Patterson, J.W.; Slingluff, C.L. Immunotype and Immunohistologic Characteristics of Tumor-Infiltrating Immune Cells Are Associated with Clinical Outcome in Metastatic Melanoma. Cancer Res. 2012, 72, 1070–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savas, P.P.; Salgado, R.; Denkert, C.; Sotiriou, C.; Darcy, P.K.P.; Smyth, M.J.M.; Loi, S. Clinical relevance of host immunity in breast cancer: From TILs to the clinic. Nat. Rev. Clin. Oncol. 2016, 13, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Santoiemma, P.P.; Powell, D.J. Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol. Ther. 2015, 16, 807–820. [Google Scholar] [CrossRef]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.M.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.; Hitre, E.; et al. Prognostic and Predictive Value of Tumor-Infiltrating Lymphocytes in a Phase III Randomized Adjuvant Breast Cancer Trial in Node-Positive Breast Cancer Comparing the Addition of Docetaxel to Doxorubicin with Doxorubicin-Based Chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef]
- Clemente, C.G.; Mihm, M.C.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Yu, P.; Fu, Y.-X. Tumor-infiltrating T lymphocytes: Friends or foes? Lab. Investig. 2006, 86, 231–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linette, G.P.; Carreno, B.M. Tumor-Infiltrating Lymphocytes in the Checkpoint Inhibitor Era. Curr. Hematol. Malign. Rep. 2019, 14, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Kwon, B.S. Role of 4-1BB in immune responses. Semin. Immunol. 1998, 10, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Cannons, J.L.; Lau, P.; Ghumman, B.; Debenedette, M.A.; Yagita, H.; Okumura, K.; Watts, T.H. 4-1BB Ligand Induces Cell Division, Sustains Survival, and Enhances Effector Function of CD4 and CD8 T Cells with Similar Efficacy. J. Immunol. 2001, 167, 1313–1324. [Google Scholar] [CrossRef] [Green Version]
- Kwon, B.S.; Weissman, S.M. cDNA sequences of two inducible T-cell genes. Proc. Natl. Acad. Sci. USA 1989, 86, 1963–1967. [Google Scholar] [CrossRef] [Green Version]
- Gramaglia, I.; Cooper, D.; Miner, K.T.; Kwon, B.S.; Croft, M. Co-stimulation of antigen-specific CD4 T cells by 4-1BB ligand. Eur. J. Immunol. 2000, 30, 392–402. [Google Scholar] [CrossRef]
- Takahashi, C.; Mittler, R.S.; Vella, A.T. Cutting edge: 4-1BB is a bona fide CD8 T cell survival signal. J. Immunol. 1999, 162, 5037–5040. [Google Scholar]
- Dawicki, W.; Watts, T.H. Expression and function of 4-1BB during CD4 versus CD8 T cell responses In Vivo. Eur. J. Immunol. 2004, 34, 743–751. [Google Scholar] [CrossRef]
- Futagawa, T.; Akiba, H.; Kodama, T.; Takeda, K.; Hosoda, Y.; Yagita, H.; Okumura, K. Expression and function of 4-1BB and 4-1BB ligand on murine dendritic cells. Int. Immunol. 2002, 14, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Taraban, V.Y.; Rowley, T.F.; O’Brien, L.; Chan, H.T.C.; Haswell, L.E.; Green, M.H.A.; Tutt, A.L.; Glennie, M.J.; Al-Shamkhani, A. Expression and costimulatory effects of the TNF receptor superfamily members CD134 (OX40) and CD137 (4-1BB), and their role in the generation of anti-tumor immune responses. Eur. J. Immunol. 2002, 32, 3617–3627. [Google Scholar] [CrossRef]
- Wen, T.; Bukczynski, J.; Watts, T.H. 4-1BB Ligand-Mediated Costimulation of Human T Cells Induces CD4 and CD8 T Cell Expansion, Cytokine Production, and the Development of Cytolytic Effector Function. J. Immunol. 2002, 168, 4897–4906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, T.H. TNF/TNFR family members in costimulation of t cell responses. Annu. Rev. Immunol. 2005, 23, 23–68. [Google Scholar] [CrossRef] [PubMed]
- Diehl, L.; Van Mierlo, G.J.D.; Boer, A.T.D.; Van Der Voort, E.; Fransen, M.; Van Bostelen, L.; Krimpenfort, P.; Melief, C.J.M.; Mittler, R.; Toes, R.E.M.; et al. In Vivo Triggering Through 4-1BB Enables Th-Independent Priming of CTL in the Presence of an Intact CD28 Costimulatory Pathway. J. Immunol. 2002, 168, 3755–3762. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, R.G.; Din, W.S.; Davis-Smith, T.; Anderson, D.M.; Gimpel, S.D.; Sato, T.A.; Maliszewski, C.R.; Brannan, C.I.; Copeland, N.G.; Jenkins, N.A.; et al. Molecular cloning of a ligand for the inducible T cell gene 4-1BB: A member of an emerging family of cytokines with homology to tumor necrosis factor. Eur. J. Immunol. 1993, 23, 2631–2641. [Google Scholar] [CrossRef] [PubMed]
- Summers, K.L.; Hock, B.D.; McKenzie, J.L.; Hart, D.N.J. Phenotypic Characterization of Five Dendritic Cell Subsets in Human Tonsils. Am. J. Pathol. 2001, 159, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Zapata, J.M.; Perez-Chacon, G.; Carr-Baena, P.; Martinez-Forero, I.; Azpilikueta, A.; Otano, I.; Melero, I. CD137 (4-1BB) Signalosome: Complexity Is a Matter of TRAFs. Front. Immunol. 2018, 9, 2618. [Google Scholar] [CrossRef]
- Arch, R.H.; Thompson, C.B. 4-1BB and Ox40 Are Members of a Tumor Necrosis Factor (TNF)-Nerve Growth Factor Receptor Subfamily That Bind TNF Receptor-Associated Factors and Activate Nuclear Factor κB. Mol. Cell. Biol. 1998, 18, 558–565. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.-O.; Kang, H.; Shin, S.-M.; Cho, K.-H.; Kwon, B.; Kwon, B.S.; Kim, S.-J.; Lee, H.-W. Cross-Linking of 4-1BB Activates TCR-Signaling Pathways in CD8+ T Lymphocytes. J. Immunol. 2005, 174, 1898–1905. [Google Scholar] [CrossRef] [Green Version]
- Cannons, J.L.; Hoeflich, K.P.; Woodgett, J.R.; Watts, T.H. Role of the stress kinase pathway in signaling via the T cell costimulatory receptor 4-1BB. J. Immunol. 1999, 163, 2990–2998. [Google Scholar]
- Saoulli, K.; Lee, S.Y.; Cannons, J.L.; Yeh, W.C.; Santana, A.; Goldstein, M.D.; Bangia, N.; Debenedette, M.A.; Mak, T.W.; Choi, Y.; et al. CD28-independent, TRAF2-dependent Costimulation of Resting T Cells by 4-1BB Ligand. J. Exp. Med. 1998, 187, 1849–1862. [Google Scholar] [CrossRef]
- Halstead, E.S.; Mueller, Y.; Altman, J.D.; Katsikis, P.D. In Vivo stimulation of CD137 broadens primary antiviral CD8+ T cell responses. Nat. Immunol. 2002, 3, 536–541. [Google Scholar] [CrossRef]
- Bertram, E.M.; Lau, P.; Watts, T.H. Temporal Segregation of 4-1BB Versus CD28-Mediated Costimulation: 4-1BB Ligand Influences T Cell Numbers Late in the Primary Response and Regulates the Size of the T Cell Memory Response Following Influenza Infection. J. Immunol. 2002, 168, 3777–3785. [Google Scholar] [CrossRef] [Green Version]
- Maus, M.V.; Thomas, A.K.; Leonard, D.G.; Allman, D.; Addya, K.; Schlienger, K.; Riley, J.L.; June, C.H. Ex Vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat. Biotechnol. 2002, 20, 143–148. [Google Scholar] [CrossRef]
- Bukczynski, J.; Wen, T.; Watts, T.H. Costimulation of human CD28 T cells by 4-1BB ligand. Eur. J. Immunol. 2003, 33, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Bukczynski, J.; Wen, T.; Ellefsen, K.; Gauldie, J.; Watts, T.H. Costimulatory ligand 4-1BBL (CD137L) as an efficient adjuvant for human antiviral cytotoxic T cell responses. Proc. Natl. Acad. Sci. USA 2004, 101, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Hurtado, J.C.; Kim, Y.J.; Kwon, B.S. Signals through 4-1BB are costimulatory to previously activated splenic T cells and inhibit activation-induced cell death. J. Immunol. 1997, 158, 2600–2609. [Google Scholar]
- May, K.F.; Chen, L.; Zheng, P.; Liu, Y. Anti-4-1BB monoclonal antibody enhances rejection of large tumor burden by promoting survival but not clonal expansion of tumor-specific CD8+ T cells. Cancer Res. 2002, 62, 3459–3465. [Google Scholar]
- Lee, H.-W.; Park, S.-J.; Choi, B.K.; Kim, H.H.; Nam, K.-O.; Kwon, B.S. 4-1BB Promotes the Survival of CD8+T Lymphocytes by Increasing Expression of Bcl-xLand Bfl-1. J. Immunol. 2002, 169, 4882–4888. [Google Scholar] [CrossRef] [Green Version]
- Menk, A.V.; Scharping, N.E.; Rivadeneira, D.B.; Calderon, M.J.; Watson, M.J.; Dunstane, D.; Watkins, S.C.; Delgoffe, G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018, 215, 1091–1100. [Google Scholar] [CrossRef] [Green Version]
- Teijeira, A.; Labiano, S.; Garasa, S.; Etxeberria, I.; Santamaría, E.; Rouzaut, A.; Enamorado, M.; Azpilikueta, A.; Inogés, S.; Bolaños, E.; et al. Mitochondrial Morphological and Functional Reprogramming Following CD137 (4-1BB) Costimulation. Cancer Immunol. Res. 2018, 6, 798–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aznar, M.A.; Labiano, S.; Diaz-Lagares, A.; Molina, C.; Garasa, S.; Azpilikueta, A.; Etxeberria, I.; Sánchez-Paulete, A.R.; Korman, A.J.; Esteller, M.; et al. CD137 (4-1BB) Costimulation Modifies DNA Methylation in CD8+ T Cell–Relevant Genes. Cancer Immunol. Res. 2018, 6, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, Z.T.; Nirschl, T.R.; Hovelson, D.H.; Johnston, R.J.; Engelhardt, J.J.; Selby, M.J.; Kochel, C.M.; Lan, R.Y.; Zhai, J.; Ghasemzadeh, A.; et al. A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. J. Clin. Investig. 2020, 130, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, I.; Glez-Vaz, J.; Teijeira, Á.; Melero, I. New emerging targets in cancer immunotherapy: CD137/4-1BB costimulatory axis. ESMO Open 2019, 4 (Suppl. 3), e000733. [Google Scholar] [CrossRef]
- Debenedette, M.A.; Wen, T.; Bachmann, M.F.; Ohashi, P.S.; Barber, B.H.; Stocking, K.L.; Peschon, J.J.; Watts, T.H. Cell Response to Influenza Virus. J. Immunol. 1999, 163, 4833–4841. [Google Scholar]
- Tan, J.T.; Whitmire, J.K.; Ahmed, R.; Pearson, T.C.; Larsen, C.P. 4-1BB ligand, a member of the TNF family, is important for the generation of antiviral CD8 T cell responses. J. Immunol. 1999, 163, 4859–4868. [Google Scholar]
- Kwon, B.S.; Hurtado, J.C.; Lee, Z.H.; Kwack, K.B.; Seo, S.K.; Choi, B.K.; Koller, B.H.; Wolisi, G.; Broxmeyer, H.E.; Vinay, D.S. Immune Responses in 4-1BB (CD137)-Deficient Mice. J. Immunol. 2002, 168, 5483–5490. [Google Scholar] [CrossRef] [Green Version]
- Melero, I.; Bach, N.; Hellström, K.E.; Aruffo, A.; Mittler, R.S.; Chen, L. Amplification of tumor immunity by gene transfer of the co-stimulatory 4-1BB ligand: Synergy with the CD28 co-stimulatory pathway. Eur. J. Immunol. 1998, 28, 1116–1121. [Google Scholar] [CrossRef]
- Wiethe, C.; Dittmar, K.; Doan, T.; Lindenmaier, W.; Tindle, R. Enhanced Effector and Memory CTL Responses Generated by Incorporation of Receptor Activator of NF-κB (RANK)/RANK Ligand Costimulatory Molecules into Dendritic Cell Immunogens Expressing a Human Tumor-Specific Antigen. J. Immunol. 2003, 171, 4121–4130. [Google Scholar] [CrossRef] [Green Version]
- Melero, I.; Shuford, W.W.; Newby, S.A.; Aruffo, A.; Ledbetter, J.A.; Hellström, K.E.; Mittler, R.S.; Chen, L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med. 1997, 3, 682–685. [Google Scholar] [CrossRef]
- Yonezawa, A.; Dutt, S.; Chester, C.; Kim, J.; Kohrt, H.E. Boosting Cancer Immunotherapy with Anti-CD137 Antibody Therapy. Clin. Cancer Res. 2015, 21, 3113–3120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Paulete, A.R.; Labiano, S.; Rodriguez-Ruiz, M.E.; Azpilikueta, A.; Etxeberria, I.; Bolaños, E.; Lang, V.; Rodriguez, M.; Aznar, M.A.; Jure-Kunkel, M.; et al. Deciphering CD137 (4-1BB) signaling in T-cell costimulation for translation into successful cancer immunotherapy. Eur. J. Immunol. 2016, 46, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.-Y.; Liu, Y. Immunotherapy of melanoma with the immune costimulatory monoclonal antibodies targeting CD137. Clin. Pharmacol. Adv. Appl. 2013, 5 (Suppl. 1), 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narazaki, H.; Zhu, Y.; Luo, L.; Zhu, G.; Chen, L. CD137 agonist antibody prevents cancer recurrence: Contribution of CD137 on both hematopoietic and nonhematopoietic cells. Blood 2010, 115, 1941–1948. [Google Scholar] [CrossRef] [PubMed]
- Gauttier, V.; Judor, J.-P.; Le Guen, V.; Cany, J.; Ferry, N.; Conchon, S. Agonistic anti-CD137 antibody treatment leads to antitumor response in mice with liver cancer. Int. J. Cancer 2014, 135, 2857–2867. [Google Scholar] [CrossRef]
- Sznol, M.; Hodi, F.S.; Margolin, K.; McDermott, D.F.; Ernstoff, M.S.; Kirkwood, J.M.; Wojtaszek, C.; Feltquate, D.; Logan, T. Phase I study of BMS-663513, a fully human anti-CD137 agonist monoclonal antibody, in patients (pts) with advanced cancer (CA). J. Clin. Oncol. 2008, 26, 3007. [Google Scholar] [CrossRef]
- Massarelli, E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. In Proceedings of the 31st Annual Meeting and Associated Programs of the Society for Immunotherapy of Cancer, National Harbor, MD, USA, 9–13 November 2016. [Google Scholar]
- Shuford, W.W.; Klussman, K.; Tritchler, D.D.; Loo, D.T.; Chalupny, J.; Siadak, A.W.; Brown, T.J.; Emswiler, J.; Raecho, H.; Larsen, C.P.; et al. 4-1BB Costimulatory Signals Preferentially Induce CD8+ T Cell Proliferation and Lead to the Amplification In Vivo of Cytotoxic T Cell Responses. J. Exp. Med. 1997, 186, 47–55. [Google Scholar] [CrossRef]
- Wehler, T.C.; Nonn, M.; Brandt, B.; Britten, C.M.; Gröne, M.; Todorova, M.; Link, I.; Khan, S.A.; Meyer, R.G.; Huber, C.; et al. Targeting the activation-induced antigen CD137 can selectively deplete alloreactive T cells from antileukemic and antitumor donor T-cell lines. Blood 2007, 109, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Wolfl, M.; Kuball, J.; Ho, W.Y.; Nguyen, H.; Manley, T.J.; Bleakley, M.; Greenberg, P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood 2007, 110, 201–210. [Google Scholar] [CrossRef]
- Watanabe, K.; Suzuki, S.; Kamei, M.; Toji, S.; Kawase, T.; Takahashi, T.; Kuzushima, K.; Akatsuka, Y. CD137-guided isolation and expansion of antigen-specific CD8 cells for potential use in adoptive immunotherapy. Int. J. Hematol. 2008, 88, 311–320. [Google Scholar] [CrossRef]
- Romero, P.; Dunbar, P.R.; Valmori, D.; Pittet, M.; Ogg, G.S.; Rimoldi, D.; Chen, J.-L.; Liénard, D.; Cerottini, J.-C.; Cerundolo, V. Ex Vivo Staining of Metastatic Lymph Nodes by Class I Major Histocompatibility Complex Tetramers Reveals High Numbers of Antigen-experienced Tumor-specific Cytolytic T Lymphocytes. J. Exp. Med. 1998, 188, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Gros, A.; Pasetto, A.; Prickett, T.; Crystal, J.S.; Robbins, P.; Rosenberg, S.A. Isolation of T-Cell Receptors Specifically Reactive with Mutated Tumor-Associated Antigens from Tumor-Infiltrating Lymphocytes Based on CD137 Expression. Clin. Cancer Res. 2017, 23, 2491–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazón, A.; Martínez-Forero, I.; Teijeira, A.; Morales-Kastresana, A.; Alfaro, C.; Sanmamed, M.F.; Perez-Gracia, J.L.; Peñuelas, I.; Hervás-Stubbs, S.; Rouzaut, A.; et al. The HIF-1α Hypoxia Response in Tumor-Infiltrating T Lymphocytes Induces Functional CD137 (4-1BB) for Immunotherapy. Cancer Discov. 2012, 2, 608–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Song, D.-G.; Poussin, M.; Yamamoto, T.; Best, A.; Li, C.; Coukos, G.; Powell, D.J. CD137 Accurately Identifies and Enriches for Naturally Occurring Tumor-Reactive T Cells in Tumor. Clin. Cancer Res. 2014, 20, 44–55. [Google Scholar] [CrossRef] [Green Version]
- Ugolini, A.; Zizzari, I.; Ceccarelli, F.; Botticelli, A.; Colasanti, T.; Strigari, L.; Rughetti, A.; Rahimi, H.; Conti, F.; Valesini, G.; et al. 4P IgM-rheumatoid factor as a novel biomarker for a reduced survival in anti-PD-1 treated NSCLC patients through the decrease of CD137+ T-cells. Ann. Oncol. 2020, 31, S1418. [Google Scholar] [CrossRef]
- Ugolini, A.; Zizzari, I.G.; Ceccarelli, F.; Botticelli, A.; Colasanti, T.; Strigari, L.; Rughetti, A.; Rahimi, H.; Conti, F.; Valesini, G.; et al. IgM-Rheumatoid factor confers primary resistance to anti-PD-1 immunotherapies in NSCLC patients by reducing CD137+ T-cells. EBioMedicine 2020, 62, 103098. [Google Scholar] [CrossRef]
- Zizzari, I.G.; Napoletano, C.; Botticelli, A.; Caponnetto, S.; Calabrò, F.; Gelibter, A.; Rughetti, A.; Ruscito, I.; Rahimi, H.; Rossi, E.; et al. TK Inhibitor Pazopanib Primes DCs by Downregulation of the β-Catenin Pathway. Cancer Immunol. Res. 2018, 6, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Zizzari, I.G.; Napoletano, C.; Di Filippo, A.; Botticelli, A.; Gelibter, A.; Calabrò, F.; Rossi, E.; Schinzari, G.; Urbano, F.; Pomati, G.; et al. Exploratory Pilot Study of Circulating Biomarkers in Metastatic Renal Cell Carcinoma. Cancers 2020, 12, 2620. [Google Scholar] [CrossRef]
- Fröhlich, A.; Loick, S.; Bawden, E.G.; Fietz, S.; Dietrich, J.; Diekmann, E.; Saavedra, G.; Fröhlich, H.; Niebel, D.; Sirokay, J.; et al. Comprehensive analysis of tumor necrosis factor receptor TNFRSF9 (4-1BB) DNA methylation with regard to molecular and clinicopathological features, immune infiltrates, and response prediction to immunotherapy in melanoma. EBioMedicine 2020, 52, 102647. [Google Scholar] [CrossRef]
- Gattinoni, L.; Powell, D.J.; Rosenberg, S.A.; Restifo, N.P. Adoptive immunotherapy for cancer: Building on success. Nat. Rev. Immunol. 2006, 6, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruella, M.; Kalos, M. Adoptive immunotherapy for cancer. Immunol. Rev. 2014, 257, 14–38. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of Tumor-Infiltrating Lymphocyte Cultures for Use in Adoptive Transfer Therapy for Melanoma Patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive Cell Transfer Therapy Following Non-Myeloablative but Lymphodepleting Chemotherapy for the Treatment of Patients with Refractory Metastatic Melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.-H.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, O.; Hovav, E.; Ziporen, Y.; Levy, D.; Kubi, A.; Zikich, D.; Hershkovitz, L.; Treves, A.J.; Shalmon, B.; Zippel, D.; et al. Establishment and Large-scale Expansion of Minimally cultured “Young” Tumor Infiltrating Lymphocytes for Adoptive Transfer Therapy. J. Immunother. 2011, 34, 212–220. [Google Scholar] [CrossRef]
- Radvanyi, L.; Bernatchez, C.; Zhang, M.; Fox, P.S.; Miller, P.; Chacon, J.; Wu, R.; Lizee, G.; Mahoney, S.; Alvarado, G.; et al. Specific Lymphocyte Subsets Predict Response to Adoptive Cell Therapy Using Expanded Autologous Tumor-Infiltrating Lymphocytes in Metastatic Melanoma Patients. Clin. Cancer Res. 2012, 18, 6758–6770. [Google Scholar] [CrossRef] [Green Version]
- Andersen, R.; Donia, M.; Ellebaek, E.; Borch, T.H.; Kongsted, P.; Iversen, T.Z.; Hölmich, L.R.; Hendel, H.W.; Met, Ö.; Andersen, M.H.; et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin. Cancer Res. 2016, 22, 3734–3745. [Google Scholar] [CrossRef] [Green Version]
- Besser, M.J.; Shapira-Frommer, R.; Treves, A.J.; Zippel, D.; Itzhaki, O.; Hershkovitz, L.; Levy, D.; Kubi, A.; Hovav, E.; Chermoshniuk, N.; et al. Clinical Responses in a Phase II Study Using Adoptive Transfer of Short-term Cultured Tumor Infiltration Lymphocytes in Metastatic Melanoma Patients. Clin. Cancer Res. 2010, 16, 2646–2655. [Google Scholar] [CrossRef] [Green Version]
- Besser, M.J.; Shapira-Frommer, R.; Itzhaki, O.; Treves, A.J.; Zippel, D.B.; Levy, D.; Kubi, A.; Shoshani, N.; Zikich, D.; Ohayon, Y.; et al. Adoptive Transfer of Tumor-Infiltrating Lymphocytes in Patients with Metastatic Melanoma: Intent-to-Treat Analysis and Efficacy after Failure to Prior Immunotherapies. Clin. Cancer Res. 2013, 19, 4792–4800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilon-Thomas, S.; Kuhn, L.; Ellwanger, S.; Janssen, W.; Royster, E.; Marzban, S.; Kudchadkar, R.; Zager, J.; Gibney, G.; Sondak, V.K.; et al. Efficacy of Adoptive Cell Transfer of Tumor-infiltrating Lymphocytes After Lymphopenia Induction for Metastatic Melanoma. J. Immunother. 2012, 35, 615–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Met, Ö.; Jensen, K.M.; Chamberlain, C.; Donia, M.; Svane, I.M. Principles of adoptive T cell therapy in cancer. Semin. Immunopathol. 2019, 41, 49–58. [Google Scholar] [CrossRef]
- Freedman, R.S.; Tomasovic, B.; Templin, S.; Atkinson, E.N.; Kudelka, A.; Edwards, C.L.; Platsoucas, C.D. Large-scale expansion in interleukin-2 of tumor-infiltrating lymphocytes from patients with ovarian carcinoma for adoptive immunotherapy. J. Immunol. Methods 1994, 167, 145–160. [Google Scholar] [CrossRef]
- Ioannides, C.G.; Freedman, R.S.; Platsoucas, C.D.; Rashed, S.; Kim, Y.P. Cytotoxic T cell clones isolated from ovarian tumor-infiltrating lymphocytes recognize multiple antigenic epitopes on autologous tumor cells. J. Immunol. 1991, 146, 1700–1707. [Google Scholar]
- Fujita, K.; Ikarashi, H.; Takakuwa, K.; Kodama, S.; Tokunaga, A.; Takahashi, T.; Tanaka, K. Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin. Cancer Res. 1995, 1, 501–507. [Google Scholar]
- Aoki, Y.; Takakuwa, K.; Kodama, S.; Tanaka, K.; Takahashi, M.; Tokunaga, A.; Takahashi, T. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res. 1991, 51, 1934–1939. [Google Scholar]
- Freedman, R.S.; Platsoucas, C.D. Immunotherapy for peritoneal ovarian carcinoma metastasis using Ex Vivo expanded tumor infiltrating lymphocytes. Cancer Treat. Res. 1996, 82, 115–146. [Google Scholar] [CrossRef]
- Webb, J.R.; Milne, K.; Watson, P.; DeLeeuw, R.J.; Nelson, B.H. Tumor-Infiltrating Lymphocytes Expressing the Tissue Resident Memory Marker CD103 Are Associated with Increased Survival in High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2014, 20, 434–444. [Google Scholar] [CrossRef] [Green Version]
- Hilders, C.G.J.M.; Ras, L.; Van Eendenburg, J.D.H.; Nooyen, Y.; Fleuren, G.J. Isolation and characterization of tumor-infiltrating lymphocytes from cervical carcinoma. Int. J. Cancer 1994, 57, 805–813. [Google Scholar] [CrossRef]
- Yannelli, J.R.; Hyatt, C.; McConnell, S.; Hines, K.; Jacknin, L.; Parker, L.; Sanders, M.; Rosenberg, S.A. Growth of tumor-infiltrating lymphocytes from human solid cancers: Summary of a 5-year experience. Int. J. Cancer 1996, 65, 413–421. [Google Scholar] [CrossRef]
- Turcotte, S.; Gros, A.; Hogan, K.; Tran, E.; Hinrichs, C.S.; Wunderlich, J.R.; Dudley, M.E.; Rosenberg, S.A. Phenotype and Function of T Cells Infiltrating Visceral Metastases from Gastrointestinal Cancers and Melanoma: Implications for Adoptive Cell Transfer Therapy. J. Immunol. 2013, 191, 2217–2225. [Google Scholar] [CrossRef] [Green Version]
- Andersen, R.; Westergaard, M.C.W.; Kjeldsen, J.W.; Mueller, A.; Pedersen, N.W.; Hadrup, S.R.; Met, Ö.; Seliger, B.; Kromann-Andersen, B.; Hasselager, T.; et al. T-cell Responses in the Microenvironment of Primary Renal Cell Carcinoma—Implications for Adoptive Cell Therapy. Cancer Immunol. Res. 2018, 6, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.Y.; Blattman, J.N.; Dossett, M.L.; Yee, C.; Greenberg, P.D. Adoptive immunotherapy: Engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell 2003, 3, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Chen, L. CD137 as a Biomarker for Tumor-Reactive T Cells: Finding Gold in the Desert. Clin. Cancer Res. 2014, 20, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Qi, X.; Ma, B.; Cao, Y.; Wang, L.; Sun, L.; Niu, H. The status, limitation and improvement of adoptive cellular immunotherapy in advanced urologic malignancies. Chin. J. Cancer Res. 2015, 27, 128–137. [Google Scholar]
- Seliktar-Ofir, S.; Merhavi-Shoham, E.; Itzhaki, O.; Yunger, S.; Markel, G.; Schachter, J.; Besser, M.J. Selection of Shared and Neoantigen-Reactive T Cells for Adoptive Cell Therapy Based on CD137 Separation. Front. Immunol. 2017, 8, 1211. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Condomines, M.; Van Der Stegen, S.J.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; Mcgettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Houot, R.; Goldstein, M.J.; Kohrt, H.E.; Myklebust, J.H.; Diehn, M.; Lin, J.T.; Irish, J.M.; Torchia, J.A.; Kolstad, A.; Chen, L.; et al. Therapeutic effect of CD137 immunomodulation in lymphoma and its enhancement by Treg depletion. Blood 2009, 114, 3431–3438. [Google Scholar] [CrossRef] [Green Version]
- Palazón, A.; Teijeira, A.; Martínez-Forero, I.; Hervás-Stubbs, S.; Roncal, C.; Peñuelas, I.; Dubrot, J.; Morales-Kastresana, A.; Pérez-Gracia, J.L.; Ochoa, M.C.; et al. Agonist Anti-CD137 mAb Act on Tumor Endothelial Cells to Enhance Recruitment of Activated T Lymphocytes. Cancer Res. 2011, 71, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Morales-Kastresana, A.; Catalán, E.; Hervas-Stubbs, S.; Palazón, A.; Azpilikueta, A.; Bolaños, E.; Anel, A.; Pardo, J.; Melero, I. Essential complicity of perforin-granzyme and FAS-L mechanisms to achieve tumor rejection following treatment with anti-CD137 mAb. J. Immunother. Cancer 2013, 1. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Siemann, D.W. Augmented antitumor effects of radiation therapy by 4-1BB antibody (BMS-469492) treatment. Anticancer. Res. 2006, 26, 3445–3453. [Google Scholar]
- Chacon, J.A.; Wu, R.C.; Sukhumalchandra, P.; Molldrem, J.J.; Sarnaik, A.; Pilon-Thomas, S.; Weber, J.; Hwu, P.; Radvanyi, L. Co-Stimulation through 4-1BB/CD137 Improves the Expansion and Function of CD8+ Melanoma Tumor-Infiltrating Lymphocytes for Adoptive T-Cell Therapy. PLoS ONE 2013, 8, e60031. [Google Scholar] [CrossRef] [Green Version]
- Melero, I.; Hirschhorn-Cymerman, D.; Morales-Kastresana, A.; Sanmamed, M.F.; Wolchok, J.D. Agonist Antibodies to TNFR Molecules That Costimulate T and NK Cells. Clin. Cancer Res. 2013, 19, 1044–1053. [Google Scholar] [CrossRef] [Green Version]
- Melero, I.; Grimaldi, A.M.; Perez-Gracia, J.L.; Ascierto, P.A. Clinical Development of Immunostimulatory Monoclonal Antibodies and Opportunities for Combination. Clin. Cancer Res. 2013, 19, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Madireddi, S.; Eun, S.-Y.; Lee, S.-W.; Nemčovičová, I.; Mehta, A.K.; Zajonc, D.M.; Nishi, N.; Niki, T.; Hirashima, M.; Croft, M. Galectin-9 controls the therapeutic activity of 4-1BB–targeting antibodies. J. Exp. Med. 2014, 211, 1433–1448. [Google Scholar] [CrossRef] [Green Version]
- Chu, D.-T.; Nguyen, D.B.; Nguyen, K.H.; Tien, N.L.B.; Van Thanh, V.; Nga, V.T.; Ngoc, V.T.N.; Dao, D.T.A.; Hoan, L.N.; Hung, N.P.; et al. An Update on Anti-CD137 Antibodies in Immunotherapies for Cancer. Int. J. Mol. Sci. 2019, 20, 1822. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Zhao, L.; Li, W.; Fan, K.; Qian, W.; Hou, S.; Wang, H.; Dai, M.; Hellstrom, I.; Hellstrom, K.E.; et al. Combinatorial PD-1 Blockade and CD137 Activation Has Therapeutic Efficacy in Murine Cancer Models and Synergizes with Cisplatin. PLoS ONE 2013, 8, e84927. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Choi, B.K.; Oh, H.S.; Kang, W.J.; Mittler, R.S.; Kwon, B.S. Mechanisms involved in synergistic anticancer effects of anti-4-1BB and cyclophosphamide therapy. Mol. Cancer Ther. 2009, 8, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westwood, J.A.; Matthews, G.M.; Shortt, J.; Faulkner, D.; Pegram, H.J.; Duong, C.P.; Chesi, M.; Bergsagel, P.L.; Sharp, L.L.; Huhn, R.D.; et al. Combination anti-CD137 and anti-CD40 antibody therapy in murine myc-driven hematological cancers. Leuk. Res. 2014, 38, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Kocak, E.; Lute, K.; Chang, X.; May, K.F.; Exten, K.R.; Zhang, H.; Abdessalam, S.F.; Lehman, A.M.; Jarjoura, D.; Zheng, P.; et al. Combination Therapy with Anti–CTL Antigen-4 and Anti-4-1BB Antibodies Enhances Cancer Immunity and Reduces Autoimmunity. Cancer Res. 2006, 66, 7276–7284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeone, E.; Ascierto, P.A. Immunomodulating antibodies in the treatment of metastatic melanoma: The experience with anti-CTLA-4, anti-CD137, and anti-PD1. J. Immunotoxicol. 2012, 9, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.; Wei, H.; Yip, Y.Y.; Feng, Q.; He, K.; Popov, V.; Hellstrom, I.; Hellstrom, K.E. Long-lasting Complete Regression of Established Mouse Tumors by Counteracting Th2 Inflammation. J. Immunother. 2013, 36, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Kalos, M.; Schaer, D.A.; Callahan, M.K.; Wolchok, J.D. Biomarkers for Immunostimulatory Monoclonal Antibodies in Combination Strategies for Melanoma and Other Tumor Types. Clin. Cancer Res. 2013, 19, 1009–1020. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Capone, M.; Urba, W.J.; Bifulco, C.; Botti, G.; Lugli, A.; Marincola, F.M.; Ciliberto, G.; Galon, J.; Fox, B.A. The additional facet of immunoscore: Immunoprofiling as a possible predictive tool for cancer treatment. J. Transl. Med. 2013, 11, 54. [Google Scholar] [CrossRef]
- Morales-Kastresana, A.; Sanmamed, M.F.; Rodriguez, I.; Palazon, A.; Martinez-Forero, I.; Labiano, S.; Hervas-Stubbs, S.; Sangro, B.; Ochoa, C.; Rouzaut, A.; et al. Combined Immunostimulatory Monoclonal Antibodies Extend Survival in an Aggressive Transgenic Hepatocellular Carcinoma Mouse Model. Clin. Cancer Res. 2013, 19, 6151–6162. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Gu, P.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Chen, S.-H. NK and CD8+ T cell-mediated eradication of poorly immunogenic B16-F10 melanoma by the combined action of IL-12 gene therapy and 4-1BB costimulation. Int. J. Cancer 2004, 109, 499–506. [Google Scholar] [CrossRef]
- Quetglas, J.I.; Dubrot, J.; Bezunartea, J.; Sanmamed, M.F.; Hervas-Stubbs, S. Immunotherapeutic Synergy Between Anti-CD137 mAb and Intratumoral Administration of a Cytopathic Semliki Forest Virus Encoding IL-12. Mol. Ther. 2012, 20, 1664–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirapu, I.; Arina, A.; Mazzolini, G.; Duarte, M.; Alfaro, C.; Feijoo, E.; Qian, C.; Chen, L.; Prieto, J.; Melero, I. Improving efficacy of interleukin-12-transfected dendritic cells injected into murine colon cancer with anti-CD137 monoclonal antibodies and alloantigens. Int. J. Cancer 2004, 110, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidehito, S.; Li, Q.; Shreiner, A.B.; Okuyama, R.; Jure-Kunkel, M.N.; Teitz-Tennenbaum, S.; Chang, A.E. Anti-CD137 Monoclonal Antibody Administration Augments the Antitumor Efficacy of Dendritic Cell-Based Vaccines. Cancer Res. 2004, 64, 8411–8419. [Google Scholar] [CrossRef] [Green Version]
- Westwood, J.A.; Hunnam, T.C.U.P.; Pegram, H.J.; Hicks, R.J.; Darcy, P.K.; Kershaw, M.H. Routes of Delivery for CpG and Anti-CD137 for the Treatment of Orthotopic Kidney Tumors in Mice. PLoS ONE 2014, 9, e95847. [Google Scholar] [CrossRef]
- Ko, E.; Luo, W.; Peng, L.; Wang, X.; Ferrone, S. Mouse Dendritic-Endothelial Cell Hybrids and 4-1BB Costimulation Elicit Antitumor Effects Mediated by Broad Antiangiogenic Immunity. Cancer Res. 2007, 67, 7875–7884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-H.; Pham-Nguyen, K.B.; Martinet, O.; Huang, Y.; Yang, W.; Thung, S.N.; Chen, L.; Mittler, R.; Woo, S.L. Rejection of Disseminated Metastases of Colon Carcinoma by Synergism of IL-12 Gene Therapy and 4-1BB Costimulation. Mol. Ther. 2000, 2, 39–46. [Google Scholar] [CrossRef]
- Li, B.; Lin, J.; VanRoey, M.; Jure-Kunkel, M.; Jooss, K. Established B16 tumors are rejected following treatment with GM-CSF-secreting tumor cell immunotherapy in combination with anti-4-1BB mAb. Clin. Immunol. 2007, 125, 76–87. [Google Scholar] [CrossRef]
- Ju, S.-A.; Cheon, S.-H.; Park, S.-M.; Tam, N.Q.; Kim, Y.M.; An, W.G.; Kim, B.-S. Eradication of established renal cell carcinoma by a combination of 5-fluorouracil and anti-4-1BB monoclonal antibody in mice. Int. J. Cancer 2008, 122, 2784–2790. [Google Scholar] [CrossRef]
- John, L.B.; Howland, L.J.; Flynn, J.K.; West, A.C.; Devaud, C.; Duong, C.P.; Stewart, T.J.; Westwood, J.A.; Guo, Z.S.; Bartlett, D.L.; et al. Oncolytic Virus and Anti–4-1BB Combination Therapy Elicits Strong Antitumor Immunity against Established Cancer. Cancer Res. 2012, 72, 1651–1660. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Doff, B.L.; Rearden, R.C.; Leggatt, G.R.; Mattarollo, S.R. NKT cell-targeted vaccination plus anti-4–1BB antibody generates persistent CD8 T cell immunity against B cell lymphoma. OncoImmunology 2015, 4, e990793. [Google Scholar] [CrossRef] [Green Version]
- Kwong, B.; Gai, S.A.; Elkhader, J.; Wittrup, K.D.; Irvine, D.J. Localized Immunotherapy via Liposome-Anchored Anti-CD137 + IL-2 Prevents Lethal Toxicity and Elicits Local and Systemic Antitumor Immunity. Cancer Res. 2013, 73, 1547–1558. [Google Scholar] [CrossRef] [Green Version]
- Curran, M.A.; Kim, M.; Montalvo, W.; Al-Shamkhani, A.; Allison, J.P. Combination CTLA-4 Blockade and 4-1BB Activation Enhances Tumor Rejection by Increasing T-Cell Infiltration, Proliferation, and Cytokine Production. PLoS ONE 2011, 6, e19499. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Hoelzinger, D.B.; Dominguez, A.L.; Van Snick, J.; Lustgarten, J. Signals through 4-1BB inhibit T regulatory cells by blocking IL-9 production enhancing antitumor responses. Cancer Immunol. Immunother. 2011, 60, 1775–1787. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, A.; Tyurin, V.A.; Tyurina, Y.Y.; Tcyganov, E.N.; Donthireddy, L.; Kagan, V.E.; Gabrilovich, D.; Veglia, F. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight 2020, 5, 138581. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Cheng, D.; Xia, Z.; Luan, M.; Wu, L.; Wang, G.; Zhang, S. Combined TIM-3 blockade and CD137 activation affords the long-term protection in a murine model of ovarian cancer. J. Transl. Med. 2013, 11, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef]
- Segal, N.H.; Logan, T.F.; Hodi, F.S.; McDermott, D.; Melero, I.; Hamid, O.; Schmidt, H.; Robert, C.; Chiarion-Sileni, V.; Ascierto, P.A.; et al. Results from an Integrated Safety Analysis of Urelumab, an Agonist Anti-CD137 Monoclonal Antibody. Clin. Cancer Res. 2017, 23, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Segal, N.H.; Gopal, A.K.; Bhatia, S.; Kohrt, H.E.; Levy, R.; Pishvaian, M.J.; Houot, R.; Bartlett, N.; Nghiem, P.; Kronenberg, S.A.; et al. A phase 1 study of PF-05082566 (anti-4-1BB) in patients with advanced cancer. J. Clin. Oncol. 2014, 32, 3007. [Google Scholar] [CrossRef]
- Segal, N.H.; He, A.R.; Doi, T.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 1816–1823. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Sznol, M.; Hu-Lieskovan, S.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Di Gravio, D.; Huang, B.; Gambhire, D.; Chen, Y.; et al. Phase Ib Study of Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 23, 5349–5357. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Sznol, M.; Hu-Lieskovan, S.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Di Gravio, D.; Huang, B.; Gambhire, D.; Chen, Y.; et al. Phase Ib study of PF-05082566 in combination with pembrolizumab in patients with advanced solid tumors. J. Clin. Oncol. 2016, 23, 5349–5357. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Pishvaian, M.J.; Shepard, D.R.; Wang, D.; Weiss, J.; Johnson, M.L.; Chung, C.H.; Chen, Y.; Huang, B.; Davis, C.B.; et al. A phase Ib study of utomilumab (PF-05082566) in combination with mogamulizumab in patients with advanced solid tumors. J. Immunother. Cancer 2019, 7, 342. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Bartlett, N.L.; Levy, R.; Houot, R.; Smith, S.D.; Segal, N.H.; Thall, A.D.; Mugundu, G.; Huang, B.; Davis, C.; et al. A phase I study of PF-05082566 (anti-4-1BB) + rituximab in patients with CD20+ NHL. J. Clin. Oncol. 2015, 33, 3004. [Google Scholar] [CrossRef]
- Hinner, M.J.; Aiba, R.S.B.; Jaquin, T.J.; Berger, S.; Dürr, M.C.; Schlosser, C.; Allersdorfer, A.; Wiedenmann, A.; Matschiner, G.; Schüler, J.; et al. Tumor-Localized Costimulatory T-Cell Engagement by the 4-1BB/HER2 Bispecific Antibody-Anticalin Fusion PRS-343. Clin. Cancer Res. 2019, 25, 5878–5889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piha-Paul, S.; Bendell, J.; Tolcher, A.; Hurvitz, S.; Patnaik, A.; Shroff, R.; Pohlmann, P.; Zettl, M.; Hahn, N.; Krishnamurthy, A.; et al. O82 A phase 1 dose escalation study of PRS-343, a HER2/4–1BB bispecific molecule, in patients with HER2-positive malignancies. J. Immunother. Cancer 2020. [Google Scholar] [CrossRef]
- Nelson, M.; Miller, R.; Nilsson, A.; Ljung, L.; Chunyk, A.; Mcmahan, C.; Bienvenue, D.; Askmyr, M.; Hernandez-Hoyos, G.; Fritzell, S. 851 Potent tumor-directed T cell activation and In Vivo tumor inhibition induced by a 4–1BB x 5T4 ADAPTIR™ bispecific antibody. J. Immunother. Cancer 2020. [Google Scholar] [CrossRef]
- Claus, C.; Ferrara, C.; Xu, W.; Sam, J.; Lang, S.; Uhlenbrock, F.; Albrecht, R.; Herter, S.; Schlenker, R.; Hüsser, T.; et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 2019, 11, eaav5989. [Google Scholar] [CrossRef]
- Compte, M.; Harwood, S.L.; Muñoz, I.G.; Navarro, R.; Zonca, M.; Perez-Chacon, G.; Erce-Llamazares, A.; Merino, N.; Tapia-Galisteo, A.; Cuesta, A.M.; et al. A tumor-targeted trimeric 4-1BB-agonistic antibody induces potent anti-tumor immunity without systemic toxicity. Nat. Commun. 2018, 9, 4809. [Google Scholar] [CrossRef]
- Nuti, M.; Zizzari, I.; Botticelli, A.; Rughetti, A.; Marchetti, P. The ambitious role of anti angiogenesis molecules: Turning a cold tumor into a hot one. Cancer Treat. Rev. 2018, 70, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Tormoen, G.; Crittenden, M.R.; Gough, M.J. Role of the immunosuppressive microenvironment in immunotherapy. Adv. Radiat. Oncol. 2018, 3, 520–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.M.; Maston, L.D.; Gough, M.J.; Ruby, C.E.; Redmond, W.L.; Crittenden, M.; Li, Y.; Puri, S.; Poehlein, C.H.; Morris, N.; et al. Signaling Through OX40 Enhances Antitumor Immunity. Semin. Oncol. 2010, 37, 524–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev. 2009, 229, 173–191. [Google Scholar] [CrossRef] [Green Version]
- Linch, S.N.; McNamara, M.J.; Redmond, W.L. OX40 Agonists and Combination Immunotherapy: Putting the Pedal to the Metal. Front. Oncol. 2015, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Lin, Q.; Zhang, Z.; Zhang, L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm. Sin. B 2020, 10, 414–433. [Google Scholar] [CrossRef]


{kind=link}
| Cancer Type | Treatment | Results | References |
|---|---|---|---|
| Metastatic NSCLC | Anti-PD-1 ICIs | Higher percentages of CD137+ T-cells in PBMC predicted a prolonged patients’ OS and PFS. | [67,68] Ugolini et al., 2020 |
| Metastatic RCCC | Anti-VEGF-R TKIs and anti-PD-1 ICIS | Percentage of CD137+ T-cells in PBMC decreased during patients’ progression. | [69] Zizzari et al., 2018 |
| Metastatic RCCC | TKIs | Higher percentages of CD137+ T-cells in PBMC were associated with responder patients. | [70] Zizzari et al., 2020 |
| Metastatic Melanoma | Anti-PD-1 ICIS | CD137 mRNA levels at the tumor site were positively associated with a prolonged OS, PFS, and a better response to the therapy. | [71] Fröhlich et al., 2020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ugolini, A.; Nuti, M. CD137+ T-Cells: Protagonists of the Immunotherapy Revolution. Cancers 2021, 13, 456. https://doi.org/10.3390/cancers13030456
Ugolini A, Nuti M. CD137+ T-Cells: Protagonists of the Immunotherapy Revolution. Cancers. 2021; 13(3):456. https://doi.org/10.3390/cancers13030456
Chicago/Turabian StyleUgolini, Alessio, and Marianna Nuti. 2021. "CD137+ T-Cells: Protagonists of the Immunotherapy Revolution" Cancers 13, no. 3: 456. https://doi.org/10.3390/cancers13030456
APA StyleUgolini, A., & Nuti, M. (2021). CD137+ T-Cells: Protagonists of the Immunotherapy Revolution. Cancers, 13(3), 456. https://doi.org/10.3390/cancers13030456