
Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives


Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Histone Acetylation and Regulation of Gene Expression
1.2. HDAC Subtypes: Structure, Function, Subcellular Localization, and Expression Patterns
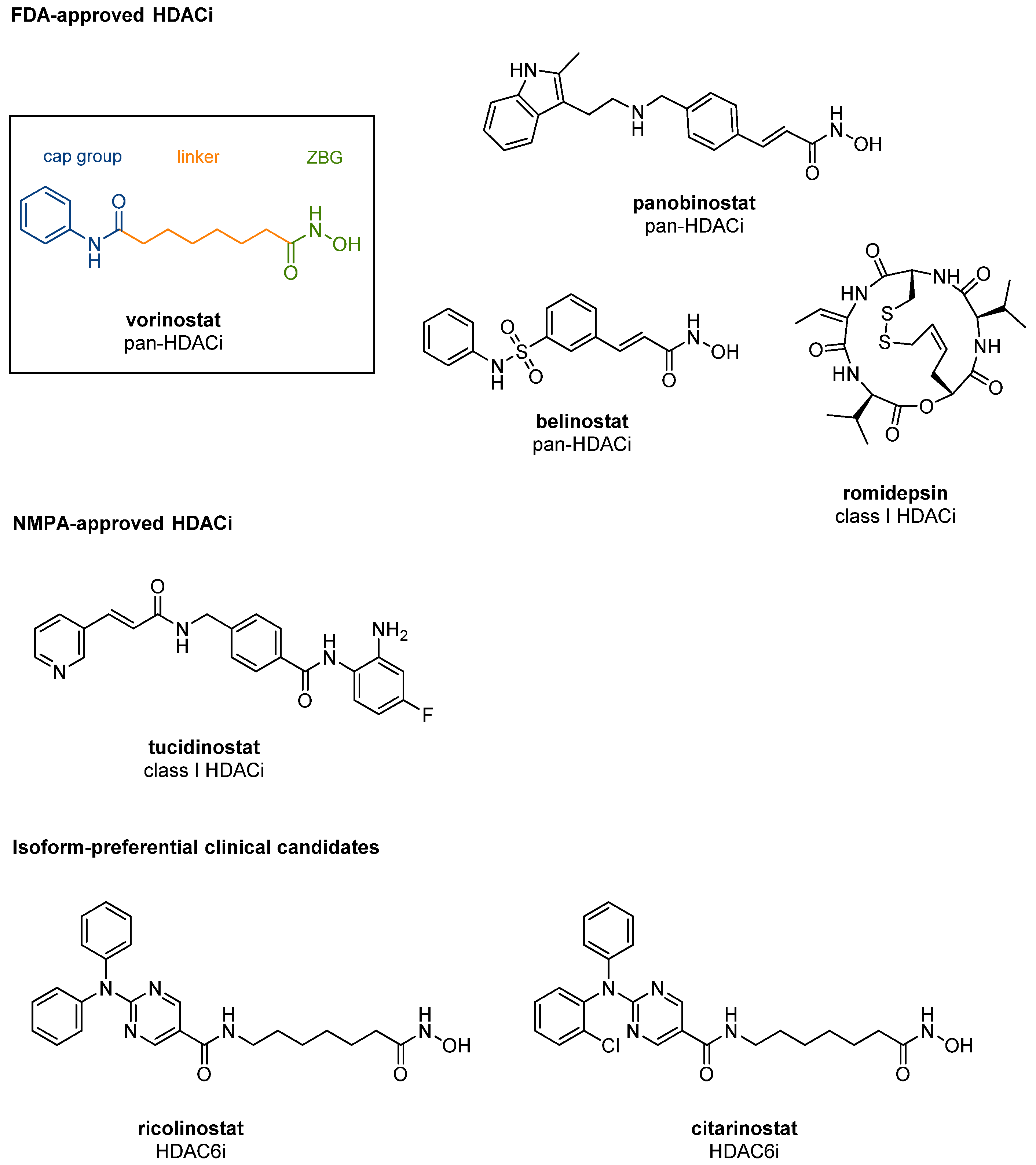
1.3. Structural Features of Zn2+-Dependent HDACs and Development of Subtype-Specific HDACis
2. Molecular Mechanisms of HDACi-Promoted Anticancer Effects
2.1. Apoptosis Induction
2.2. Autophagy Induction
2.3. Senescence Induction
2.4. Effects on DNA Damage
2.5. Effects on Hormone Signalling
2.6. Immune Effects
3. Combination Strategies
3.1. Combination with mTOR Inhibitors
3.2. Combination with Kinase Inhibitors (EGFR, PI3K)
3.3. Combination with Selective Estrogen Receptor Modulators (SERMs) and Antiestrogens
3.4. Combination with Immune Checkpoint Inhibitors
4. Bifunctional HDAC Inhibitors for Cancer Therapy
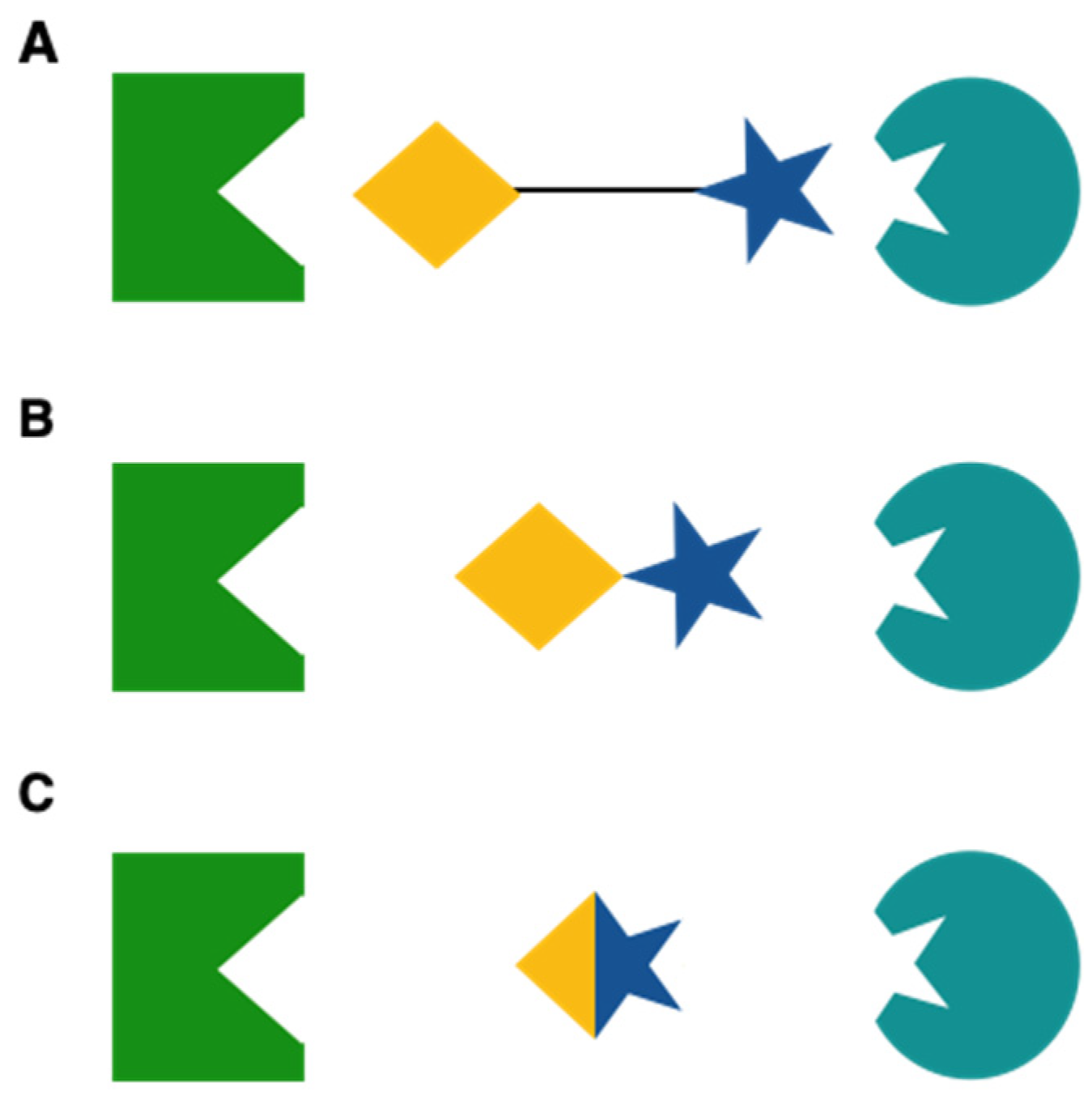
4.1. Multitarget Drugs: Advantages and Disadvantages
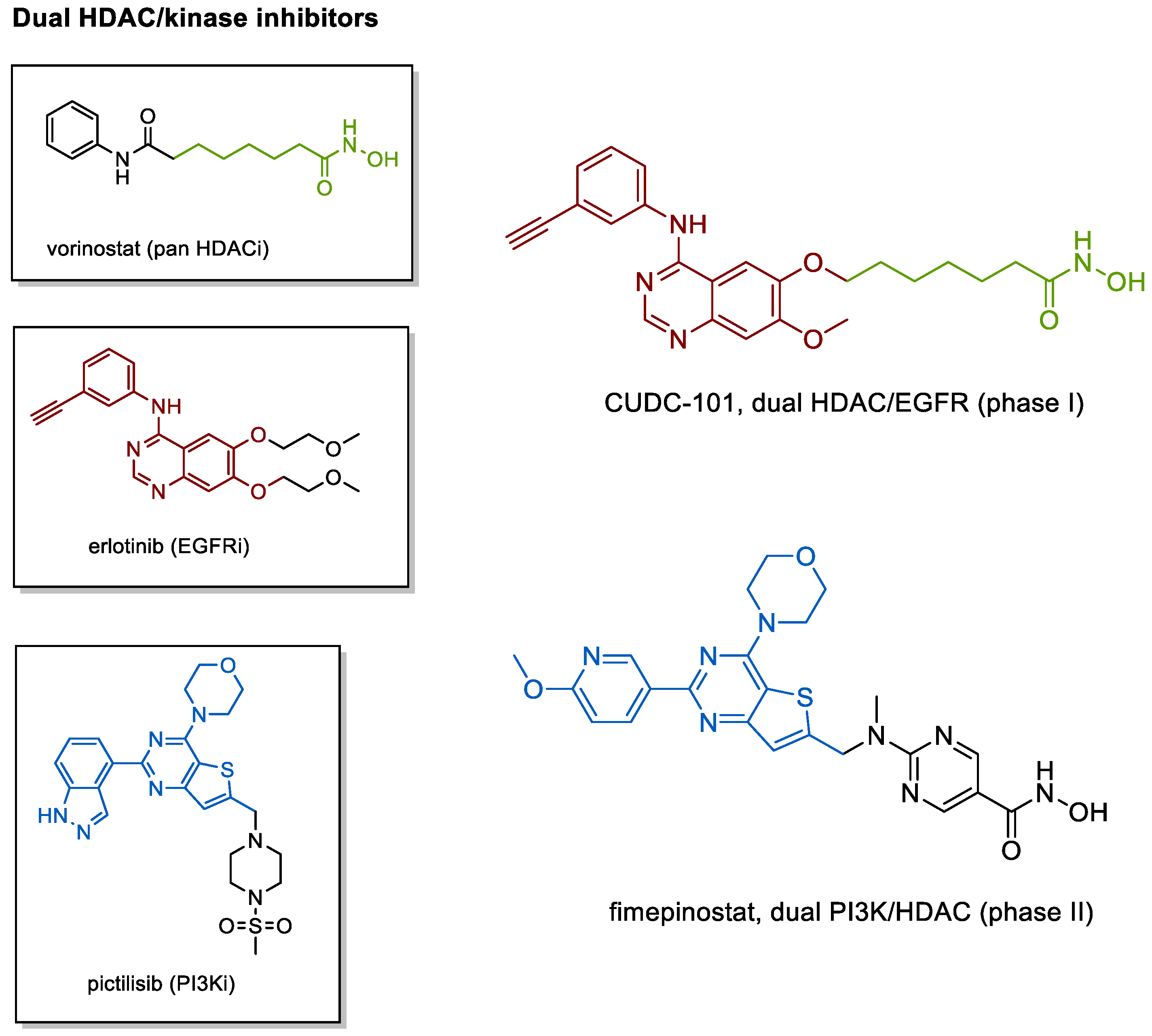
4.2. Kinase Inhibiting HDACis
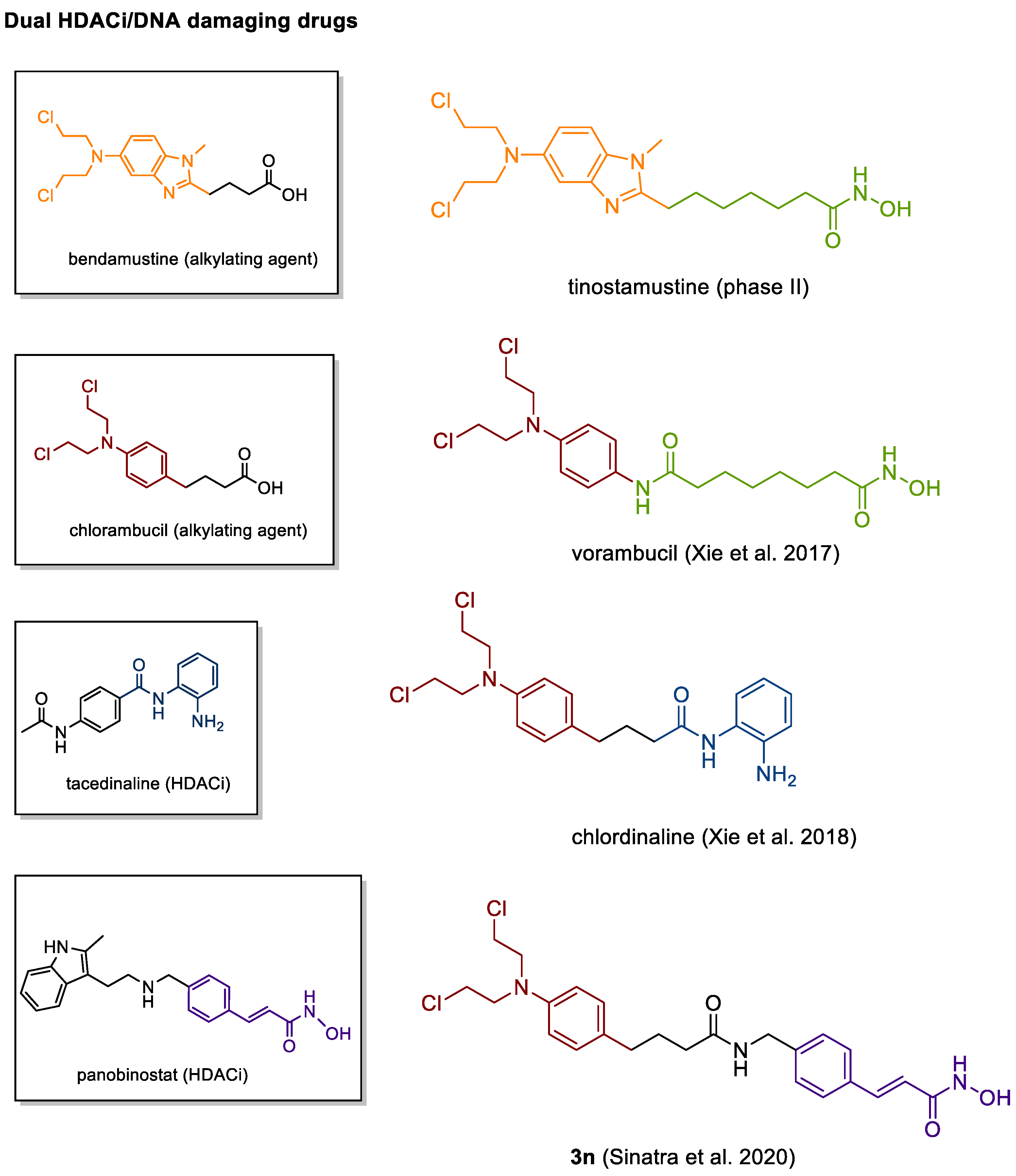
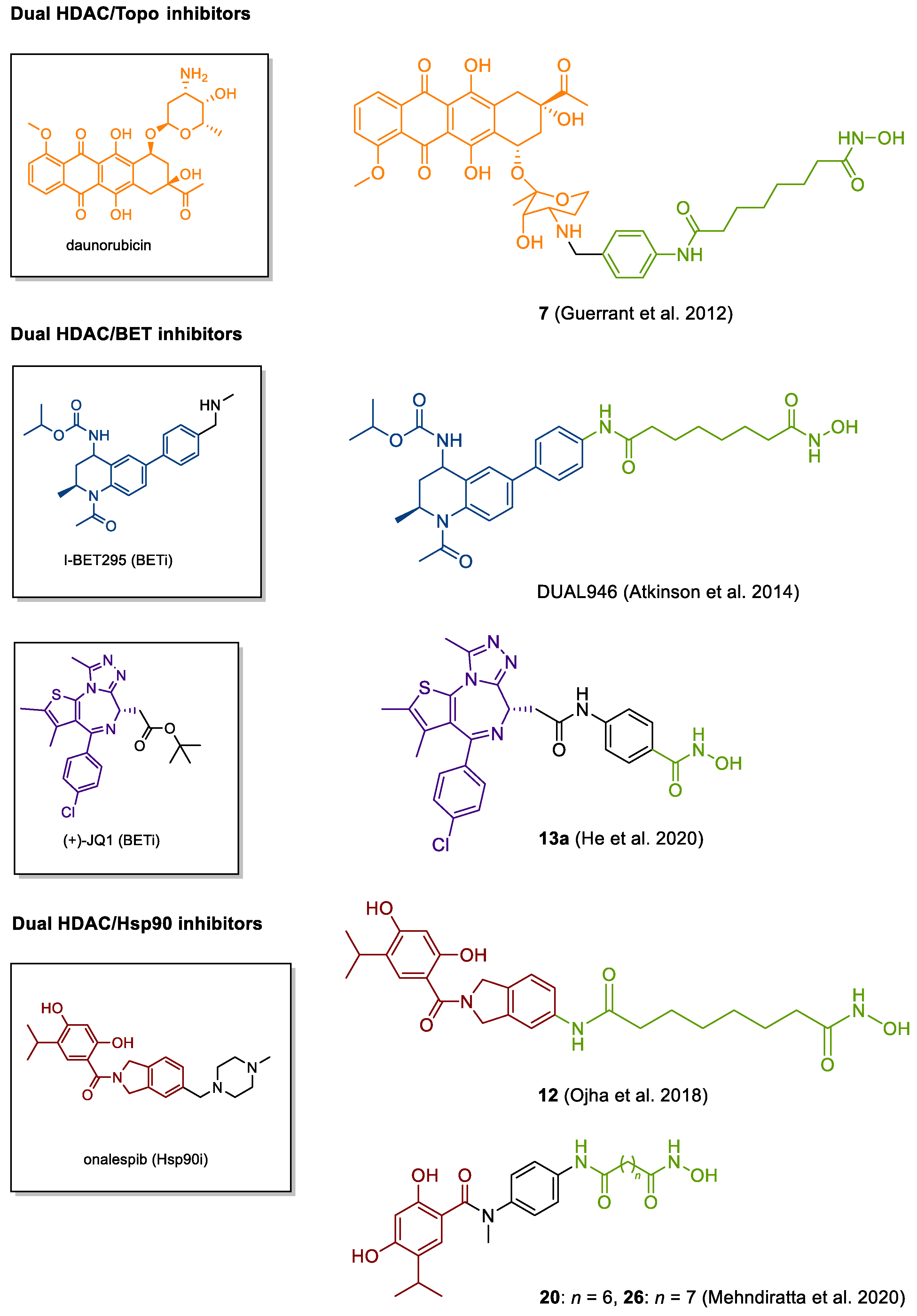
4.3. DNA Damaging HDACis
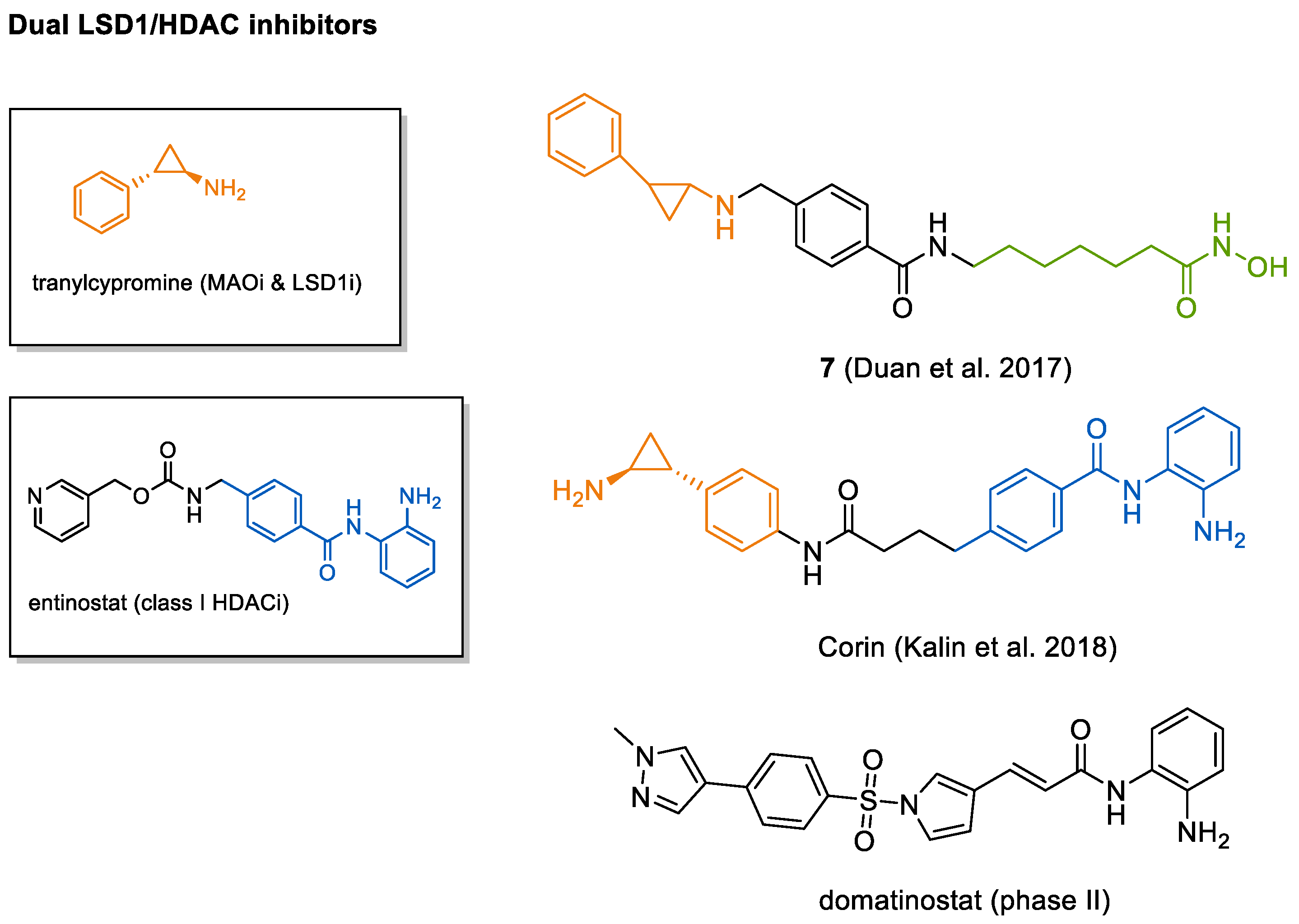
4.4. HDAC-LSD1 Inhibitor
4.5. Other Emerging Targets
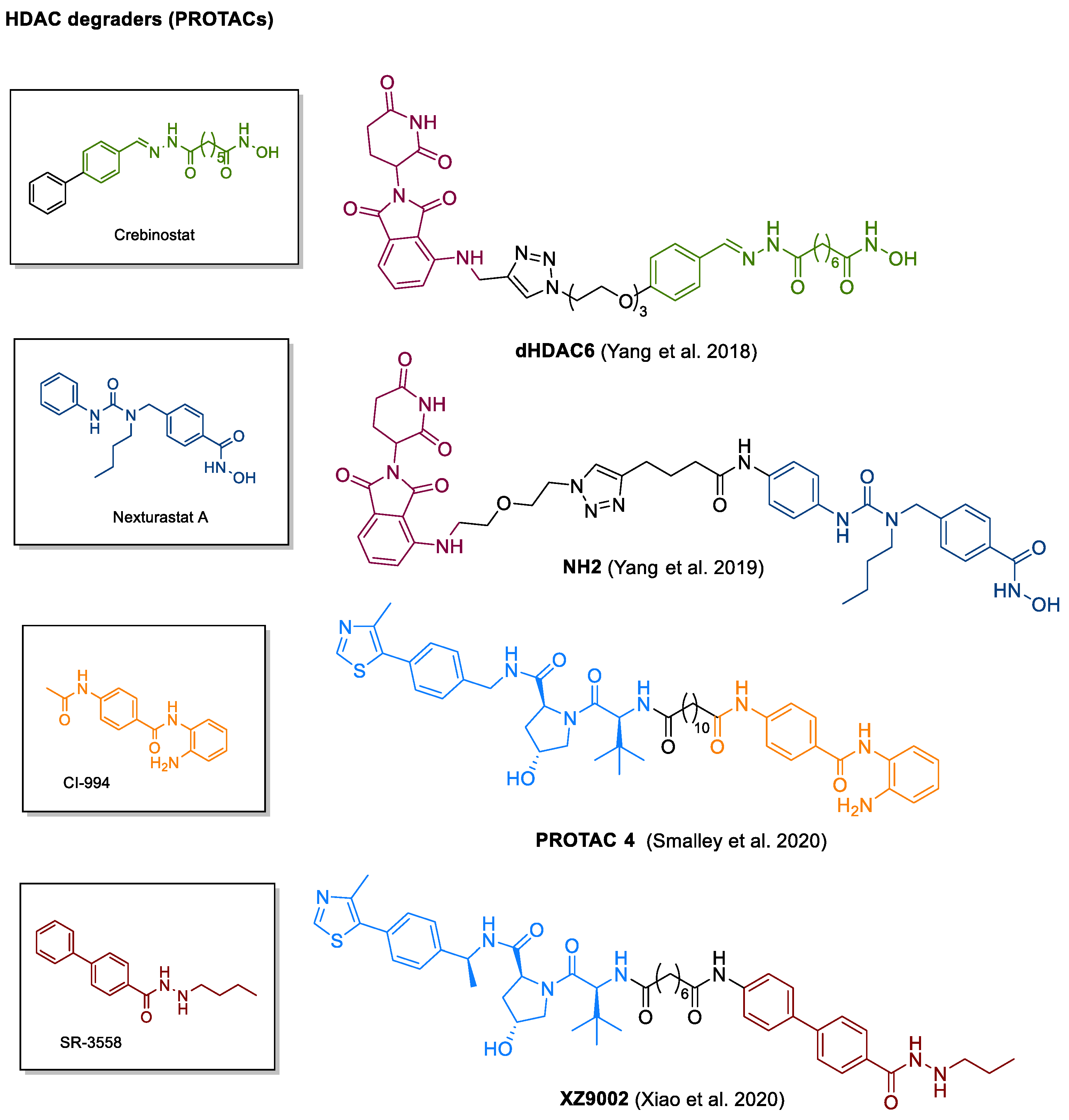
4.6. PROTACs
5. Outlook
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Combination Partner | Entity | Clin. Trial Phase | Best Response | Most Common Grade 3 or 4 Tox. | Ref. |
|---|---|---|---|---|---|---|
| Abexinostat | Pazopanib | Renal cell carcinoma, solid tumors | I | 51 pts, ORR 21%, median duration 9.1 months, 70% with pazopanib-refractory disease had tumor regression | fatigue, thrombocyto-penia, neutropenia | [291] |
| Doxorubicin | Sarcoma | I | 21 pts, PR + SD 47,7% | neutropenia | [292] | |
| Radiotherapy | Solid tumors | I | 51 pts, CR+PR 8%, SD 53% | thrombocytopenia | [293] | |
| Solid tumors | I | --- | thrombocytopenia | [294] | ||
| Entinostat | Exemestane + Entinostat (EE) vs. Entinostat + Placebo (EP) | Breast cancer | II | 130 pts, PFS 4.3 vs. 2.3 month (EE vs. EP) p = 0.11, OS 28.1 vs. 19.8 months (EE vs. EP) p = 0.036 | fatigue, neutropenia | [234] |
| Erlotinib + Entinostat (EE) vs. Erlotinib + Placebo (EP) | NSCLC | II | 130 pts, 4-month PFS comparable, OS 8.9 vs. 6,7 months (EE vs. EP) p = 0.39 | rash, fatigue, nausea | [225] | |
| Solid tumors (+lymphoma) | I | 29 pts, no CR or PR, 15 pts with SD (62 to 309 days) | nausea, vomiting | [295] | ||
| Interleukin 2 | Renal cell carcinoma | I/II | 47 pts, ORR 37%, median PFS 13.8 months, median OS 65.3 months | hypophosphatemia, lymphopenia | [237] | |
| 13-cis retinoic acid | Solid tumors | I | 19 pts, no ORR, 7 pts with SD (14 to 63 weeks) | hyponatremia, neutropenia, anemia | [296] | |
| Sorafenib | Solid tumors | I | 31 pts, 1pt PR, 2pts SD | muscle weakness, skin rash, fatigue | [297] | |
| Lapatinib | Breast cancer | Ib | 35pts, 3 pts CR, 3pts PR, 1pt SD | diarrhea, thrombocytopenia, neutropenia | [221] | |
| Solid tumors | I | Well tolerated, no PR or CR | hypophosphatemia, hyponatremia | [298] | ||
| Solid tumors | I | 27 pts, 2 pts w/PR, 6 pts w/SD | hypophosphatemia, hyponatremia | [299] | ||
| 5-Azacitidine | Breast cancer | II | 40 pts, ORR 4% | neutropenia, leukopenia | [300] | |
| 5-Azacitidine | mCRC | II | 47 pts, no response | lymphopenia, leukopenia | [301] | |
| 5-Azacitidine | NSCLC | I/II | 42 pts, 1 pt CR, 1 pt PR, 10 pts SD | fatigue, anemia | [302] | |
| Romidepsin | CRC | II | 25 pts, no ORR, 4 pts w/SD | fatigue | [303] | |
| SCLC | II | 16 pts, no ORR, 3pts w/SD | lymphopenia, nausea | [304] | ||
| CRPC | II | 35 pts, 2 PR | fatigue, nausea | [305] | ||
| Solid tumors | I | No ORR, 11xSD (median 30 weeks) | lymphopenia, nausea | [306] | ||
| Gemcitabine | Solid tumors | I | 27 pts, 2 PR, 14 SD | thrombocytopenia | [307] | |
| Glioma | I/II | 35 pts, no ORR | [308] | |||
| SCCHN | II | 13 pts, no ORR, 2 SD | fatigue, anemia | [309] | ||
| Erlotinib | NSCLC | I | 13 pts, 6 SD | fatigue, nausea | [310] | |
| Azacitidine | Solid tumors | I | 14 pts, 5 SD | fatigue, nausea | [311] | |
| Tucidinostat | Solid tumors (lymphoma) | I | 31 pts (22 w/solid tumors) 5 PR (4× lymphoma) 11 SD (1 lymphoma) | fatigue, myelosuppression | [312] | |
| Paclitaxel, carboplatin | NSCLC | I | 10 pts, 1 PR. 4 SD, 2 intracranial CR | thrombocytopenia, neutropenia | [313] | |
| Exemestane + Tucidinostat (ET) vs Exemestane + Placebo (EP) | Breast cancer | III | 365 pts, PFS 7.4 vs. 3.8 months (ET vs. EP) p = 0.033 | neutropenia, thrombocytopenia | [236] | |
| Belinostat | HCC | I/II | 42 pts (Phase II) 1 PR, 19 SD | elevated transaminases, bilirubinemia | [314] | |
| Cisplatin, doxorubicin, cyclophosphamid | Thymic tumors | I/II | 26 pts, 1 CR, 2 PR, 9 PR, 14 SD | myelosuppression, nausea | [315] | |
| Solid tumors | I | 72 pts, 13 SD | lymphopenia, fatigue | [316] | ||
| Solid tumors | I | 46 pts, 18 SD | nausea | [317] | ||
| Doxorubicin | Sarcoma, solid tumors | I/II | Phase I: 25 pts. 2 PR 16 SD, phase II 16 pts 1 CR 1 PR 9 SD | neutropenia | [318] | |
| Mesothelioma | II | 13 pts, no ORR, 2 SD | fatigue, hyponatremia | [319] | ||
| Ovarian cancer | II | 32 pts, 1 PR, 10 SD | thrombosis | [320] | ||
| 13-cis retinoic acid | Solid tumors | I | 51 pts, 2 PR, 10 SD | allergic reaction | [321] | |
| Carboplatin or paclitaxel | Solid tumors | I | 23 pts, 2 PR, 8 SD | myelosuppresion | [322] | |
| Thymic tumors | II | 41 pts, 2 PR, 25 SD | lymphopenia, QTc prolongation | [323] | ||
| Carboplatin and paclitaxel | Ovarian cancer | II | 35 pts, 3 CR, 12 PR, ORR 44% | leukopenia, fatigue | [324] | |
| Carboplatin | Ovarian cancer | II | 27 pts, 1 CR, 1 PR, 12 SD | leukopenia, thrombocytopenia, vomiting | [325] | |
| Cisplatin and etoposide | Mostly NET | I | 28 pts, PR 11, SD 13 | myelosuppresion | [326] | |
| Panobinostat | Everolimus | Renal cell carcinoma | I | 21 pts, no ORR, 13 SD | thrombocytopenia | [196] |
| Solid tumors | I | 9 pts, no ORR, 1 SD | thrombocytopenia | [327] | ||
| Bortezomib | Pancreas | II | 7 pts, no ORR | thrombocytopenia | [328] | |
| Epirubicin | Sarcoma, solid tumors | I | 40 pts, 4 PR, 11 SD | neutropenia, thrombocytopenia | [329] | |
| Carboplatin and etoposide | Lung cancer | I | 6 pts, no tolerable dose | thrombocytopenia | [330] | |
| Letrozole | Breast cancer | I | 12 pts, 2 PR, 5 SD | thrombocytopenia | [233] | |
| Bevacizumab + everolimus | Solid tumors | I | 12 pts, 1 PR, 3 SD | thrombocytopenia, hypertension | [331] | |
| Solid tumors | I | 25 pts, 4 SD | Nausea, thrombocytopenia | [332] | ||
| Radiation | Gliomas | I | 12 pts, increasing PFS and OS with increasing dose | neutropenia | [333] | |
| Solid tumors | I | 37 pts, 1 PR, 7 SD | Fatigue, thrombocytopenia | [334] | ||
| CRPC | II | 35 pts, no ORR | Fatigue, thrombocytopenia | [335] | ||
| CRPC | I | 16 pts, 5 of 8 with PSA decrease | neutropenia | [336] | ||
| SCLC | II | 21 pts, no ORR, 5 SD | nausea | [337] | ||
| Bevacizumab | Glioma | II | 24 pts, no PFS difference vs. bevacizumab historically | myelosuppression, hypophopsphatemia | [338] | |
| Paclitaxel and carboplatin | Solid tumors | I | 21 pts, 3 PR, 11 SD | myelosuppresion | [339] | |
| NET | II | 15 pts, 100% SD | thrombocytopenia, fatigue | [340] | ||
| Melanoma | I | 15 pts, 4 SD | myelosuppression | [341] | ||
| Radiotherapy +/− cisplatin and etoposide | NSCLC | I | 12 pts, radiation group DCR (SD + PR) 66%, chemoradiation group 100% PR | myelosuppression | [342] | |
| Renal cell carcinoma | II | 20 pts, 5 SD | thrombocytopenia | [343] | ||
| Erlotinib | NSCLC + HNSCC | I | 42 pts, 3 PR, 14 SD | nausea, thrombocytopenia | [223] | |
| Bevacizumab | Glioma | I | 12 pts, 3 PR, 7 SD | thrombocytopenia | [344] | |
| Sarcoma | II | 47 pts, no ORR, 17 SD | thrombocytopenia | [345] | ||
| Imatinib | GIST | I | 12 pts, 1 PR, 7 SD | thrombocytopenia | [346] | |
| Vorinostat | Ridaforolimus | Renal cell carcinoma | I | 15 pts, no ORR, 4 SD | thrombocytopenia, anemia | [197] |
| Capecitabine, cisplatin | Gastric cancer | I | 18 pts, 9 PR, 5 SD | thrombocytopenia, fatigue | [347] | |
| Capecitabine, cisplatin | Gastric cancer | II | 45 pts, 19 PR, 23 SD | neutropenia, fatigue | [348] | |
| 5-FU | CRC | I/II | 10 pts, 2 SD | thrombocytopenia, fatigue | [349] | |
| Solid tumors | II | 16 pts, 8 SD | myelosuppression, nausea | [350] | ||
| Paclitaxel, doxorubicin, cyclophosphamide | Breast cancer | I/II | 55 pts, comparable pCR rates as standard of care | [351] | ||
| Radiation | Brain metastasis | I | Safe administration | thrombocytopenia, fatigue | [352] | |
| Docetaxel | Solid tumors | I | 12 pts, no ORR | neutropenia | [353] | |
| Sarcoma | II | 40 pts, 9 SD | myelosuppression, fatigue | [354] | ||
| Erlotinib | NSCLC | II | 33 pts, no ORR | anemia, fatigue | [355] | |
| Paclitaxel, bevacizumab | Breast cancer | I/II | 53 pts, 2 CR, 24 PR, 16 SD | diarrhea, fatigue | [356] | |
| Carboplatin, paclitaxel +/− Placebo | NSCLC | II | 94 pts, ORR 34% w/ Vorinostat vs. 12.5 w/ Placebo | thrombocytopenia | [357] | |
| Solid tumors | I | 57 pts, 1 PR, 12 SD | thrombocytopenia | [358] | ||
| Bevacizumab | Renal cell carcinoma | I/II | 36 pts, 1 CR, 5 PR, 19 SD | thrombocytopenia | [359] | |
| Temozolomide | Gliomas | I/II | 39 pts, 2 CR, 15 PR, 19 SD | fatigue, thrombocytopenia | [360] | |
| Tamoxifen | Breast cancer | II | 43 pts, 8 PR, 9 SD | myelosuppression | [235] | |
| 13-cis retinoic acid | Renal cell car | I | 14 pts, 1 PR, 9 SD | fatigue, nausea | [361] | |
| Ovarian cancer | II | 27 pts, 1 PR, 9 SD | neutropenia | [362] | ||
| Carboplatin, gemcitabine | Ovarian cancer | I | 15 pts, 6 PR, 1 SD | myelosuppression | [363] | |
| Breast cancer | II | 14 pts, no ORR, 4 SD | lymphopenia | [364] | ||
| Mesothelioma | III | 661 pts, OS 30.7 vs. 27.1 weeks (Vorinostat vs. Placebo) p = 0.86 | fatigue | [365] | ||
| Solid tumors | I | 73 pts, 1 CR, 3 PR, 18 SD | fatigue | [366] | ||
| Bortezomib | NSCLC | II | 18 pts, no ORR, 5 SD | thrombocytopenia | [367] | |
| Gefitinib | NSCLC | I/II | 52 pts, 16 PR, 6 SD | anorexia, diarrhea | [224] | |
| Melanoma | II | 32 pts, 2 PR, 16 SD | fatigue | [368] | ||
| Pembrolizumab | NSCLC | I | 33, 4 PR, 16 SD | fatigue | [238] | |
| Sorafenib | HCC | I | 16 pts, no ORR, 10 SD | tash, hypokalemia | [369] | |
| Adenocystic | II | 30 pts, 2 PR, 27 SD | lymphopenia | [370] | ||
| Trastuzumab | Breast cancer | I/II | 16 pts, no ORR | thrombocytopenia | [222] | |
| Temozolomide | Glioma | I/II | Acceptable tolerability, OS endpoint not met | myelosuppression | [371] | |
| Glioma | II | 66 pts, 2 objective responses, efficacy endpoint met | thrombocytopenia | [372] | ||
| 5-FU | Solid tumors | I | 43 pts, 1 PR, 24 SD | fatigue, hand-foot-syndrom | [373] | |
| 5-FU, Oxaliplatin | CRC | I | 21 pts, no ORR. 11 SD | fatigue | [374] | |
| 5-FU | CRC | II | Not enough activity, accrual halted | [375] | ||
| GI cancer | I | 16 pts, no ORR, 8 SD | thrombocytopenia | [376] | ||
| Chemoradiation (5-FU) | Pancreatic cancer | I | 21 pts, 19 SD | lymphopenia | [377] | |
| HNSCC | II | 13 pts, 1 PR, 3 SD | thrombocytopenia | [363] |
References
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of Human Tumour Cells with Defined Genetic Elements. Nature 1999. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Review Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Ohm, J.E. Epigenetic Gene Silencing in Cancer—A Mechanism for Early Oncogenic Pathway Addiction? Nat. Rev. Cancer 2006, 6, 107–116. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Shilatifard, A. Epigenetic Modifications of Histones in Cancer. Genome Biol. 2019, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–32. [Google Scholar] [CrossRef]
- Thiagalingam, S.; Cheng, K.H.; Lee, H.J.; Mineva, N.; Thiagalingam, A.; Ponte, J.F. Histone Deacetylases: Unique Players in Shaping the Epigenetic Histone Code. Ann. N. Y. Acad. Sci. 2003. [Google Scholar] [CrossRef]
- Lund, A.H.; Van Lohuizen, M. Epigenetics and Cancer. Genes Dev. 2004. [Google Scholar] [CrossRef] [Green Version]
- Allfrey, V.G.; Mirsky, A.E. Structural Modifications of Histones and Their Possible Role in the Regulation of RNA Synthesis. Science 1964, 144, 559. [Google Scholar] [CrossRef]
- Grunstein, M. Histone Acetylation in Chromatin Structure and Transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of Acetylation at Lys16 and Trimethylation at Lys20 of Histone H4 Is a Common Hallmark of Human Cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and Nuclear Recruitment of HDACl in Hormone Refractory Prostate Cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, F.R.; Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Scholman, K.; Denkert, C.; Dietel, M.; et al. Class I Histone Deacetylases 1, 2 and 3 Are Highly Expressed in Renal Cell Cancer. BMC Cancer 2008, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyet, C.; Jentsch, B.; Hermanns, T.; Schweckendiek, D.; Seifert, H.H.; Schmidtpeter, M.; Sulser, T.; Moch, H.; Wild, P.J.; Kristiansen, G. Expression of Histone Deacetylases 1, 2 and 3 in Urothelial Bladder Cancer. BMC Clin. Pathol. 2014, 14, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Winzer, K.J.; Dietel, M.; Weichert, W.; Denkert, C. Differential Expression of Histone Deacetylases HDAC1, 2 and 3 in Human Breast Cancer—Overexpression of HDAC2 and HDAC3 Is Associated with Clinicopathological Indicators of Disease Progression. BMC Cancer 2013, 13, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Motoyama, S.; Ogawa, J. Strong Expression of HDAC3 Correlates with a Poor Prognosis in Patients with Adenocarcinoma of the Lung. Tumour Biol. 2010, 31, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer Activities of Histone Deacetylase Inhibitors. Nature Rev. Drug Discov. 2006. [Google Scholar] [CrossRef]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 Trial of Oral Vorinostat (Suberoylanilide Hydroxamic Acid, SAHA) for Refractory Cutaneous T-Cell Lymphoma (CTCL). Blood 2007. [Google Scholar] [CrossRef]
- Andrews, J.M.; Schmidt, J.A.; Carson, K.R.; Musiek, A.C.; Mehta-Shah, N.; Payton, J.E. Novel Cell Adhesion/Migration Pathways Are Predictive Markers of HDAC Inhibitor Resistance in Cutaneous T Cell Lymphoma. EBioMedicine 2019, 46, 170–183. [Google Scholar] [CrossRef] [Green Version]
- Qu, K.; Zaba, L.C.; Satpathy, A.T.; Giresi, P.G.; Li, R.; Jin, Y.; Armstrong, R.; Jin, C.; Schmitt, N.; Rahbar, Z.; et al. Chromatin Accessibility Landscape of Cutaneous T Cell Lymphoma and Dynamic Response to HDAC Inhibitors. Cancer Cell 2017, 32, 27–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A Mammalian Histone Deacetylase Related to the Yeast Transcriptional Regulator Rpd3p. Science 1996. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, H.; Dessain, S.K.; Eaton, E.N.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. HSIR2SIRT1 Functions as an NAD-Dependent P53 Deacetylase. Cell 2001. [Google Scholar] [CrossRef] [Green Version]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a Histone Deacetylase Homologue Bound to the TSA and SAHA Inhibitors. Nature 1999. [Google Scholar] [CrossRef]
- Micelli, C.; Rastelli, G. Histone Deacetylases: Structural Determinants of Inhibitor Selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef]
- Hansen, B.K.; Gupta, R.; Baldus, L.; Lyon, D.; Narita, T.; Lammers, M.; Choudhary, C.; Weinert, B.T. Analysis of Human Acetylation Stoichiometry Defines Mechanistic Constraints on Protein Regulation. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.M.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 Regulates Hsp90 Acetylation and Chaperone-Dependent Activation of Glucocorticoid Receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The Acetylation of Tau Inhibits Its Function and Promotes Pathological Tau Aggregation. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 Is a Microtubule-Associated Deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.; Dent, S.R.; et al. Article HDAC6 Modulates Cell Motility by Altering the Acetylation Level of Cortactin. Mol. Cell 2007, 197–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelières, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone Deacetylase 6 Inhibition Compensates for the Transport Deficit in Huntington’s Disease by Increasing Tubulin Acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef] [PubMed]
- Simões-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a Target for Neurodegenerative Diseases: What Makes It Different from the Other HDACs? Mol. Neurodegener. 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Hai, Y.; Shinsky, S.A.; Porter, N.J.; Christianson, D.W. Histone Deacetylase 10 Structure and Molecular Function as a Polyamine Deacetylase. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Kozikowski, A.P. Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors—What Some May Have Forgotten or Would Rather Forget? ChemMedChem 2016, 11, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, Y.; Boukari, H.; Banerjee, I.; Sbalzarini, I.F.; Horvath, P.; Helenius, A. Histone Deacetylase 8 Is Required for Centrosome Cohesion and Influenza a Virus Entry. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, I.; Miyake, Y.; Philip Nobs, S.; Schneider, C.; Horvath, P.; Kopf, M.; Matthias, P.; Helenius, A.; Yamauchi, Y. Influenza A Virus Uses the Aggresome Processing Machinery for Host Cell Entry. Science 2014, 346, 473–477. [Google Scholar] [CrossRef]
- Brindisi, M.; Saraswati, A.P.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Old but Gold: Tracking the New Guise of Histone Deacetylase 6 (HDAC6) Enzyme as a Biomarker and Therapeutic Target in Rare Diseases. J. Med. Chem. 2020, 63, 23–39. [Google Scholar] [CrossRef]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Ebert, M.P.A.; Pross, M.; Dietel, M.; Denkert, C.; Röcken, C. Association of Patterns of Class I Histone Deacetylase Expression with Patient Prognosis in Gastric Cancer: A Retrospective Analysis. Lancet Oncol. 2008. [Google Scholar] [CrossRef]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; De Francesco, R.; Gallinari, P.; et al. Crystal Structure of a Eukaryotic Zinc-Dependent Histone Deacetylase, Human HDAC8, Complexed with a Hydroxamic Acid Inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressi, J.C.; Jennings, A.J.; Skene, R.; Wu, Y.; Melkus, R.; De Jong, R.; O’Connell, S.; Grimshaw, C.E.; Navre, M.; Gangloff, A.R. Exploration of the HDAC2 Foot Pocket: Synthesis and SAR of Substituted N-(2-Aminophenyl)Benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 3142–3145. [Google Scholar] [CrossRef]
- Millard, C.J.; Watson, P.J.; Celardo, I.; Gordiyenko, Y.; Cowley, S.M.; Robinson, C.V.; Fairall, L.; Schwabe, J.W.R. Class I HDACs Share a Common Mechanism of Regulation by Inositol Phosphates. Mol. Cell 2013, 51, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Watson, P.J.; Fairall, L.; Santos, G.M.; Schwabe, J.W.R. Structure of HDAC3 Bound to Co-Repressor and Inositol Tetraphosphate. Nature 2012, 481, 335–340. [Google Scholar] [CrossRef]
- Hai, Y.; Christianson, D.W. Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, M.; Hoffmann, K.; Brosch, G.; Loidl, P. Analogues of Trichostatin A and Trapoxin B as Histone Deacetylase Inhibitors. Bioorg. Med. Chem. Lett. 1997, 7, 1655–1658. [Google Scholar] [CrossRef]
- Roche, J.; Bertrand, P. Inside HDACs with More Selective HDAC Inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Wambua, M.K.; Nalawansha, D.A.; Negmeldin, A.T.; Pflum, M.K.H. Mutagenesis Studies of the 14 Å internal Cavity of Histone Deacetylase 1: Insights toward the Acetate-Escape Hypothesis and Selective Inhibitor Design. J. Med. Chem. 2014, 57, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Methot, J.L.; Chakravarty, P.K.; Chenard, M.; Close, J.; Cruz, J.C.; Dahlberg, W.K.; Fleming, J.; Hamblett, C.L.; Hamill, J.E.; Harrington, P.; et al. Exploration of the Internal Cavity of Histone Deacetylase (HDAC) with Selective HDAC1/HDAC2 Inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Paris, M.; Porcelloni, M.; Binaschi, M.; Fattori, D. Histone Deacetylase Inhibitors: From Bench to Clinic. J. Med. Chem. 2008, 51, 1505–1529. [Google Scholar] [CrossRef]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfí, A.; et al. Unraveling the Hidden Catalytic Activity of Vertebrate Class IIa Histone Deacetylases. Proc. Natl. Acad. Sci. USA 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bürli, R.W.; Luckhurst, C.A.; Aziz, O.; Matthews, K.L.; Yates, D.; Lyons, K.A.; Beconi, M.; McAllister, G.; Breccia, P.; Stott, A.J.; et al. Design, Synthesis, and Biological Evaluation of Potent and Selective Class IIa Histone Deacetylase (HDAC) Inhibitors as a Potential Therapy for Huntington’s Disease. J. Med. Chem. 2013, 56, 9934–9954. [Google Scholar] [CrossRef]
- Schuetz, A.; Min, J.; Allali-Hassani, A.; Schapira, M.; Shuen, M.; Loppnau, P.; Mazitschek, R.; Kwiatkowski, N.P.; Lewis, T.A.; Maglathin, R.L.; et al. Human HDAC7 Harbors a Class IIa Histone Deacetylase-Specific Zinc Binding Motif and Cryptic Deacetylase Activity. J. Biol. Chem. 2008, 283, 11355–11363. [Google Scholar] [CrossRef] [Green Version]
- Hook, S.S.; Orian, A.; Cowley, S.M.; Eisenman, R.N. Histone Deacetylase 6 Binds Polyubiquitin through Its Zinc Finger (PAZ Domain) and Copurifies with Deubiquitinating Enzymes. Proc. Natl. Acad. Sci. USA 2002. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, P. Inside HDAC with HDAC Inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef]
- Sanaei, M.; Kavoosi, F. Histone Deacetylases and Histone Deacetylase Inhibitors: Molecular Mechanisms of Action in Various Cancers. Adv. Biomed. Res. 2019, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Wang, Y.; Jing, Y. Apoptosis Induction Byhistone Deacetylase Inhibitors in Cancer Cells: Role of Ku70. Int. J. Mol. Sci. 2019, 20, 1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Zhang, J.; Zhong, Q. Histone Deacetylase Inhibitors and Cell Death. Cell. Mol. Life Sci. 2014. [Google Scholar] [CrossRef]
- Bolden, J.E.; Shi, W.; Jankowski, K.; Kan, C.Y.; Cluse, L.; Martin, B.P.; MacKenzie, K.L.; Smyth, G.K.; Johnstone, R.W. HDAC Inhibitors Induce Tumor-Cell-Selective pro-Apoptotic Transcriptional Responses. Cell Death Dis. 2013. [Google Scholar] [CrossRef] [Green Version]
- Lagneaux, L.; Gillet, N.; Stamatopoulos, B.; Delforge, A.; Dejeneffe, M.; Massy, M.; Meuleman, N.; Kentos, A.; Martiat, P.; Willems, L.; et al. Valproic Acid Induces Apoptosis in Chronic Lymphocytic Leukemia Cells through Activation of the Death Receptor Pathway and Potentiates TRAIL Response. Exp. Hematol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Gillenwater, A.M.; Zhong, M.; Lotan, R. Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Induces Apoptosis through Both Mitochondrial and Fas (Cd95) Signaling in Head and Neck Squamous Carcinoma Cells. Mol. Cancer Ther. 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlisi, D.; Lauricella, M.; D’Anneo, A.; Emanuele, S.; Angileri, L.; Di Fazio, P.; Santulli, A.; Vento, R.; Tesoriere, G. The Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Sensitises Human Hepatocellular Carcinoma Cells to TRAIL-Induced Apoptosis by TRAIL-DISC Activation. Eur. J. Cancer 2009. [Google Scholar] [CrossRef] [PubMed]
- VanOosten, R.L.; Moore, J.M.; Karacay, B.; Griffith, T.S. Histone Deacetylase Inhibitors Modulate Renal Cell Carcinoma Sensitivity to TRAIL/Apo-2L-Induced Apoptosis by Enhancing TRAIL-R2 Expression. Cancer Biol. Ther. 2005. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.Y.; Omar, H.A.; Chiu, C.F.; Chi, Z.P.; Hu, J.L.; Weng, J.R. Antitumor Effects of (S)-HDAC42, a Phenylbutyrate-Derived Histone Deacetylase Inhibitor, in Multiple Myeloma Cells. Cancer Chemother. Pharmacol. 2011, 68, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Iacomino, G.; Medici, M.C.; Russo, G.L. Valproic Acid Sensitizes K562 Erythroleukemia Cells to TRAIL/Apo2L-Induced Apoptosis. Anticancer Res. 2008, 28, 855–864. [Google Scholar]
- Zhang, X.D.; Gillespie, S.K.; Borrow, J.M.; Hersey, P. The Histone Deacetylase Inhibitor Suberic Bishydroxamate Regulates the Expression of Multiple Apoptotic Mediators and Induces Mitochondria-Dependent Apoptosis of Melanoma Cells. Mol. Cancer Ther. 2004, 3, 425–435. [Google Scholar] [PubMed]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The Histone Deacetylase Inhibitor MS-275 Promotes Differentiation or Apoptosis in Human Leukemia Cells through a Process Regulated by Generation of Reactive Oxygen Species and Induction of P21CIP1/WAF1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Ruefli, A.A.; Ausserlechner, M.J.; Bernhard, D.; Sutton, V.R.; Tainton, K.M.; Kofler, R.; Smyth, M.J.; Johnstone, R.W. The Histone Deacetylase Inhibitor and Chemotherapeutic Agent Suberoylanilide Hydroxamic Acid (SAHA) Induces a Cell-Death Pathway Characterized by Cleavage of Bid and Production of Reactive Oxygen Species. Proc. Natl. Acad. Sci. USA 2001. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, H.; Hiraku, Y.; Tada-Oikawa, S.; Murata, M.; Ikemura, K.; Iwamoto, T.; Kagawa, Y.; Okuda, M.; Kawanishi, S. Romidepsin (FK228), a Potent Histone Deacetylase Inhibitor, Induces Apoptosis through the Generation of Hydrogen Peroxide. Cancer Sci. 2010. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Kim, S.H.; Park, W.H. Reactive Oxygen Species, Glutathione, and Thioredoxin Influence Suberoyl Bishydroxamic Acid-Induced Apoptosis in A549 Lung Cancer Cells. Tumor Biol. 2015. [Google Scholar] [CrossRef]
- Marks, P.A. Thioredoxin in Cancer-Role of Histone Deacetylase Inhibitors. Semin. Cancer Biol. 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosato, R.R.; Almenara, J.A.; Maggio, S.C.; Coe, S.; Atadja, P.; Dent, P.; Grant, S. Role of Histone Deacetylase Inhibitor-Induced Reactive Oxygen Species and DNA Damage in LAQ-824/Fludarabine Antileukemic Interactions. Mol. Cancer Ther. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Friday, B.B.; Lai, J.P.; McCollum, A.; Atadja, P.; Roberts, L.R.; Adjei, A.A. Abrogation of MAPK and Akt Signaling by AEE788 Synergistically Potentiates Histone Deacetylase Inhibitor-Induced Apoptosis through Reactive Oxygen Species Generation. Clin. Cancer Res. 2007. [Google Scholar] [CrossRef] [Green Version]
- Pei, X.Y.; Dai, Y.; Grant, S. Synergistic Induction of Oxidative Injury and Apoptosis in Human Multiple Myeloma Cells by the Proteasome Inhibitor Bortezomib and Histone Deacetylase Inhibitors. Clin. Cancer Res. 2004. [Google Scholar] [CrossRef] [Green Version]
- You, B.R.; Park, W.H. Suberoyl Bishydroxamic Acid-Induced Apoptosis in HeLa Cells via ROS-Independent, GSH-Dependent Manner. Mol. Biol. Rep. 2013. [Google Scholar] [CrossRef]
- Lucas, D.M.; Davis, M.E.; Parthun, M.R.; Mone, A.P.; Kitada, S.; Cunningham, K.D.; Flax, E.L.; Wickham, J.; Reed, J.C.; Byrd, J.C.; et al. The Histone Deacetylase Inhibitor MS-275 Induces Caspase-Dependent Apoptosis in B-Cell Chronic Lymphocytic Leukemia Cells. Leukemia 2004. [Google Scholar] [CrossRef]
- Jahr, H. HDACi and Nrf2: Not from Alpha to Omega but from Acetylation to OA. Arthritis Res. Ther. 2015. [Google Scholar] [CrossRef] [Green Version]
- Cai, D.; Yin, S.; Yang, J.; Jiang, Q.; Cao, W. Histone Deacetylase Inhibition Activates Nrf2 and Protects against Osteoarthritis. Arthritis Res. Ther. 2015. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Luo, D.; Xia, W.; Gu, C.; Xu, X.; Qiu, Q.; Zhang, Z. Nuclear Factor (Erythroid-Derived 2)-Like 2 (Nrf2) Contributes to the Neuroprotective Effects of Histone Deacetylase Inhibitors In Retinal Ischemia–Reperfusion Injury. Neuroscience 2019, 418, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhu, X.; Kim, Y.; Li, J.; Huang, S.; Saleem, S.; Li, R.C.; Xu, Y.; Dore, S.; Cao, W. Histone Deacetylase Inhibition Activates Transcription Factor Nrf2 and Protects against Cerebral Ischemic Damage. Free Radic. Biol. Med. 2012, 52, 928–936. [Google Scholar] [CrossRef]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The Role of Nrf2. Curr. Cancer Drug Targets 2017. [Google Scholar] [CrossRef] [PubMed]
- Paunkov, A.; Chartoumpekis, D.V.; Ziros, P.G.; Sykiotis, G.P. A Bibliometric Review of the Keap1/Nrf2 Pathway and Its Related Antioxidant Compounds. Antioxidants 2019, 8, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Campbell, K.H.; MacLeod, A.K.; McLaughlin, L.A.; Henderson, C.J.; Roland Wolf, C. HDAC Inhibitors Increase NRF2-Signaling in Tumour Cells and Blunt the Efficacy of Co-Adminstered Cytotoxic Agents. PLoS ONE 2014. [Google Scholar] [CrossRef] [Green Version]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. P53 at the Crossroads between Different Types of Hdac Inhibitor-Mediated Cancer Cell Death. Int. J. Mol. Sci. 2019, 20, 2415. [Google Scholar] [CrossRef] [Green Version]
- Poulaki, V.; Mitsiades, C.S.; Kotoula, V.; Negri, J.; McMullan, C.; Miller, J.W.; Marks, P.A.; Mitsiades, N. Molecular Sequelae of Histone Deacetylase Inhibition in Human Retinoblastoma Cell Lines: Clinical Implications. Investig. Ophthalmol. Vis. Sci. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greve, G.; Schiffmann, I.; Pfeifer, D.; Pantic, M.; Schüler, J.; Lübbert, M. The Pan-HDAC Inhibitor Panobinostat Acts as a Sensitizer for Erlotinib Activity in EGFR-Mutated and -Wildtype Non-Small Cell Lung Cancer Cells. BMC Cancer 2015, 15. [Google Scholar] [CrossRef] [Green Version]
- Mahalakshmi, R.; Husayn Ahmed, P.; Mahadevan, V. HDAC Inhibitors Show Differential Epigenetic Regulation and Cell Survival Strategies on P53 Mutant Colon Cancer Cells. J. Biomol. Struct. Dyn. 2018. [Google Scholar] [CrossRef]
- Sonnemann, J.; Marx, C.; Becker, S.; Wittig, S.; Palani, C.D.; Krämer, O.H.; Beck, J.F. P53-Dependent and P53-Independent Anticancer Effects of Different Histone Deacetylase Inhibitors. Br. J. Cancer 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palani, C.D.; Beck, J.F.; Sonnemann, J. Histone Deacetylase Inhibitors Enhance the Anticancer Activity of Nutlin-3 and Induce P53 Hyperacetylation and Downregulation of MDM2 and MDM4 Gene Expression. Investig. New Drugs 2012. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.M.; Quelle, D.E. P53 Acetylation: Regulation and Consequences. Cancers 2014, 7, 30. [Google Scholar] [CrossRef]
- Gu, W.; Luo, J.; Brooks, C.L.; Nikolaev, A.Y.; Li, M. Dynamics of the P53 Acetylation Pathway. In Reversible Protein Acetylation: Novartis Foundation Symposium; Wiley: Hoboken, NJ, USA, 2004. [Google Scholar] [CrossRef]
- Diss, E.; Nalabothula, N.; Nguyen, D.; Chang, E.; Kwok, Y.; Carrier, F. Vorinostat SAHA Promotes Hyper-Radiosensitivity in Wild Type P53 Human Glioblastoma Cells. J. Clin. Oncol. Res. 2014, 2, 1–16. [Google Scholar]
- Di Gennaro, E.; Bruzzese, F.; Pepe, S.; Leone, A.; Delrio, P.; Subbarayan, P.R.; Avallone, A.; Budillon, A. Modulation of Thymidilate Synthase and P53 Expression by HDAC Inhibitor Vorinostat Resulted in Synergistic Antitumor Effect in Combination with 5FU or Raltitrexed. Cancer Biol. Ther. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, W.B.; Zhu, J.; Eilers, G.; Li, X.; Kuang, Y.; Liu, L.; Mariño-Enríquez, A.; Yan, Z.; Li, H.; Meng, F.; et al. HDACi Inhibits Liposarcoma via Targeting of the MDM2-P53 Signaling Axis and PTEN, Irrespective of P53 Mutational Status. Oncotarget 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and Autophagic Cell Death Induced by Histone Deacetylase Inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.H.; Yang, N.; Lin, Q.; Xia, D.; Shen, H.M. Histone Deacetylase Inhibitors Induce Autophagy through FOXO1-Dependent Pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Thurn, K.T.; Biçaku, E.; Marchion, D.C.; Münster, P.N. Addition of a Histone Deacetylase Inhibitor Redirects Tamoxifen-Treated Breast Cancer Cells into Apoptosis, Which Is Opposed by the Induction of Autophagy. Breast Cancer Res. Treat. 2011, 130, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Mazzitelli, C.; Monti, P.; Degan, P.; Miele, M.; Fronza, G.; Menichini, P. Autophagy Induced by SAHA Affects Mutant P53 Degradation and Cancer Cell Survival. Biosci. Rep. 2019, 39, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Guan, F.; Wang, Y.; Zhang, Z.; Li, Y.; Cui, Y.; Li, Z.; Liu, H.; Zhang, Y.; Wang, Y.; et al. MS-275 Combined with Cisplatin Exerts Synergistic Antitumor Effects in Human Esophageal Squamous Cell Carcinoma Cells. Toxicol. Appl. Pharmacol. 2020, 395. [Google Scholar] [CrossRef]
- Chiao, M.T.; Cheng, W.Y.; Yang, Y.C.; Shen, C.C.; Ko, J.L. Suberoylanilide Hydroxamic Acid (SAHA) Causes Tumor Growth Slowdown and Triggers Autophagy in Glioblastoma Stem Cells. Autophagy 2013, 9, 1509–1526. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy Potentiates the Anti-Cancer Effects of the Histone Deacetylase Inhibitors in Hepatocellular Carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Won, A.J.; Lee, J.; Jung, J.H.; Yoon, S.; Lee, B.M.; Kim, H.S. Molecular Mechanism of SAHA on Regulation of Autophagic Cell Death in Tamoxifen-Resistant MCF-7 Breast Cancer Cells. Int. J. Med. Sci. 2012, 9, 881–893. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.; Balusu, R.; Fiskus, W.; Mudunuru, U.; Venkannagari, S.; Chauhan, L.; Smith, J.E.; Hembruff, S.L.; Ha, K.; Atadja, P.; et al. Combination of Pan-Histone Deacetylase Inhibitor and Autophagy Inhibitor Exerts Superior Efficacy against Triple-Negative Human Breast Cancer Cells. Mol. Cancer Ther. 2012, 11, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of Autophagy in Histone Deacetylase Inhibitor-Induced Apoptotic and Nonapoptotic Cell Death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fazio, P.; Waldegger, P.; Jabari, S.; Lingelbach, S.; Montalbano, R.; Ocker, M.; Slater, E.P.; Bartsch, D.K.; Illig, R.; Neureiter, D.; et al. Autophagy-Related Cell Death by Pan-Histone Deacetylase Inhibition in Liver Cancer. Oncotarget 2016, 7, 28998–29010. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Tanaka, K.; Sakimura, R.; Okada, T.; Nakamura, T.; Li, Y.; Takasaki, M.; Nakabeppu, Y.; Iwamoto, Y. Suberoylanilide Hydroxamic Acid (SAHA) Induces Apoptosis or Autophagy-Associated Cell Death in Chondrosarcoma Cell Lines. Anticancer Res. 2008, 28, 1585–1591. [Google Scholar] [PubMed]
- Del Bufalo, D.; Desideri, M.; De Luca, T.; Di Martile, M.; Gabellini, C.; Monica, V.; Busso, S.; Eramo, A.; De Maria, R.; Milella, M.; et al. Histone Deacetylase Inhibition Synergistically Enhances Pemetrexed Cytotoxicity through Induction of Apoptosis and Autophagy in Non-Small Cell Lung Cancer. Mol. Cancer 2014, 13, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Yao, R.; Zhao, D.; Zhou, L.; Wu, Y.; Yang, Y.; Sun, Y.; Lu, L.; Gao, W. Trichostatin A Reverses the Chemoresistance of Lung Cancer with High IGFBP2 Expression through Enhancing Autophagy. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Mancera, P.A.; Young, A.R.J.; Narita, M. Inside and out: The Activities of Senescence in Cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Ghayad, S.E.; Rammal, G.; Sarkis, O.; Basma, H.; Ghamloush, F.; Fahs, A.; Karam, M.; Harajli, M.; Rabeh, W.; Mouawad, J.E.; et al. The Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid (SAHA) as a Therapeutic Agent in Rhabdomyosarcoma. Cancer Biol. Ther. 2019, 20, 272–283. [Google Scholar] [CrossRef] [Green Version]
- Kaletsch, A.; Pinkerneil, M.; Hoffmann, M.J.; Jaguva Vasudevan, A.A.; Wang, C.; Hansen, F.K.; Wiek, C.; Hanenberg, H.; Gertzen, C.; Gohlke, H.; et al. Effects of Novel HDAC Inhibitors on Urothelial Carcinoma Cells. Clin. Epigenet. 2018, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- An, H.M.; Xue, Y.F.; Shen, Y.L.; Du, Q.; Hu, B. Sodium Valproate Induces Cell Senescence in Human Hepatocarcinoma Cells. Molecules 2013, 18, 14935–14947. [Google Scholar] [CrossRef]
- Freese, K.; Seitz, T.; Dietrich, P.; Lee, S.M.L.; Thasler, W.E.; Bosserhoff, A.; Hellerbrand, C. Histone Deacetylase Expressions in Hepatocellular Carcinoma and Functional Effects of Histone Deacetylase Inhibitors on Liver Cancer Cells in Vitro. Cancers 2019, 11, 1587. [Google Scholar] [CrossRef] [Green Version]
- Almeida, L.O.; Guimarães, D.M.; Martins, M.D.; Martins, M.A.T.; Warner, K.A.; Nör, J.E.; Castilho, R.M.; Squarize, C.H. Unlocking the Chromatin of Adenoid Cystic Carcinomas Using HDAC Inhibitors Sensitize Cancer Stem Cells to Cisplatin and Induces Tumor Senescence. Stem Cell Res. 2017, 21, 94–105. [Google Scholar] [CrossRef]
- Cho, J.H.; Dimri, M.; Dimri, G.P. MicroRNA-31 Is a Transcriptional Target of Histone Deacetylase Inhibitors and a Regulator of Cellular Senescence. J. Biol. Chem. 2015, 290, 10555–10567. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.C.; Chang, W.C.; Hsu, T.I.; Liu, J.J.; Yeh, S.H.; Wang, J.Y.; Liou, J.P.; Ko, C.Y.; Chang, K.Y.; Chuang, J.Y. Suberoylanilide Hydroxamic Acid Represses Glioma Stem-like Cells. J. Biomed. Sci. 2016, 23, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H.M. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Robert, C.; Rassool, F.V. HDAC Inhibitors: Roles of DNA Damage and Repair, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 116. [Google Scholar] [CrossRef]
- Kolbinger, F.R.; Koeneke, E.; Ridinger, J.; Heimburg, T.; Müller, M.; Bayer, T.; Sippl, W.; Jung, M.; Gunkel, N.; Miller, A.K.; et al. The HDAC6/8/10 Inhibitor TH34 Induces DNA Damage-Mediated Cell Death in Human High-Grade Neuroblastoma Cell Lines. Arch. Toxicol. 2018, 92, 2649–2664. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.; Walmsley, R. Histone-Deacetylase Inhibitors Produce Positive Results in the GADD45a-GFP GreenScreen HC Assay. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2013. [Google Scholar] [CrossRef] [PubMed]
- Olaharski, A.J.; Ji, Z.; Woo, J.Y.; Lim, S.; Hubbard, A.E.; Zhang, L.; Smith, M.T. The Histone Deacetylase Inhibitor Trichostatin a Has Genotoxic Effects in Human Lymphoblasts in Vitro. Toxicol. Sci. 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, A.; Assmann, A.S.; Schumacher, L.; Stuijvenberg, J.V.; Kassack, M.U.; Schulz, W.A.; Roos, W.P.; Hansen, F.K.; Pflieger, M.; Kurz, T.; et al. In Vitro Assessment of the Genotoxic Hazard of Novel Hydroxamic Acid-and Benzamide-Type Histone Deacetylase Inhibitors (HDACi). Int. J. Mol. Sci. 2020, 21, 4747. [Google Scholar] [CrossRef]
- Attia, S.M.; Al-Khalifa, M.K.; Al-Hamamah, M.A.; Alotaibi, M.R.; Attia, M.S.M.; Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A. Vorinostat Is Genotoxic and Epigenotoxic in the Mouse Bone Marrow Cells at the Human Equivalent Doses. Toxicology 2020. [Google Scholar] [CrossRef]
- Attia, S.M.; Al-Hamamah, M.A.; Alotaibi, M.R.; Harisa, G.I.; Attia, M.M.; Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A. Investigation of Belinostat-Induced Genomic Instability by Molecular Cytogenetic Analysis and Pathway-Focused Gene Expression Profiling. Toxicol. Appl. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Al-Hamamah, M.A.; Alotaibi, M.R.; Ahmad, S.F.; Ansari, M.A.; Attia, M.S.M.; Nadeem, A.; Bakheet, S.A.; As Sobeai, H.M.; Attia, S.M. Genetic and Epigenetic Alterations Induced by the Small-Molecule Panobinostat: A Mechanistic Study at the Chromosome and Gene Levels. DNA Repair 2019. [Google Scholar] [CrossRef]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone Deacetylase Inhibitor Induces DNA Damage, Which Normal but Not Transformed Cells Can Repair. Proc. Natl. Acad. Sci. USA 2010. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.S.; Galloway, S.; Lagrutta, A.; Armstrong, M.; Miller, T.; Richon, V.M.; Andrews, P.A. Nonclinical Safety Assessment of the Histone Deacetylase Inhibitor Vorinostat. Int. J. Toxicol. 2010. [Google Scholar] [CrossRef]
- Petruccelli, L.A.; Dupéré-Richer, D.; Pettersson, F.; Retrouvey, H.; Skoulikas, S.; Miller, W.H. Vorinostat Induces Reactive Oxygen Species and Dna Damage in Acute Myeloid Leukemia Cells. PLoS ONE 2011. [Google Scholar] [CrossRef] [Green Version]
- Martirosyan, A.; Leonard, S.; Shi, X.; Griffith, B.; Gannett, P.; Strobl, J. Actions of a Histone Deacetylase Inhibitor NSC3852 (5-Nitroso-8-Quinolinol) Link Reactive Oxygen Species to Cell Differentiation and Apoptosis in MCF-7 Human Mammary Tumor Cells. J. Pharmacol. Exp. Ther. 2006. [Google Scholar] [CrossRef] [Green Version]
- Jia, M.; Zhang, H.; Lin, Y.; Chen, D.; Chen, Y.; Xia, Y. Consecutive Lossen Rearrangement/Transamidation Reaction of Hydroxamic Acids under Catalyst- and Additive-Free Conditions. Org. Biomol. Chem. 2018, 16, 3615–3624. [Google Scholar] [CrossRef]
- Lee, M.S.; Isobe, M. Metabolic Activation of the Potent Mutagen, 2-Naphthohydroxamic Acid, in Salmonella Typhimurium TA98. Cancer Res. 1990, 50, 4300–4307. [Google Scholar] [PubMed]
- Ducháčková, L.; Roithová, J. The Interaction of Zinc(II) and Hydroxamic Acids and a Metal-Triggered Lossen Rearrangement. Chem. A Eur. J. 2009. [Google Scholar] [CrossRef]
- Samuni, Y.; Wink, D.A.; Krishna, M.C.; Mitchell, J.B.; Goldstein, S. Suberoylanilide Hydroxamic Acid Radiosensitizes Tumor Hypoxic Cells in Vitro through the Oxidation of Nitroxyl to Nitric Oxide. Free Radic. Biol. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrov, R.; Hristova, R.; Stoynov, S.; Gospodinov, A. The Chromatin Response to Double-Strand DNA Breaks and Their Repair. Cells 2020, 9, 1853. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, S.Y.; Miller, K.M. Preserving Genome Integrity and Function: The DNA Damage Response and Histone Modifications. Crit. Rev. Biochem. Mol. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Gursoy-Yuzugullu, O.; Parasuram, R.; Price, B.D. The Tale of a Tail: Histone H4 Acetylation and the Repair of DNA Breaks. Philos. Trans. R. Soc. B Biol. Sci. 2017. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Krumm, A. Survey and Summary: The Multifaceted Influence of Histone Deacetylases on DNA Damage Signalling and DNA Repair. Nucleic Acids Res. 2016, 44, 10017–10030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, P.; Wyrick, J.J. Organization of DNA Damage, Excision Repair, and Mutagenesis in Chromatin: A Genomic Perspective. DNA Repair 2019. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Biol. 2018, 34, 265–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauer, M.H.; Gasser, S.M. Chromatin and Nucleosome Dynamics in DNA Damage and Repair. Genes Dev. 2017. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Shi, B.; Liu, X.; An, H.X. Acetylation and Deacetylation of DNA Repair Proteins in Cancers. Front. Oncol. 2020. [Google Scholar] [CrossRef]
- Pao, P.C.; Patnaik, D.; Watson, L.A.; Gao, F.; Pan, L.; Wang, J.; Adaikkan, C.; Penney, J.; Cam, H.P.; Huang, W.C.; et al. HDAC1 Modulates OGG1-Initiated Oxidative DNA Damage Repair in the Aging Brain and Alzheimer’s Disease. Nat. Commun. 2020. [Google Scholar] [CrossRef]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 Function in the DNA-Damage Response to Promote DNA Nonhomologous End-Joining. Nat. Struct. Mol. Biol. 2010. [Google Scholar] [CrossRef]
- Reid, G.; Métivier, R.; Lin, C.Y.; Denger, S.; Ibberson, D.; Ivacevic, T.; Brand, H.; Benes, V.; Liu, E.T.; Gannon, F. Multiple Mechanisms Induce Transcriptional Silencing of a Subset of Genes, Including Oestrogen Receptor α, in Response to Deacetylase Inhibition by Valproic Acid and Trichostatin A. Oncogene 2005, 24, 4894–4907. [Google Scholar] [CrossRef] [Green Version]
- De Los Santos, M.; Martínez-Iglesias, O.; Aranda, A. Anti-Estrogenic Actions of Histone Deacetylase Inhibitors in MCF-7 Breast Cancer Cells. Endocr. Relat. Cancer 2007, 14, 1021–1028. [Google Scholar] [CrossRef] [Green Version]
- Margueron, R.; Duong, V.; Castet, A.; Cavaillès, V. Histone Deacetylase Inhibition and Estrogen Signalling in Human Breast Cancer Cells. Biochem. Pharmacol. 2004, 68, 1239–1246. [Google Scholar] [CrossRef]
- Urbinati, G.; Marsaud, V.; Plassat, V.; Fattal, E.; Lesieur, S.; Renoir, J.M. Liposomes Loaded with Histone Deacetylase Inhibitors for Breast Cancer Therapy. Int. J. Pharm. 2010, 397, 184–193. [Google Scholar] [CrossRef]
- Urbinati, G.; Marsaud, V.; Vergnaud-Gauduchon, J.; Renoir, J.M.; Urbinati, G.; Marsaud, V.; Nicolas, V.; Vergnaud-Gauduchon, J.; Renoir, J.M.; Renoir, J.M. Liposomal Trichostatin A: Therαpeutic Potential in Hormone-Dependent and -Independent Breast Cancer Xenograft Models. Horm. Mol. Biol. Clin. Investig. 2011, 6, 215–225. [Google Scholar] [CrossRef]
- Noh, H.; Park, J.; Shim, M.; Lee, Y.J. Trichostatin A Enhances Estrogen Receptor-Alpha Repression in MCF-7 Breast Cancer Cells under Hypoxia. Biochem. Biophys. Res. Commun. 2016, 470, 748–752. [Google Scholar] [CrossRef]
- Fiskus, W.; Ren, Y.; Mohapatra, A.; Bali, P.; Mandawat, A.; Rao, R.; Herger, B.; Yang, Y.; Atadja, P.; Wu, J.; et al. Hydroxamic Acid Analogue Histone Deacetylase Inhibitors Attenuate Estrogen Receptor-α Levels and Transcriptional Activity: A Result of Hyperacetylation and Inhibition of Chaperone Function of Heat Shock Protein 90. Clin. Cancer Res. 2007, 13, 4882–4890. [Google Scholar] [CrossRef] [Green Version]
- Hideshima, T.; Mazitschek, R.; Qi, J.; Mimura, N.; Tseng, J.C.; Kung, A.L.; Bradner, J.E.; Anderson, K.C. HDAC6 Inhibitor WT161 Downregulates Growth Factor Receptors in Breast Cancer. Oncotarget 2017, 8, 80109–80123. [Google Scholar] [CrossRef] [Green Version]
- Tu, Z.; Li, H.; Ma, Y.; Tang, B.; Tian, J.; Akers, W.; Achilefu, S.; Gu, Y. The Enhanced Antiproliferative Response to Combined Treatment of Trichostatin A with Raloxifene in MCF-7 Breast Cancer Cells and Its Relevance to Estrogen Receptor β Expression. Mol. Cell. Biochem. 2012, 366, 111–122. [Google Scholar] [CrossRef]
- Sabnis, G.J.; Goloubeva, O.; Chumsri, S.; Nguyen, N.; Sukumar, S.; Brodie, A.M.H. Functional Activation of the Estrogen Receptor-α and Aromatase by the HDAC Inhibitor Entinostat Sensitizes ER-Negative Tumors to Letrozole. Cancer Res. 2011, 71, 1893–1903. [Google Scholar] [CrossRef] [Green Version]
- Jang, E.R.; Lim, S.J.; Lee, E.S.; Jeong, G.; Kim, T.Y.; Bang, Y.J.; Lee, J.S. The Histone Deacetylase Inhibitor Trichostatin a Sensitizes Estrogen Receptor α-Negative Breast Cancer Cells to Tamoxifen. Oncogene 2004, 23, 1724–1736. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Ferguson, A.T.; Nass, S.J.; Phillips, D.L.; Butash, K.A.; Wang, S.M.; Herman, J.G.; Davidson, N.E. Transcriptional Activation of Estrogen Receptor α in Human Breast Cancer Cells by Histone Deacetylase Inhibition. Cancer Res. 2000, 60, 6890–6894. [Google Scholar]
- Shi, Y.; Jia, Y.; Zhao, W.; Zhou, L.; Xie, X.; Tong, Z. Histone Deacetylase Inhibitors Alter the Expression of Molecular Markers in Breast Cancer Cells via MicroRNAs. Int. J. Mol. Med. 2018, 42, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Nouriemamzaden, F.; Word, B.; Cotton, E.; Hawkins, A.; Littlejohn, K.; Moore, R.; Miranda-Carbon, G.; Orish, C.N.; Lyn-Cook, B. Modulation of Estrogen α and Progesterone Receptors in Triple Negative Breast Cancer Cell Lines: The Effects of Vorinostat and Indole-3-Carbinol in Vitro. Anticancer Res. 2020, 40, 3669–3683. [Google Scholar] [CrossRef]
- Plotkin, A.; Volmar, C.H.; Wahlestedt, C.; Ayad, N.; El-Ashry, D. Transcriptional Repression of ER through HMAPK Dependent Histone Deacetylation by Class I HDACs. Breast Cancer Res. Treat. 2014, 147, 249–263. [Google Scholar] [CrossRef]
- de Cremoux, P.; Dalvai, M.; N’Doye, O.; Moutahir, F.; Rolland, G.; Chouchane-Mlik, O.; Assayag, F.; Lehmann-Che, J.; Kraus-Berthie, L.; Nicolas, A.; et al. HDAC Inhibition Does Not Induce Estrogen Receptor in Human Triple-Negative Breast Cancer Cell Lines and Patient-Derived Xenografts. Breast Cancer Res. Treat. 2015, 149, 81–89. [Google Scholar] [CrossRef]
- Kavlashvili, T.; Jia, Y.; Dai, D.; Meng, X.; Thiel, K.W.; Leslie, K.K.; Yang, S. Inverse Relationship between Progesterone Receptor and Myc in Endometrial Cancer. PLoS ONE 2016, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xiao, X.; Jia, Y.; Liu, X.; Zhang, Y.; Wang, X.; Winters, C.; Devor, E.; Meng, X.; Thiel, K.; et al. Epigenetic Modification Restores Functional PR Expression in Endometrial Cancer Cells. Curr. Pharm. Des. 2014, 20, 1874–1880. [Google Scholar] [CrossRef] [Green Version]
- Rokhlin, O.W.; Glover, R.B.; Guseva, N.V.; Taghiyev, A.F.; Kohlgraf, K.G.; Cohen, M.B. Mechanisms of Cell Death Induced by Histone Deacetylase Inhibitors in Androgen Receptor-Positive Prostate Cancer Cells. Mol. Cancer Res. 2006, 4, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Welsbie, D.S.; Xu, J.; Chen, Y.; Borsu, L.; Scher, H.I.; Rosen, N.; Sawyers, C.L. Histone Deacetylases Are Required for Androgen Receptor Function in Hormone-Sensitive and Castrate-Resistant Prostate Cancer. Cancer Res. 2009, 69, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Katsushima, K.; Shinjo, K.; Hatanaka, A.; Ohka, F.; Suzuki, S.; Naiki-Ito, A.; Soga, N.; Takahashi, S.; Kondo, Y. Histone Deacetylase Inhibition in Prostate Cancer Triggers MiR-320-Mediated Suppression of the Androgen Receptor. Cancer Res. 2016, 76, 4192–4202. [Google Scholar] [CrossRef] [Green Version]
- Gravina, G.L.; Marampon, F.; Giusti, I.; Carosa, E.; Di Sante, S.; Ricevuto, E.; Dolo, V.; Tombolini, V.; Jannini, E.A.; Festuccia, C. Differential Effects of PXD101 (Belinostat) on Androgen-Dependent and Androgen-Independent Prostate Cancer Models. Int. J. Oncol. 2012, 40, 711–720. [Google Scholar] [CrossRef]
- Björkman, M.; Iljin, K.; Halonen, P.; Sara, H.; Kaivanto, E.; Nees, M.; Kallioniemi, O.P. Defining the Molecular Action of HDAC Inhibitors and Synergism with Androgen Deprivation in ERG-Positive Prostate Cancer. Int. J. Cancer 2008, 123, 2774–2781. [Google Scholar] [CrossRef]
- McCaw, T.R.; Randall, T.D.; Forero, A.; Buchsbaum, D.J. Modulation of Antitumor Immunity with Histone Deacetylase Inhibitors. Immunotherapy 2017, 9, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhao, W.; Yan, C.; Watson, C.C.; Massengill, M.; Xie, M.; Massengill, C.; Noyes, D.R.; Martinez, G.V.; Afzal, R.; et al. HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2016, 22, 4119–4132. [Google Scholar] [CrossRef] [Green Version]
- Burke, B.; Eden, C.; Perez, C.; Belshoff, A.; Hart, S.; Plaza-Rojas, L.; Delos Reyes, M.; Prajapati, K.; Voelkel-Johnson, C.; Henry, E.; et al. Inhibition of Histone Deacetylase (HDAC) Enhances Checkpoint Blockade Efficacy by Rendering Bladder Cancer Cells Visible for T Cell-Mediated Destruction. Front. Oncol. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Christmas, B.J.; Rafie, C.I.; Hopkins, A.C.; Scott, B.A.; Ma, H.S.; Cruz, K.A.; Woolman, S.; Armstrong, T.D.; Connolly, R.M.; Azad, N.A.; et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol. Res. 2018, 6, 1561–1577. [Google Scholar] [CrossRef] [Green Version]
- Bretz, A.C.; Parnitzke, U.; Kronthaler, K.; Dreker, T.; Bartz, R.; Hermann, F.; Ammendola, A.; Wulff, T.; Hamm, S. Domatinostat Favors the Immunotherapy Response by Modulating the Tumor Immune Microenvironment (TIME). J. Immunother. Cancer 2019, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.D.; Park, S.M.; Ha, H.C.; Lee, A.R.; Won, H.; Cha, H.; Cho, S.; Cho, J.M. HDAC Inhibitor, CG-745, Enhances the Anti-Cancer Effect of Anti-PD-1 Immune Checkpoint Inhibitor by Modulation of the Immune Microenvironment. J. Cancer 2020, 11, 4059–4072. [Google Scholar] [CrossRef] [Green Version]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC Inhibition Potentiates Immunotherapy in Triple Negative Breast Cancer. Oncotarget 2017, 8, 114156–114172. [Google Scholar] [CrossRef] [Green Version]
- Briere, D.; Sudhakar, N.; Woods, D.M.; Hallin, J.; Engstrom, L.D.; Aranda, R.; Chiang, H.; Sodré, A.L.; Olson, P.; Weber, J.S.; et al. The Class I/IV HDAC Inhibitor Mocetinostat Increases Tumor Antigen Presentation, Decreases Immune Suppressive Cell Types and Augments Checkpoint Inhibitor Therapy. Cancer Immunol. Immunother. 2018, 67, 381–392. [Google Scholar] [CrossRef]
- Shen, L.; Ciesielski, M.; Ramakrishnan, S.; Miles, K.M.; Ellis, L.; Sotomayor, P.; Shrikant, P.; Fenstermaker, R.; Pili, R. Class I Histone Deacetylase Inhibitor Entinostat Suppresses Regulatory T Cells and Enhances Immunotherapies in Renal and Prostate Cancer Models. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Hicks, K.C.; Fantini, M.; Donahue, R.N.; Schwab, A.; Knudson, K.M.; Tritsch, S.R.; Jochems, C.; Clavijo, P.E.; Allen, C.T.; Hodge, J.W.; et al. Epigenetic Priming of Both Tumor and NK Cells Augments Antibody-Dependent Cellular Cytotoxicity Elicited by the Anti-PD-L1 Antibody Avelumab against Multiple Carcinoma Cell Types. Oncoimmunology 2018, 7, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Kirkwood, J.; Dent, P. HDAC Inhibitors Enhance the Immunotherapy Response of Melanoma Cells. Oncotarget 2017, 8, 83155–83170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.C.; Lin, Y.C.; Liao, Y.H.; Liou, J.P.; Chen, C.H. MPT0G612, a Novel HDAC6 Inhibitor, Induces Apoptosis and Suppresses Ifn-Γ-Induced Programmed Death-Ligand 1 in Human Colorectal Carcinoma Cells. Cancers 2019, 11, 1617. [Google Scholar] [CrossRef] [Green Version]
- Knox, T.; Sahakian, E.; Banik, D.; Hadley, M.; Palmer, E.; Noonepalle, S.; Kim, J.; Powers, J.; Gracia-Hernandez, M.; Oliveira, V.; et al. Author Correction: Selective HDAC6 Inhibitors Improve Anti-PD-1 Immune Checkpoint Blockade Therapy by Decreasing the Anti-Inflammatory Phenotype of Macrophages and down-Regulation of Immunosuppressive Proteins in Tumor Cells. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Woods, D.M.; Sodré, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, S.S.; Cranmer, L.D. Vorinostat Synergizes with Ridaforolimus and Abrogates the Ridaforolimus-Induced Activation of AKT in Synovial Sarcoma Cells. BMC Res. Notes 2014, 7, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalingam, D.; Medina, E.C.; Esquivel, J.A.; Espitia, C.M.; Smith, S.; Oberheu, K.; Swords, R.; Kelly, K.R.; Mita, M.M.; Mita, A.C.; et al. Vorinostat Enhances the Activity of Temsirolimus in Renal Cell Carcinoma through Suppression of Survivin Levels. Clin. Cancer Res. 2010, 16, 141–153. [Google Scholar] [CrossRef] [Green Version]
- Thelen, P.; Krahn, L.; Bremmer, F.; Strauss, A.; Brehm, R.; Loertzer, H. Synergistic Effects of Histone Deacetylase Inhibitor in Combination with MTOR Inhibitor in the Treatment of Prostate Carcinoma. Int. J. Mol. Med. 2013, 31, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Wedel, S.; Hudak, L.; Seibel, J.M.; Makarević, J.; Juengel, E.; Tsaur, I.; Wiesner, C.; Haferkamp, A.; Blaheta, R.A. Impact of Combined HDAC and MTOR Inhibition on Adhesion, Migration and Invasion of Prostate Cancer Cells. Clin. Exp. Metastasis 2011, 28, 479–491. [Google Scholar] [CrossRef]
- Piao, J.; Chen, L.; Quan, T.; Li, L.; Quan, C.; Piao, Y.; Jin, T.; Lin, Z. Superior Efficacy of Co-Treatment with the Dual PI3K/MTOR Inhibitor BEZ235 and Histone Deacetylase Inhibitor Trichostatin A against NSCLC. Oncotarget 2016, 7, 60169–60180. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Ku, S.Y.; Ramakrishnan, S.; Lasorsa, E.; Azabdaftari, G.; Godoy, A.; Pili, R. Combinatorial Antitumor Effect of HDACs and the PI3K-Akt-MTOR Pathway Inhibition in a Pten Deficient Model of Prostate Cancer. Oncotarget 2013, 4, 2225–2236. [Google Scholar] [CrossRef] [Green Version]
- Makarević, J.; Rutz, J.; Juengel, E.; Maxeiner, S.; Mani, J.; Vallo, S.; Tsaur, I.; Roos, F.; Chun, F.; Blaheta, R. HDAC Inhibition Counteracts Metastatic Re-Activation of Prostate Cancer Cells Induced by Chronic MTOR Suppression. Cells 2018, 7, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juengel, E.; Najafi, R.; Rutz, J.; Maxeiner, S.; Makarevic, J.; Roos, F.; Tsaur, I.; Haferkamp, A.; Blaheta, R.A. HDAC Inhibition as a Treatment Concept to Combat Temsirolimusresistant Bladder Cancer Cells. Oncotarget 2017, 8, 110016–110028. [Google Scholar] [CrossRef] [Green Version]
- Wilson-Edell, K.A.; Yevtushenko, M.A.; Rothschild, D.E.; Rogers, A.N.; Benz, C.C. MTORC1/C2 and Pan-HDAC Inhibitors Synergistically Impair Breast Cancer Growth by Convergent AKT and Polysome Inhibiting Mechanisms. Breast Cancer Res. Treat. 2014, 144, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Sulaiman, A.; McGarry, S.; Lam, K.M.; El-Sahli, S.; Chambers, J.; Kaczmarek, S.; Li, L.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; et al. Co-Inhibition of MTORC1, HDAC and ESR1α Retards the Growth of Triple-Negative Breast Cancer and Suppresses Cancer Stem Cells. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malone, C.F.; Emerson, C.; Ingraham, R.; Barbosa, W.; Guerra, S.; Yoon, H.; Liu, L.L.; Michor, F.; Haigis, M.; Macleod, K.F.; et al. MTOR and HDAC Inhibitors Converge on the TXNIP/Thioredoxin Pathway to Cause Catastrophic Oxidative Stress and Regression of RAS-Driven Tumors. Cancer Discov. 2017, 7, 1450–1463. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Garrido-Laguna, I.; Naing, A.; Fu, S.; Falchook, G.S.; Piha-Paul, S.A.; Wheler, J.J.; Hong, D.S.; Tsimberidou, A.M.; Subbiah, V.; et al. Phase I Dose-Escalation Study of the MTOR Inhibitor Sirolimus and the HDAC Inhibitor Vorinostat in Patients with Advanced Malignancy. Oncotarget 2016, 7, 67521–67531. [Google Scholar] [CrossRef] [Green Version]
- Wood, A.; George, S.; Adra, N.; Chintala, S.; Damayanti, N.; Pili, R. Phase I Study of the MTOR Inhibitor Everolimus in Combination with the Histone Deacetylase Inhibitor Panobinostat in Patients with Advanced Clear Cell Renal Cell Carcinoma. Investig. New Drugs 2020, 38, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Zibelman, M.; Wong, Y.N.; Devarajan, K.; Malizzia, L.; Corrigan, A.; Olszanski, A.J.; Denlinger, C.S.; Roethke, S.K.; Tetzlaff, C.H.; Plimack, E.R. Phase i Study of the MTOR Inhibitor Ridaforolimus and the HDAC Inhibitor Vorinostat in Advanced Renal Cell Carcinoma and Other Solid Tumors. Investig. New Drugs 2015, 33, 1040–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Yuan, X.; Zhang, W.; Tang, M.; Zheng, L.; Wang, F.; Yan, W.; Yang, S.; Wei, Y.; He, J.; et al. Discovery of Novel Dual Histone Deacetylase and Mammalian Target of Rapamycin Target Inhibitors as a Promising Strategy for Cancer Therapy. J. Med. Chem. 2019, 62, 1577–1592. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E.; Kim, D.E.; Kim, M.J.; Lee, J.S.; Rho, J.K.; Jeong, S.Y.; Choi, E.K.; Kim, C.S.; Hwang, J.J. Vorinostat Enhances Gefitinib-Induced Cell Death through Reactive Oxygen Species-Dependent Cleavage of HSP90 and Its Clients in Non-Small Cell Lung Cancer with the EGFR Mutation. Oncol. Rep. 2019, 41, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Tanimoto, A.; Takeuchi, S.; Arai, S.; Fukuda, K.; Yamada, T.; Roca, X.; Ong, S.T.; Yano, S. Histone Deacetylase 3 Inhibition Overcomes BIM Deletion Polymorphism-Mediated Osimertinib Resistance in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2017, 23, 3139–3149. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Liang, C.; Song, W.; Tao, D.; Yao, J.; Wang, S.; Ma, L.; Shi, Y.; Han, X. Antitumor Activity of Histone Deacetylase Inhibitor Chidamide Alone or in Combination with Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Icotinib in NSCLC. J. Cancer 2019, 10, 1275–1287. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.; Chen, P.C.; Wu, W.S.; Wu, H.C.; Lan, C.H.; Huang, Y.H.; Cheng, C.H.; Chen, K.C.; Lin, C.W. Panobinostat Sensitizes KRAS-Mutant Non-Small-Cell Lung Cancer to Gefitinib by Targeting TAZ. Int. J. Cancer 2017, 141, 1921–1931. [Google Scholar] [CrossRef]
- Cai, X.; Zhai, H.X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-Ethynylphenylamino)-7-Methoxyquinazolin-6-Yloxy)-N- Hydroxyheptanamide (CUDC-101) as a Potent Multi-Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef]
- Lai, C.J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a Multitargeted Inhibitor of Histone Deacetylase, Epidermal Growth Factor Receptor, and Human Epidermal Growth Factor Receptor 2, Exerts Potent Anticancer Activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, Y.; Mehta, A.; Boufraqech, M.; Davis, S.; Wang, J.; Tian, Z.; Yu, Z.; Boxer, M.B.; Kiefer, J.A.; et al. Dual Inhibition of HDAC and EGFR Signaling with CUDC-101 Induces Potent Suppression of Tumor Growth and Metastasis in Anaplastic Thyroid Cancer. Oncotarget 2015, 6, 9073–9085. [Google Scholar] [CrossRef] [Green Version]
- Moertl, S.; Payer, S.; Kell, R.; Winkler, K.; Anastasov, N.; Atkinson, M.J. Comparison of Radiosensitization by HDAC Inhibitors CUDC-101 and SAHA in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3259. [Google Scholar] [CrossRef] [Green Version]
- Ji, M.; Li, Z.; Lin, Z.; Chen, L. Antitumor Activity of the Novel HDAC Inhibitor CUDC-101 Combined with Gemcitabine in Pancreatic Cancer. Am. J. Cancer Res. 2018, 8, 2402–2418. [Google Scholar]
- Citro, S.; Bellini, A.; Miccolo, C.; Ghiani, L.; Carey, T.E.; Chiocca, S. Synergistic Antitumour Activity of HDAC Inhibitor SAHA and EGFR Inhibitor Gefitinib in Head and Neck Cancer: A Key Role for ΔNp63α. Br. J. Cancer 2019, 120, 658–667. [Google Scholar] [CrossRef] [Green Version]
- Liffers, K.; Kolbe, K.; Westphal, M.; Lamszus, K.; Schulte, A. Histone Deacetylase Inhibitors Resensitize EGFR/EGFRvIII-Overexpressing, Erlotinib-Resistant Glioblastoma Cells to Tyrosine Kinase Inhibition. Target. Oncol. 2016, 11, 29–40. [Google Scholar] [CrossRef]
- Qian, C.; Lai, C.J.; Bao, R.; Wang, D.G.; Wang, J.; Xu, G.X.; Atoyan, R.; Qu, H.; Yin, L.; Samson, M.; et al. Cancer Network Disruption by a Single Molecule Inhibitor Targeting Both Histone Deacetylase Activity and Phosphatidylinositol 3-Kinase Signaling. Clin. Cancer Res. 2012, 18, 4104–4113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaselli, D.; Lucidi, A.; Rotili, D.; Mai, A. Epigenetic Polypharmacology: A New Frontier for Epi-Drug Discovery. Med. Res. Rev. 2020, 40, 190–244. [Google Scholar] [CrossRef] [PubMed]
- Ranganna, K.; Selvam, C.; Shivachar, A.; Yousefipour, Z. Histone Deacetylase Inhibitors as Multitarget-Directed Epi-Drugs in Blocking Pi3k Oncogenic Signaling: A Polypharmacology Approach. Int. J. Mol. Sci. 2020, 21, 8198. [Google Scholar] [CrossRef]
- Hu, C.; Xia, H.; Bai, S.; Zhao, J.; Edwards, H.; Li, X.; Yang, Y.; Lyu, J.; Wang, G.; Zhan, Y.; et al. CUDC-907, a Novel Dual PI3K and HDAC Inhibitor, in Prostate Cancer: Antitumour Activity and Molecular Mechanism of Action. J. Cell. Mol. Med. 2020, 24, 7239–7253. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.A.; Pinheiro, P.S.M.; Fraga, C.A.M. Multitarget Inhibition of Histone Deacetylase (HDAC) and Phosphatidylinositol-3-Kinase (PI3K): Current and Future Prospects. ChemMedChem 2020. [Google Scholar] [CrossRef]
- Fu, X.H.; Zhang, X.; Yang, H.; Xu, X.W.; Hu, Z.L.; Yan, J.; Zheng, X.L.; Wei, R.R.; Zhang, Z.Q.; Tang, S.R.; et al. CUDC-907 Displays Potent Antitumor Activity against Human Pancreatic Adenocarcinoma in Vitro and in Vivo through Inhibition of HDAC6 to Downregulate c-Myc Expression. Acta Pharmacol. Sin. 2019, 40, 677–688. [Google Scholar] [CrossRef]
- Kotian, S.; Zhang, L.; Boufraqech, M.; Gaskins, K.; Gara, S.K.; Quezado, M.; Nilubol, N.; Kebebew, E. Dual Inhibition of HDAC and Tyrosine Kinase Signaling Pathways with CUDC-907 Inhibits Thyroid Cancer Growth and Metastases. Clin. Cancer Res. 2017, 23, 5044–5054. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Bian, X.; Lin, W. The Dual HDAC-PI3K Inhibitor CUDC-907 Displays Single-Agent Activity and Synergizes with PARP Inhibitor Olaparib in Small Cell Lung Cancer. J. Exp. Clin. Cancer Res. 2020, 39, 1–14. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.W.; Fu, L.W. CUDC-907, a Dual HDAC and PI3K Inhibitor, Reverses Platinum Drug Resistance. Investig. New Drugs 2018, 36, 10–19. [Google Scholar] [CrossRef]
- Shimizu, T.; LoRusso, P.M.; Papadopoulos, K.P.; Patnaik, A.; Beeram, M.; Smith, L.S.; Rasco, D.W.; Mays, T.A.; Chambers, G.; Ma, A.; et al. Phase I First-in-Human Study of CUDC-101, a Multitargeted Inhibitor of HDACs, EGFR, and HER2 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2014, 20, 5032–5040. [Google Scholar] [CrossRef] [Green Version]
- Galloway, T.J.; Wirth, L.J.; Colevas, A.D.; Gilbert, J.; Bauman, J.E.; Saba, N.F.; Raben, D.; Mehra, R.; Ma, A.W.; Atoyan, R.; et al. A Phase I Study of CUDC-101, a Multitarget Inhibitor of HDACs, EGFR, and HER2, in Combination with Chemoradiation in Patients with Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 1566–1573. [Google Scholar] [CrossRef] [Green Version]
- Lim, B.; Murthy, R.K.; Lee, J.; Jackson, S.A.; Iwase, T.; Davis, D.W.; Willey, J.S.; Wu, J.; Shen, Y.; Tripathy, D.; et al. A Phase Ib Study of Entinostat plus Lapatinib with or without Trastuzumab in Patients with HER2-Positive Metastatic Breast Cancer That Progressed during Trastuzumab Treatment. Br. J. Cancer 2019, 120, 1105–1112. [Google Scholar] [CrossRef]
- Goldstein, L.J.; Zhao, F.; Wang, M.; Swaby, R.F.; Sparano, J.A.; Meropol, N.J.; Bhalla, K.N.; Pellegrino, C.M.; Katherine Alpaugh, R.; Falkson, C.I.; et al. A Phase I/II Study of Suberoylanilide Hydroxamic Acid (SAHA) in Combination with Trastuzumab (Herceptin) in Patients with Advanced Metastatic and/or Local Chest Wall Recurrent HER2-Amplified Breast Cancer: A Trial of the ECOG-ACRIN Cancer Research Group. Breast Cancer Res. Treat. 2017, 165, 375–382. [Google Scholar] [CrossRef]
- Gray, J.E.; Haura, E.; Chiappori, A.; Tanvetyanon, T.; Williams, C.C.; Pinder-Schenck, M.; Kish, J.A.; Kreahling, J.; Lush, R.; Neuger, A.; et al. A Phase i, Pharmacokinetic, and Pharmacodynamic Study of Panobinostat, an HDAC Inhibitor, Combined with Erlotinib in Patients with Advanced Aerodigestive Tract Tumors. Clin. Cancer Res. 2014, 20, 1644–1655. [Google Scholar] [CrossRef] [Green Version]
- Han, J.Y.; Lee, S.H.; Lee, G.K.; Yun, T.; Lee, Y.J.; Hwang, K.H.; Kim, J.Y.; Kim, H.T. Phase I/II Study of Gefitinib (Iressa®) and Vorinostat (IVORI) in Previously Treated Patients with Advanced Non-Small Cell Lung Cancer. Cancer Chemother. Pharmacol. 2015, 75, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Witta, S.E.; Jotte, R.M.; Konduri, K.; Neubauer, M.A.; Spira, A.I.; Ruxer, R.L.; Varella-Garcia, M.; Bunn, P.A.; Hirsch, F.R. Randomized Phase II Trial of Erlotinib with and without Entinostat in Patients with Advanced Non-Small-Cell Lung Cancer Who Progressed on Prior Chemotherapy. J. Clin. Oncol. 2012, 30, 2248–2255. [Google Scholar] [CrossRef]
- Brodie, A.M.H.; Sabnis, G.J.; Chumsri, S.; Brodie, A.M.H.; Chumsri, S.; Sukumar, S.; Brodie, A.M.H. Extending Aromatase Inhibitor Sensitivity in Hormone Resistant Breast Cancer. Horm. Mol. Biol. Clin. Investig. 2011, 5, 97–103. [Google Scholar] [CrossRef]
- Sabnis, G.J.; Goloubeva, O.G.; Kazi, A.A.; Shah, P.; Brodie, A.H. HDAC Inhibitor Entinostat Restores Responsiveness of Letrozole-Resistant MCF-7Ca Xenografts to Aromatase Inhibitors through Modulation of Her-2. Mol. Cancer Ther. 2013, 12, 2804–2816. [Google Scholar] [CrossRef] [Green Version]
- Hodges-Gallagher, L.; Valentine, C.D.; El Bader, S.; Kushner, P.J. Inhibition of Histone Deacetylase Enhances the Anti-Proliferative Action of Antiestrogens on Breast Cancer Cells and Blocks Tamoxifen-Induced Proliferation of Uterine Cells. Breast Cancer Res. Treat. 2007, 105, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Kanaya, N.; Petrossian, K.; Ye, J.; Warden, C.; Liu, Z.; Nishimura, R.; Osako, T.; Okido, M.; Shimada, K.; et al. Inhibition of the Proliferation of Acquired Aromatase Inhibitor-Resistant Breast Cancer Cells by Histone Deacetylase Inhibitor LBH589 (Panobinostat). Breast Cancer Res. Treat. 2013, 137, 93–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza-Sanchez, R.; Cotnoir-White, D.; Kulpa, J.; Jutras, I.; Pottel, J.; Moitessier, N.; Mader, S.; Gleason, J.L. Design, Synthesis and Evaluation of Antiestrogen and Histone Deacetylase Inhibitor Molecular Hybrids. Bioorg. Med. Chem. 2015, 23, 7597–7606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Li, C.; Zhang, S.; Hu, Z.; Wu, J.; Dong, C.; Huang, J.; Zhou, H.B. Novel Bioactive Hybrid Compound Dual Targeting Estrogen Receptor and Histone Deacetylase for the Treatment of Breast Cancer. J. Med. Chem. 2015, 58, 4550–4572. [Google Scholar] [CrossRef]
- Gryder, B.E.; Rood, M.K.; Johnson, K.A.; Patil, V.; Raftery, E.D.; Yao, L.P.D.; Rice, M.; Azizi, B.; Doyle, D.F.; Oyelere, A.K. Histone Deacetylase Inhibitors Equipped with Estrogen Receptor Modulation Activity. J. Med. Chem. 2013, 56, 5782–5796. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.W.; Allred, J.B.; Moreno-Aspitia, A.; Northfelt, D.W.; Ingle, J.N.; Goetz, M.P.; Perez, E.A. Phase i Study of Panobinostat (LBH589) and Letrozole in Postmenopausal Metastatic Breast Cancer Patients. Clin. Breast Cancer 2016, 16, 82–86. [Google Scholar] [CrossRef] [Green Version]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized Phase II, Double-Blind, Placebo-Controlled Study of Exemestane with or without Entinostat in Postmenopausal Women with Locally Recurrent or Metastatic Estrogen Receptor-Positive Breast Cancer Progressing on Treatment with a Nonsteroidal Aromata. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar] [CrossRef] [Green Version]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A Phase II Study of the Histone Deacetylase Inhibitor Vorinostat Combined with Tamoxifen for the Treatment of Patients with Hormone Therapy-Resistant Breast Cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus Exemestane for Postmenopausal Patients with Advanced, Hormone Receptor-Positive Breast Cancer (ACE): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Pili, R.; Quinn, D.I.; Hammers, H.J.; Monk, P.; George, S.; Dorff, T.B.; Olencki, T.; Shen, L.; Orillion, A.; Lamonica, D.; et al. Immunomodulation by Entinostat in Renal Cell Carcinoma Patients Receiving High-Dose Interleukin 2: A Multicenter, Single-Arm, Phase I/II Trial (NCI-CTEP#7870). Clin. Cancer Res. 2017, 23, 7199–7208. [Google Scholar] [CrossRef] [Green Version]
- Gray, J.E.; Saltos, A.; Tanvetyanon, T.; Haura, E.B.; Creelan, B.; Antonia, S.J.; Shafique, M.; Zheng, H.; Dai, W.; Saller, J.J.; et al. Phase I/Ib Study of Pembrolizumab plus Vorinostat in Advanced/Metastatic Non–Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6623–6632. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.P.; Wu, Q.; Voutsinas, J.; Fromm, J.R.; Jiang, X.; Pillarisetty, V.G.; Lee, S.M.; Santana-Davila, R.; Goulart, B.; Baik, C.S.; et al. A Phase II Trial of Pembrolizumab and Vorinostat in Recurrent Metastatic Head and Neck Squamous Cell Carcinomas and Salivary Gland Cancer. Clin. Cancer Res. 2020, 26, 837–845. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Janne, P.A.; Opyrchal, M.; Hafez, N.; Raez, L.E.; Gabrilovich, D.I.; Wang, F.; Trepel, J.B.; Lee, M.-J.; Yuno, A.; et al. Entinostat plus Pembrolizumab in Patients with Metastatic NSCLC Previously Treated with Anti-PD-(L)1 Therapy. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Van Tilburg, C.M.; Van Tilburg, C.M.; Van Tilburg, C.M.; Witt, R.; Witt, R.; Heiss, M.; Heiss, M.; Pajtler, K.W.; Pajtler, K.W.; Pajtler, K.W.; et al. INFORM2 NivEnt: The First Trial of the INFORM2 Biomarker Driven Phase I/II Trial Series: The Combination of Nivolumab and Entinostat in Children and Adolescents with Refractory High-Risk Malignancies. BMC Cancer 2020, 20, 1–11. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Scripture, C.D.; Figg, W.D. Drug Interactions in Cancer Therapy. Nat. Rev. Cancer 2006, 6, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Kay, C.; Rankovic, Z. From Magic Bullets to Designed Multiple Ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A Perspective on Multi-Target Drug Discovery and Design for Complex Diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Lötsch, J.; Geisslinger, G. Low-Dose Drug Combinations along Molecular Pathways Could Maximize Therapeutic Effectiveness While Minimizing Collateral Adverse Effects. Drug Discov. Today 2011, 16, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- De Lera, A.R.; Ganesan, A. Two-Hit Wonders: The Expanding Universe of Multitargeting Epigenetic Agents. Curr. Opin. Chem. Biol. 2020, 57, 135–154. [Google Scholar] [CrossRef]
- De Lera, A.R.; Ganesan, A. Epigenetic Polypharmacology: From Combination Therapy to Multitargeted Drugs. Clin. Epigenet. 2016, 8, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smalley, J.P.; Cowley, S.M.; Hodgkinson, J.T. Bifunctional HDAC Therapeutics: One Drug to Rule Them All? Molecules 2020, 25, 4394. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Luan, Y.; Li, J.; Bernatchez, J.A.; Li, R. Kinase and Histone Deacetylase Hybrid Inhibitors for Cancer Therapy. J. Med. Chem. 2019, 62, 3171–3183. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wan, Y.; Xiao, Y.; Xia, C.; Duan, G. Dual-Target Inhibitors Based on HDACs: Novel Antitumor Agents for Cancer Therapy. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Li, Y.; Tang, P.; Yuan, Q. Rational Design, Synthesis and Preliminary Antitumor Activity Evaluation of a Chlorambucil Derivative with Potent DNA/HDAC Dual-Targeting Inhibitory Activity. Bioorg. Med. Chem. Lett. 2017, 27, 4415–4420. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Tang, P.; Yuan, Q. Rational Design and Characterization of a DNA/HDAC Dual-Targeting Inhibitor Containing Nitrogen Mustard and 2-Aminobenzamide Moieties. MedChemComm 2018, 9, 344–352. [Google Scholar] [CrossRef]
- Sinatra, L.; Bandolik, J.J.; Roatsch, M.; Sönnichsen, M.; Schoeder, C.T.; Hamacher, A.; Schöler, A.; Borkhardt, A.; Meiler, J.; Bhatia, S.; et al. Hydroxamic Acids Immobilized on Resins (HAIRs): Synthesis of Dual-Targeting HDAC Inhibitors and HDAC Degraders (PROTACs). Angew. Chem. Int. Ed. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhijun, H.; Shusheng, W.; Han, M.; Jianping, L.; Li-sen, Q.; Dechun, L. Pre-Clinical Characterization of 4SC-202, a Novel Class I HDAC Inhibitor, against Colorectal Cancer Cells. Tumor Biol. 2016, 37, 10257–10267. [Google Scholar] [CrossRef]
- Morera, L.; Lübbert, M.; Jung, M. Targeting Histone Methyltransferases and Demethylases in Clinical Trials for Cancer Therapy. Clin. Epigenet. 2016, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Wobser, M.; Weber, A.; Glunz, A.; Tauch, S.; Seitz, K.; Butelmann, T.; Hesbacher, S.; Goebeler, M.; Bartz, R.; Kohlhof, H.; et al. Elucidating the Mechanism of Action of Domatinostat (4SC-202) in Cutaneous T Cell Lymphoma Cells. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.C.; Ma, Y.C.; Qin, W.P.; Ding, L.N.; Zheng, Y.C.; Zhu, Y.L.; Zhai, X.Y.; Yang, J.; Ma, C.Y.; Guan, Y.Y. Design and Synthesis of Tranylcypromine Derivatives as Novel LSD1/HDACs Dual Inhibitors for Cancer Treatment. Eur. J. Med. Chem. 2017. [Google Scholar] [CrossRef]
- Kalin, J.H.; Wu, M.; Gomez, A.V.; Song, Y.; Das, J.; Hayward, D.; Adejola, N.; Wu, M.; Panova, I.; Chung, H.J.; et al. Targeting the CoREST Complex with Dual Histone Deacetylase and Demethylase Inhibitors. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Guerrant, W.; Patil, V.; Canzoneri, J.C.; Oyelere, A.K. Dual Targeting of Histone Deacetylase and Topoisomerase II with Novel Bifunctional Inhibitors. J. Med. Chem. 2012, 55, 1465–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrant, W.; Patil, V.; Canzoneri, J.C.; Yao, L.P.; Hood, R.; Oyelere, A.K. Dual-Acting Histone Deacetylase-Topoisomerase i Inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 3283–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cincinelli, R.; Musso, L.; Artali, R.; Guglielmi, M.B.; La Porta, I.; Melito, C.; Colelli, F.; Cardile, F.; Signorino, G.; Fucci, A.; et al. Hybrid Topoisomerase i and HDAC Inhibitors as Dual Action Anticancer Agents. PLoS ONE 2018, 13, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.H. Dual Inhibitors against Topoisomerases and Histone Deacetylases. J. Cancer Prev. 2015, 20, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.J.; Lin, Y.J.; Chao, M.W.; Sung, T.Y.; Wu, Y.W.; Chen, Y.Y.; Lin, M.H.; Liou, J.P.; Pan, S.L.; Yang, C.R. The Anticancer Effects of MPT0G211, a Novel HDAC6 Inhibitor, Combined with Chemotherapeutic Agents in Human Acute Leukemia Cells. Clin. Epigenet. 2018, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Reßing, N.; Sönnichsen, M.; Osko, J.; Schöler, A.; Schliehe-Diecks, J.; Skerhut, A.J.; Borkhardt, A.; Hauer, J.; Kassack, M.U.; Christianson, D.W.; et al. Multicomponent Synthesis, Binding Mode and Structure-Activity Relationships of Selective Histone Deacetylase 6 (HDAC6) Inhibitors with Bifurcated Capping Groups. J. Med. Chem. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, S.J.; Soden, P.E.; Angell, D.C.; Bantscheff, M.; Chung, C.W.; Giblin, K.A.; Smithers, N.; Furze, R.C.; Gordon, L.; Drewes, G.; et al. The Structure Based Design of Dual HDAC/BET Inhibitors as Novel Epigenetic Probes. MedChemComm 2014, 5, 342–351. [Google Scholar] [CrossRef]
- Tang, F.; Yang, Z.; Tan, Y.; Li, Y. Super-Enhancer Function and Its Application in Cancer Targeted Therapy. NPJ Precis. Oncol. 2020, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Hou, S.; Chen, H.; Ran, T.; Jiang, F.; Bian, Y.; Zhang, D.; Zhi, Y.; Wang, L.; Zhang, L.; et al. Targeting Epigenetic Reader and Eraser: Rational Design, Synthesis and in Vitro Evaluation of Dimethylisoxazoles Derivatives as BRD4/HDAC Dual Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 2931–2935. [Google Scholar] [CrossRef]
- Noguchi-Yachide, T.; Sakai, T.; Hashimoto, Y.; Yamaguchi, T. Discovery and Structure-Activity Relationship Studies of N6-Benzoyladenine Derivatives as Novel BRD4 Inhibitors. Bioorg. Med. Chem. 2015, 23, 953–959. [Google Scholar] [CrossRef]
- He, S.; Dong, G.; Li, Y.; Wu, S.; Wang, W.; Sheng, C. Potent Dual BET/HDAC Inhibitors for Efficient Treatment of Pancreatic Cancer. Angew. Chem. Int. Ed. 2020, 59, 3028–3032. [Google Scholar] [CrossRef]
- Pinzi, L.; Benedetti, R.; Altucci, L.; Rastelli, G. Design of Dual Inhibitors of Histone Deacetylase 6 and Heat Shock Protein 90. ACS Omega 2020, 5, 11473–11480. [Google Scholar] [CrossRef]
- Ojha, R.; Huang, H.L.; HuangFu, W.C.; Wu, Y.W.; Nepali, K.; Lai, M.J.; Su, C.J.; Sung, T.Y.; Chen, Y.L.; Pan, S.L.; et al. 1-Aroylindoline-Hydroxamic Acids as Anticancer Agents, Inhibitors of HSP90 and HDAC. Eur. J. Med. Chem. 2018, 150, 667–677. [Google Scholar] [CrossRef]
- Mehndiratta, S.; Lin, M.H.; Wu, Y.W.; Chen, C.H.; Wu, T.Y.; Chuang, K.H.; Chao, M.W.; Chen, Y.Y.; Pan, S.L.; Chen, M.C.; et al. N-Alkyl-Hydroxybenzoyl Anilide Hydroxamates as Dual Inhibitors of HDAC and HSP90, Downregulating IFN-γ Induced PD-L1 Expression. Eur. J. Med. Chem. 2020, 185, 111725. [Google Scholar] [CrossRef] [PubMed]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.T.; Leisten, E.D.; Tang, W. Development of the First Small Molecule Histone Deacetylase 6 (HDAC6) Degraders. Bioorg. Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yang, K.; Zhang, Z.; Leisten, E.D.; Li, Z.; Xie, H.; Liu, J.; Smith, K.A.; Novakova, Z.; Barinka, C.; et al. Development of Multifunctional Histone Deacetylase 6 Degraders with Potent Antimyeloma Activity. J. Med. Chem. 2019, 62, 7042–7057. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Lv, W.; Su, S.; Wu, W.; Rao, Y. Developing Potent PROTACs Tools for Selective Degradation of HDAC6 Protein. Protein Cell 2019, 10, 606–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Lv, W.; He, M.; Deng, H.; Li, H.; Wu, W.; Rao, Y. Plasticity in Designing PROTACs for Selective and Potent Degradation of HDAC6. Chem. Commun. 2019, 55, 14848–14851. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhao, Y.; Nie, X.; Wu, H.; Wang, B.; Almodovar-Rivera, C.M.; Xie, H.; Tang, W. A Cell-Based Target Engagement Assay for the Identification of Cereblon E3 Ubiquitin Ligase Ligands and Their Application in HDAC6 Degraders. Cell Chem. Biol. 2020, 1–11. [Google Scholar] [CrossRef]
- Yang, K.; Wu, H.; Zhang, Z.; Leisten, E.D.; Nie, X.; Liu, B.; Wen, Z.; Zhang, J.; Cunningham, M.D.; Tang, W. Development of Selective Histone Deacetylase 6 (HDAC6) Degraders Recruiting von Hippel-Lindau (VHL) E3 Ubiquitin Ligase. ACS Med. Chem. Lett. 2020, 11, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Smalley, J.P.; Adams, G.E.; Millard, C.J.; Song, Y.; Norris, J.K.S.; Schwabe, J.W.R.; Cowley, S.M.; Hodgkinson, J.T. PROTAC-Mediated Degradation of Class I Histone Deacetylase Enzymes in Corepressor Complexes. Chem. Commun. 2020, 56, 4476–4479. [Google Scholar] [CrossRef]
- Roatsch, M.; Vogelmann, A.; Herp, D.; Jung, M.; Olsen, C.A. Proteolysis-Targeting Chimeras (PROTACs) Based on Macrocyclic Tetrapeptides Selectively Degrade Class I Histone Deacetylases 1–3. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Cao, F.; de Weerd, S.; Chen, D.; Zwinderman, M.R.H.; van der Wouden, P.E.; Dekker, F.J. Induced Protein Degradation of Histone Deacetylases 3 (HDAC3) by Proteolysis Targeting Chimera (PROTAC). Eur. J. Med. Chem. 2020, 208, 112800. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, J.; Zhao, L.Y.; Chen, X.; Zheng, G.; Zhang, X.; Liao, D. Discovery of Histone Deacetylase 3 (HDAC3)-Specific PROTACs. Chem. Commun. 2020, 56, 9866–9869. [Google Scholar] [CrossRef]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted Protein Degradation: Expanding the Toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.H.R.; Rasmusson, T.; Robinson, J.; Pachl, F.; Read, J.; Kawatkar, S.; O’ Donovan, D.H.; Bagal, S.; Code, E.; Rawlins, P.; et al. EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem. Biol. 2020, 27, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.Y.; Zhao, Y.T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Thomas, S.; Pawlowska, N.; Bartelink, I.; Grabowsky, J.; Jahan, T.; Cripps, A.; Harb, A.; Leng, J.; Reinert, A.; et al. Inhibiting Histone Deacetylase as a Means to Reverse Resistance to Angiogenesis Inhibitors: Phase i Study of Abexinostat plus Pazopanib in Advanced Solid Tumor Malignancies. J. Clin. Oncol. 2017, 35, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.; Flamand, Y.; Balasubramanian, S.; Butrynski, J.E.; Harmon, D.C.; George, S.; Cote, G.M.; Wagner, A.J.; Morgan, J.A.; Sirisawad, M.; et al. Phase 1 Study of Oral Abexinostat, a Histone Deacetylase Inhibitor, in Combination with Doxorubicin in Patients with Metastatic Sarcoma. Cancer 2015, 121, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Moyal, E.C.J.; Gregorc, V.; Zucali, P.A.; Menard, J.; Soria, J.C.; Kloos, I.; Hsu, J.; Luan, Y.; Liu, E.; et al. A Phase 1 Dose-Escalation Study of the Oral Histone Deacetylase Inhibitor Abexinostat in Combination with Standard Hypofractionated Radiotherapy in Advanced Solid Tumors. Oncotarget 2017, 8, 56199–56209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouliard, S.; Robert, R.; Jacquet-Bescond, A.; Du Rieu, Q.C.; Balasubramanian, S.; Loury, D.; Loriot, Y.; Hollebecque, A.; Kloos, I.; Soria, J.C.; et al. Pharmacokinetic/Pharmacodynamic Modelling-Based Optimisation of Administration Schedule for the Histone Deacetylase Inhibitor Abexinostat (S78454/PCI-24781) in Phase I. Eur. J. Cancer 2013, 49, 2791–2797. [Google Scholar] [CrossRef]
- Ryan, Q.C.; Headlee, D.; Acharya, M.; Sparreboom, A.; Trepel, J.B.; Ye, J.; Figg, W.D.; Hwang, K.; Chung, E.J.; Murgo, A.; et al. Phase I and Pharmacokinetic Study of MS-275, a Histone Deacetylase Inhibitor, in Patients with Advanced and Refractory Solid Tumors or Lymphoma. J. Clin. Oncol. 2005, 23, 3912–3922. [Google Scholar] [CrossRef] [PubMed]
- Pili, R.; Salumbides, B.; Zhao, M.; Altiok, S.; Qian, D.; Zwiebel, J.; Carducci, M.A.; Rudek, M.A. Phase i Study of the Histone Deacetylase Inhibitor Entinostat in Combination with 13-Cis Retinoic Acid in Patients with Solid Tumours. Br. J. Cancer 2012, 106, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Ngamphaiboon, N.; Dy, G.K.; Ma, W.W.; Zhao, Y.; Reungwetwattana, T.; Depaolo, D.; Ding, Y.; Brady, W.; Fetterly, G.; Adjei, A.A. A Phase i Study of the Histone Deacetylase (HDAC) Inhibitor Entinostat, in Combination with Sorafenib in Patients with Advanced Solid Tumors. Investig. New Drugs 2015, 33, 225–232. [Google Scholar] [CrossRef]
- Wang, S.; Huang, J.; Lyu, H.; Lee, C.K.; Tan, J.; Wang, J.; Liu, B. Functional Cooperation of MiR-125a, MiR-125b, and MiR-205 in Entinostat-Induced Downregulation of ErbB2/ErbB3 and Apoptosis in Breast Cancer Cells. Cell Death Dis. 2013. [Google Scholar] [CrossRef] [Green Version]
- Kummar, S.; Gutierrez, M.; Gardner, E.R.; Donovan, E.; Hwang, K.; Eun, J.C.; Lee, M.J.; Maynard, K.; Kalnitskiy, M.; Chen, A.; et al. Phase I Trial of MS-275, a Histone Deacetylase Inhibitor, Administered Weekly in Refractory Solid Tumors and Lymphoid Malignancies. Clin. Cancer Res. 2007, 13, 5411–5417. [Google Scholar] [CrossRef] [Green Version]
- Connolly, R.M.; Li, H.; Jankowitz, R.C.; Zhang, Z.; Rudek, M.A.; Jeter, S.C.; Slater, S.A.; Powers, P.; Wolff, A.C.; Fetting, J.H.; et al. Combination Epigenetic Therapy in Advanced Breast Cancer with 5-Azacitidine and Entinostat: A Phase II National Cancer Institute/Stand up to Cancer Study. Clin. Cancer Res. 2017, 23, 2691–2701. [Google Scholar] [CrossRef] [Green Version]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and Regional Mortality from 235 Causes of Death for 20 Age Groups in 1990 and 2010: A Systematic Analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Azad, N.S.; El-Khoueiry, A.; Yin, J.; Oberg, A.L.; Flynn, P.; Adkins, D.; Sharma, A.; Weisenberger, D.J.; Brown, T.; Medvari, P.; et al. Combination Epigenetic Therapy in Metastatic Colorectal Cancer (MCRC) with Subcutaneous 5-Azacitidine and Entinostat; a Phase 2 Consortium/Stand Up 2 Cancer Study. Oncotarget 2017, 8, 35326–35338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, R.P.; Rankin, C.; Hoff, P.M.G.; Gold, P.J.; Billingsley, K.G.; Chapman, R.A.; Wong, L.; Ward, J.H.; Abbruzzese, J.L.; Blanke, C.D. Phase II Trial of Romidepsin (NSC-630176) in Previously Treated Colorectal Cancer Patients with Advanced Disease: A Southwest Oncology Group Study (S0336). Investig. New Drugs 2009, 27, 469–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otterson, G.A.; Hodgson, L.; Pang, H.; Vokes, E.E. Phase II Study of the Histone Deacetylase Inhibitor Romidepsin in Relapsed Small Cell Lung Cancer (Cancer and Leukemia Group B 30304). J. Thorac. Oncol. 2010, 5, 1644–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molife, L.R.; Attard, G.; Fong, P.C.; Karavasilis, V.; Reid, A.H.M.; Patterson, S.; Riggs, C.E.; Higano, C.; Stadler, W.M.; McCulloch, W.; et al. Phase II, Two-Stage, Single-Arm Trial of the Histone Deacetylase Inhibitor (HDACi) Romidepsin in Metastatic Castration-Resistant Prostate Cancer (CRPC). Ann. Oncol. 2009, 21, 109–113. [Google Scholar] [CrossRef]
- Amiri-Kordestani, L.; Luchenko, V.; Peer, C.J.; Ghafourian, K.; Reynolds, J.; Draper, D.; Frye, R.; Woo, S.; Venzon, D.; Wright, J.; et al. Phase i Trial of a New Schedule of Romidepsin in Patients with Advanced Cancers. Clin. Cancer Res. 2013, 19, 4499–4507. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.F.; Infante, J.R.; Spigel, D.R.; Peacock, N.W.; Thompson, D.S.; Greco, F.A.; McCulloch, W.; Burris, H.A., III. Phase 1 Results from a Study of Romidepsin in Combination with Gemcitabine in Patients with Advanced Solid Tumors. Cancer Invest. 2012, 30, 481–486. [Google Scholar] [CrossRef]
- Iwamoto, F.M.; Lamborn, K.R.; Kuhn, J.G.; Wen, P.Y.; Alfred Yung, W.K.; Gilbert, M.R.; Chang, S.M.; Lieberman, F.S.; Prados, M.D.; Fine, H.A. A Phase I/II Trial of the Histone Deacetylase Inhibitor Romidepsin for Adults with Recurrent Malignant Glioma: North American Brain Tumor Consortium Study 03-03. Neuro-Oncol. 2011, 13, 509–516. [Google Scholar] [CrossRef]
- Haigentz, M.; Kim, M.; Sarta, C.; Lin, J.; Keresztes, R.S.; Culliney, B.; Gaba, A.G.; Smith, R.V.; Shapiro, G.I.; Chirieac, L.R.; et al. Phase II Trial of the Histone Deacetylase Inhibitor Romidepsin in Patients with Recurrent/Metastatic Head and Neck Cancer. Oral Oncol. 2012, 48, 1281–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, D.E.; Boothman, D.A.; Fattah, F.J.; Dong, Y.; Zhu, H.; Skelton, R.A.; Priddy, L.L.; Vo, P.; Dowell, J.E.; Sarode, V.; et al. Phase 1 Study of Romidepsin plus Erlotinib in Advanced Non-Small Cell Lung Cancer. Lung Cancer 2015, 90, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, S.L.; Zahurak, M.; Sharma, A.; Durham, J.N.; Reiss, K.A.; Sartorius-Mergenthaler, S.; Downs, M.; Anders, N.M.; Ahuja, N.; Rudek, M.A.; et al. A Phase 1 Trial of the Oral DNA Methyltransferase Inhibitor CC-486 and the Histone Deacetylase Inhibitor Romidepsin in Advanced Solid Tumors. Cancer 2019, 125, 2837–2845. [Google Scholar] [CrossRef]
- Dong, M.; Ning, Z.Q.; Xing, P.Y.; Xu, J.L.; Cao, H.X.; Dou, G.F.; Meng, Z.Y.; Shi, Y.K.; Lu, X.P.; Feng, F.Y. Phase i Study of Chidamide (CS055/HBI-8000), a New Histone Deacetylase Inhibitor, in Patients with Advanced Solid Tumors and Lymphomas. Cancer Chemother. Pharmacol. 2012, 69, 1413–1422. [Google Scholar] [CrossRef]
- Hu, X.; Wang, L.; Lin, L.; Han, X.; Dou, G.; Meng, Z.; Shi, Y. A Phase i Trial of an Oral Subtype-Selective Histone Deacetylase Inhibitor, Chidamide, in Combination with Paclitaxel and Carboplatin in Patients with Advanced Non-Small Cell Lung Cancer. Chin. J. Cancer Res. 2016, 28, 444–451. [Google Scholar] [CrossRef] [Green Version]
- Yeo, W.; Chung, H.C.; Chan, S.L.; Wang, L.Z.; Lim, R.; Picus, J.; Boyer, M.; Mo, F.K.F.; Koh, J.; Rha, S.Y.; et al. Epigenetic Therapy Using Belinostat for Patients with Unresectable Hepatocellular Carcinoma: A Multicenter Phase I/II Study with Biomarker and Pharmacokinetic Analysis of Tumors from Patients in the Mayo Phase II Consortium and the Cancer Therapeutics Res. J. Clin. Oncol. 2012, 30, 3361–3367. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Rajan, A.; Szabo, E.; Tomita, Y.; Carter, C.A.; Scepura, B.; Lopez-Chavez, A.; Lee, M.J.; Redon, C.E.; Frosch, A.; et al. A Phase I/II Trial of Belinostat in Combination with Cisplatin, Doxorubicin, and Cyclophosphamide in Thymic Epithelial Tumors: A Clinical and Translational Study. Clin. Cancer Res. 2014, 20, 5392–5402. [Google Scholar] [CrossRef] [Green Version]
- Takebe, N.; Beumer, J.H.; Kummar, S.; Kiesel, B.F.; Dowlati, A.; O’Sullivan Coyne, G.; Piekarz, R.; Rubinstein, L.; Fogli, L.K.; Vaishampayan, U.; et al. A Phase I Pharmacokinetic Study of Belinostat in Patients with Advanced Cancers and Varying Degrees of Liver Dysfunction. Br. J. Clin. Pharmacol. 2019, 85, 2499–2511. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.L.; Plumb, J.A.; Vidal, L.; Tjrørnelund, J.; Knoblauch, P.; Rasmussen, A.; Chean, E.O.; Buhl-Jensen, P.; Brown, R.; Evans, T.R.J.; et al. A Phase 1 Pharmacokinetic and Pharmacodynamic Study of the Histone Deacetylase Inhibitor Belinostat in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2008, 14, 804–810. [Google Scholar] [CrossRef] [Green Version]
- Vitfell-Rasmussen, J.; Judson, I.; Safwat, A.; Jones, R.L.; Rossen, P.B.; Lind-Hansen, M.; Knoblauch, P.; Krarup-Hansen, A. A Phase I/II Clinical Trial of Belinostat (PXD101) in Combination with Doxorubicin in Patients with Soft Tissue Sarcomas. Sarcoma 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, S.S.; Belani, C.P.; Ruel, C.; Frankel, P.; Gitlitz, B.; Koczywas, M.; Espinoza-Delgado, I.; Gandara, D. Phase II Study of Belinostat (PXD101), a Histone Deacetylase Inhibitor, for Second Line Therapy of Advanced Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2009, 4, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, H.J.; Hirte, H.; Colgan, T.; Covens, A.; MacAlpine, K.; Grenci, P.; Wang, L.; Mason, J.; Pham, P.A.; Tsao, M.S.; et al. Phase II Trial of the Histone Deacetylase Inhibitor Belinostat in Women with Platinum Resistant Epithelial Ovarian Cancer and Micropapillary (LMP) Ovarian Tumours. Eur. J. Cancer 2010, 46, 1573–1579. [Google Scholar] [CrossRef] [Green Version]
- Luu, T.; Frankel, P.; Beumer, J.H.; Lim, D.; Cristea, M.; Appleman, L.J.; Lenz, H.J.; Gandara, D.R.; Kiesel, B.F.; Piekarz, R.L.; et al. Phase I Trial of Belinostat in Combination with 13-Cis-Retinoic Acid in Advanced Solid Tumor Malignancies: A California Cancer Consortium NCI/CTEP Sponsored Trial. Cancer Chemother. Pharmacol. 2019, 84, 1201–1208. [Google Scholar] [CrossRef]
- Lassen, U.; Molife, L.R.; Sorensen, M.; Engelholm, S.A.; Vidal, L.; Sinha, R.; Penson, R.T.; Buhl-Jensen, P.; Crowley, E.; Tjornelund, J.; et al. A Phase i Study of the Safety and Pharmacokinetics of the Histone Deacetylase Inhibitor Belinostat Administered in Combination with Carboplatin and/or Paclitaxel in Patients with Solid Tumours. Br. J. Cancer 2010, 103, 12–17. [Google Scholar] [CrossRef]
- Giaccone, G.; Rajan, A.; Berman, A.; Kelly, R.J.; Szabo, E.; Lopez-Chavez, A.; Trepel, J.; Lee, M.J.; Cao, L.; Espinoza-Delgado, I.; et al. Phase II Study of Belinostat in Patients with Recurrent or Refractory Advanced Thymic Epithelial Tumors. J. Clin. Oncol. 2011, 29, 2052–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dizon, D.S.; Damstrup, L.; Finkler, N.J.; Lassen, U.; Celano, P.; Glasspool, R.; Crowley, E.; Lichenstein, H.S.; Knoblach, P.; Penson, R.T. Phase II Activity of Belinostat (PXD-101), Carboplatin, and Paclitaxel in Women with Previously Treated Ovarian Cancer. Int. J. Gynecol. Cancer 2012, 22, 979–986. [Google Scholar] [CrossRef]
- Dizon, D.S.; Blessing, J.A.; Penson, R.T.; Drake, R.D.; Walker, J.L.; Johnston, C.M.; Disilvestro, P.A.; Fader, A.N. A Phase II Evaluation of Belinostat and Carboplatin in the Treatment of Recurrent or Persistent Platinum-Resistant Ovarian, Fallopian Tube, or Primary Peritoneal Carcinoma: A Gynecologic Oncology Group Study. Gynecol. Oncol. 2012, 125, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Balasubramaniam, S.; Redon, C.E.; Peer, C.J.; Bryla, C.; Lee, M.J.; Trepel, J.B.; Tomita, Y.; Rajan, A.; Giaccone, G.; Bonner, W.M.; et al. Phase I Trial of Belinostat with Cisplatin and Etoposide in Advanced Solid Tumors, with a Focus on Neuroendocrine and Small Cell Cancers of the Lung. Anticancer Drugs 2018, 29, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.J.; Strong, R.; McArthur, G.A.; Michael, M.; Algar, E.; Muscat, A.; Rigby, L.; Ferguson, M.; Ashley, D.M. A Phase I Study of Panobinostat in Pediatric Patients with Refractory Solid Tumors, Including CNS Tumors. Cancer Chemother. Pharmacol. 2018, 82, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cao, Q.; Dudek, A.Z. Phase II Study of Panobinostat and Bortezomib in Patients with Pancreatic Cancer Progressing on Gemcitabine-Based Therapy. Anticancer Res. 2012, 32, 1027–1031. [Google Scholar]
- Thomas, S.; Aggarwal, R.; Jahan, T.; Ryan, C.; Troung, T.; Cripps, A.M.; Raha, P.; Thurn, K.T.; Chen, S.; Grabowsky, J.A.; et al. A Phase I Trial of Panobinostat and Epirubicin in Solid Tumors with a Dose Expansion in Patients with Sarcoma. Ann. Oncol. 2016, 27, 947–952. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Zahoor, H.; Mclaughlin, B.; Gooding, W.E.; Schmitz, J.C.; Siegfried, J.M.; Socinski, M.A.; Argiris, A. Phase I Trial of Carboplatin and Etoposide in Combination with Panobinostat in Patients with Lung Cancer. Anticancer Res. 2013, 33, 4475–4482. [Google Scholar] [PubMed]
- Strickler, J.H.; Starodub, A.N.; Jia, J.; Meadows, K.L.; Nixon, A.B.; Dellinger, A.; Morse, M.A.; Uronis, H.E.; Marcom, P.K.; Zafar, S.Y.; et al. Phase I Study of Bevacizumab, Everolimus, and Panobinostat (LBH-589) in Advanced Solid Tumors. Cancer Chemother. Pharmacol. 2012, 70, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Slingerland, M.; Hess, D.; Clive, S.; Sharma, S.; Sandstrom, P.; Loman, N.; Porro, M.G.; Mu, S.; Waldron, E.; Valera, S.Z.; et al. A Phase I, Open-Label, Multicenter Study to Evaluate the Pharmacokinetics and Safety of Oral Panobinostat in Patients with Advanced Solid Tumors and Various Degrees of Hepatic Function. Cancer Chemother. Pharmacol. 2014, 74, 1089–1098. [Google Scholar] [CrossRef]
- Shi, W.; Palmer, J.D.; Werner-Wasik, M.; Andrews, D.W.; Evans, J.J.; Glass, J.; Kim, L.; Bar-Ad, V.; Judy, K.; Farrell, C.; et al. Phase I Trial of Panobinostat and Fractionated Stereotactic Re-Irradiation Therapy for Recurrent High Grade Gliomas. J. Neurooncol. 2016, 127, 535–539. [Google Scholar] [CrossRef]
- Sharma, S.; Witteveen, P.O.; Lolkema, M.P.; Hess, D.; Gelderblom, H.; Hussain, S.A.; Porro, M.G.; Waldron, E.; Valera, S.Z.; Mu, S. A Phase I, Open-Label, Multicenter Study to Evaluate the Pharmacokinetics and Safety of Oral Panobinostat in Patients with Advanced Solid Tumors and Varying Degrees of Renal Function. Cancer Chemother. Pharmacol. 2015, 75, 87–95. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Picus, J.; Hussain, A.; Ellard, S.; Chi, K.N.; Nydam, T.; Allen-Freda, E.; Mishra, K.K.; Porro, M.G.; Scher, H.I.; et al. A Phase 2 Study of Intravenous Panobinostat in Patients with Castration-Resistant Prostate Cancer. Cancer Chemother. Pharmacol. 2013, 72, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Rathkopf, D.; Wong, B.Y.; Ross, R.W.; Anand, A.; Tanaka, E.; Woo, M.M.; Hu, J.; Dzik-Jurasz, A.; Yang, W.; Scher, H.I. A Phase I Study of Oral Panobinostat Alone and in Combination with Docetaxel in Patients with Castration-Resistant Prostate Cancer. Cancer Chemother. Pharmacol. 2010, 66, 181–189. [Google Scholar] [CrossRef]
- De Marinis, F.; Atmaca, A.; Tiseo, M.; Giuffreda, L.; Rossi, A.; Gebbia, V.; D’Antonio, C.; Zotto, L.D.; Al-Batran, S.E.; Marsoni, S.; et al. A Phase II Study of the Histone Deacetylase Inhibitor Panobinostat (LBH589) in Pretreated Patients with Small-Cell Lung Cancer. J. Thorac. Oncol. 2013, 8, 1091–1094. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Q.; Reardon, D.A.; Schiff, D.; Drappatz, J.; Muzikansky, A.; Grimm, S.A.; Norden, A.D.; Nayak, L.; Beroukhim, R.; Rinne, M.L.; et al. Phase II Study of Panobinostat in Combination with Bevacizumab for Recurrent Glioblastoma and Anaplastic Glioma. Neuro Oncol. 2015, 17, 862–867. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.F.; Infante, J.R.; Thompson, D.S.; Mohyuddin, A.; Bendell, J.C.; Yardley, D.A.; Burris, H.A. A Phase i Trial of Oral Administration of Panobinostat in Combination with Paclitaxel and Carboplatin in Patients with Solid Tumors. Cancer Chemother. Pharmacol. 2012, 70, 471–475. [Google Scholar] [CrossRef]
- Results, C.T. Clinical Trial Results A Phase II Trial of a Histone Deacetylase Inhibitor Panobinostat in Patients With Low-Grade Neuroendocrine Tumors. Oncologist 2016, 21, 785–786. [Google Scholar]
- Ibrahim, N.; Buchbinder, E.I.; Granter, S.R.; Rodig, S.J.; Giobbie-Hurder, A.; Becerra, C.; Tsiaras, A.; Gjini, E.; Fisher, D.E.; Hodi, F.S. A Phase I Trial of Panobinostat (LBH589) in Patients with Metastatic Melanoma. Cancer Med. 2016, 5, 3041–3050. [Google Scholar] [CrossRef] [PubMed]
- Takhar, H.S.; Singhal, N.; Gowda, R.; Penniment, M.; Takhar, P.; Brown, M.P. Phase i Study Evaluating the Safety and Efficacy of Oral Panobinostat in Combination with Radiotherapy or Chemoradiotherapy in Patients with Inoperable Stage III Non-Small-Cell Lung Cancer. Anticancer Drugs 2015, 26, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Infante, J.R.; Spigel, D.R.; Arrowsmith, E.R.; Boccia, R.V.; Burris, H.A. A Phase II Trial of Panobinostat, a Histone Deacetylase Inhibitor, in the Treatment of Patients with Refractory Metastatic Renal Cell Carcinoma. Cancer Invest. 2011, 29, 451–455. [Google Scholar] [CrossRef]
- Drappatz, J.; Lee, E.Q.; Hammond, S.; Grimm, S.A.; Norden, A.D.; Beroukhim, R.; Gerard, M.; Schiff, D.; Chi, A.S.; Batchelor, T.T.; et al. Phase i Study of Panobinostat in Combination with Bevacizumab for Recurrent High-Grade Glioma. J. Neurooncol. 2012, 107, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Cassier, P.A.; Lefranc, A.; Amela, E.Y.; Chevreau, C.; Bui, B.N.; Lecesne, A.; Ray-Coquard, I.; Chabaud, S.; Penel, N.; Berge, Y.; et al. A Phase II Trial of Panobinostat in Patients with Advanced Pretreated Soft Tissue Sarcoma. A Study from the French Sarcoma Group. Br. J. Cancer 2013, 109, 909–914. [Google Scholar] [CrossRef]
- Bauer, S.; Hilger, R.A.; Mühlenberg, T.; Grabellus, F.; Nagarajah, J.; Hoiczyk, M.; Reichardt, A.; Ahrens, M.; Reichardt, P.; Grunewald, S.; et al. Phase i Study of Panobinostat and Imatinib in Patients with Treatment-Refractory Metastatic Gastrointestinal Stromal Tumors. Br. J. Cancer 2014, 110, 1155–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, C.; Ryu, M.H.; Na, Y.S.; Ryoo, B.Y.; Lee, C.W.; Maeng, J.; Kim, S.Y.; Koo, D.H.; Park, I.; Kang, Y.K. Phase i and Pharmacodynamic Study of Vorinostat Combined with Capecitabine and Cisplatin as First-Line Chemotherapy in Advanced Gastric Cancer. Investig. New Drugs 2014, 32, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.; Ryu, M.H.; Na, Y.S.; Ryoo, B.Y.; Lee, C.W.; Kang, Y.K. Vorinostat in Combination with Capecitabine plus Cisplatin as a First-Line Chemotherapy for Patients with Metastatic or Unresectable Gastric Cancer: Phase II Study and Biomarker Analysis. Br. J. Cancer 2016, 114, 1185–1190. [Google Scholar] [CrossRef] [Green Version]
- Wilson, P.M.; El-Khoueiry, A.; Iqbal, S.; Fazzone, W.; Labonte, M.J.; Groshen, S.; Yang, D.; Danenberg, K.D.; Cole, S.; Kornacki, M.; et al. A Phase I/II Trial of Vorinostat in Combination with 5-Fluorouracil in Patients with Metastatic Colorectal Cancer Who Previously Failed 5-FU-Based Chemotherapy. Cancer Chemother. Pharmacol. 2010, 65, 979–988. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Van Cutsem, E.; Dumez, H.; Chen, C.; Ricker, J.L.; Randolph, S.S.; Schöffski, P. Early Phase II Trial of Oral Vorinostat in Relapsed or Refractory Breast, Colorectal, or Non-Small Cell Lung Cancer. Investig. New Drugs 2008, 26, 483–488. [Google Scholar] [CrossRef]
- Tu, Y.; Hershman, D.L.; Bhalla, K.; Fiskus, W.; Pellegrino, C.M.; Andreopoulou, E.; Makower, D.; Kalinsky, K.; Fehn, K.; Fineberg, S.; et al. A Phase I-II Study of the Histone Deacetylase Inhibitor Vorinostat plus Sequential Weekly Paclitaxel and Doxorubicin-Cyclophosphamide in Locally Advanced Breast Cancer. Breast Cancer Res. Treat. 2014, 146, 145–152. [Google Scholar] [CrossRef]
- Shi, W.; Lawrence, Y.R.; Choy, H.; Werner-Wasik, M.; Andrews, D.W.; Evans, J.J.; Judy, K.D.; Farrell, C.J.; Moshel, Y.; Berger, A.C.; et al. Vorinostat as a Radiosensitizer for Brain Metastasis: A Phase I Clinical Trial. J. Neurooncol. 2014, 118, 313–319. [Google Scholar] [CrossRef]
- Schneider, B.J.; Kalemkerian, G.P.; Bradley, D.; Smith, D.C.; Egorin, M.J.; Daignault, S.; Dunn, R.; Hussain, M. Phase I Study of Vorinostat (Suberoylanilide Hydroxamic Acid, NSC 701852) in Combination with Docetaxel in Patients with Advanced and Relapsed Solid Malignancies. Investig. New Drugs 2012, 30, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, T.; Mayer-Steinacker, R.; Mayer, F.; Grünwald, V.; Schütte, J.; Hartmann, J.T.; Kasper, B.; Hüsing, J.; Hajda, J.; Ottawa, G.; et al. Vorinostat in Refractory Soft Tissue Sarcomas—Results of a Multi-Centre Phase II Trial of the German Soft Tissue Sarcoma and Bone Tumour Working Group (AIO). Eur. J. Cancer 2016, 64, 74–82. [Google Scholar] [CrossRef]
- Reguart, N.; Rosell, R.; Cardenal, F.; Cardona, A.F.; Isla, D.; Palmero, R.; Moran, T.; Rolfo, C.; Pallarès, M.C.; Insa, A.; et al. Phase I/II Trial of Vorinostat (SAHA) and Erlotinib for Non-Small Cell Lung Cancer (NSCLC) Patients with Epidermal Growth Factor Receptor (EGFR) Mutations after Erlotinib Progression. Lung Cancer 2014, 84, 161–167. [Google Scholar] [CrossRef]
- Ramaswamy, B.; Fiskus, W.; Cohen, B.; Pellegrino, C.; Hershman, D.L.; Chuang, E.; Luu, T.; Somlo, G.; Goetz, M.; Swaby, R.; et al. Phase I-II Study of Vorinostat plus Paclitaxel and Bevacizumab in Metastatic Breast Cancer: Evidence for Vorinostat-Induced Tubulin Acetylation and Hsp90 Inhibition in Vivo. Breast Cancer Res. Treat. 2012, 132, 1063–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; et al. Carboplatin and Paclitaxel in Combination with Either Vorinostat or Placebo for First-Line Therapy of Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, S.S.; Kummar, S.; Sarantopoulos, J.; Shibata, S.; LoRusso, P.; Yerk, M.; Holleran, J.; Lin, Y.; Beumer, J.H.; Harvey, R.D.; et al. Phase I Study of Vorinostat in Patients with Advanced Solid Tumors and Hepatic Dysfunction: A National Cancer Institute Organ Dysfunction Working Group Study. J. Clin. Oncol. 2010, 28, 4507–4512. [Google Scholar] [CrossRef] [Green Version]
- Pili, R.; Liu, G.; Chintala, S.; Verheul, H.; Rehman, S.; Attwood, K.; Lodge, M.A.; Wahl, R.; Martin, J.I.; Miles, K.M.; et al. Combination of the Histone Deacetylase Inhibitor Vorinostat with Bevacizumab in Patients with Clear-Cell Renal Cell Carcinoma: A Multicentre, Single-Arm Phase I/II Clinical Trial. Br. J. Cancer 2017, 116, 874–883. [Google Scholar] [CrossRef] [Green Version]
- Peters, K.B.; Lipp, E.S.; Miller, E.; Herndon, J.E.; McSherry, F.; Desjardins, A.; Reardon, D.A.; Friedman, H.S. Phase I/II Trial of Vorinostat, Bevacizumab, and Daily Temozolomide for Recurrent Malignant Gliomas. J. Neurooncol. 2018, 137, 349–356. [Google Scholar] [CrossRef]
- Molina, A.M.; van der Mijn, J.C.; Christos, P.; Wright, J.; Thomas, C.; Dutcher, J.P.; Nanus, D.M.; Tagawa, S.T.; Gudas, L.J. NCI 6896: A Phase I Trial of Vorinostat (SAHA) and Isotretinoin (13-Cis Retinoic Acid) in the Treatment of Patients with Advanced Renal Cell Carcinoma. Investig. New Drugs 2020, 38, 1383–1389. [Google Scholar] [CrossRef]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P. A Phase II Study of Vorinostat in the Treatment of Persistent or Recurrent Epithelial Ovarian or Primary Peritoneal Carcinoma: A Gynecologic Oncology Group Study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef]
- Matulonis, U.; Berlin, S.; Lee, H.; Whalen, C.; Obermayer, E.; Penson, R.; Liu, J.; Campos, S.; Krasner, C.; Horowitz, N. Phase I Study of Combination of Vorinostat, Carboplatin, and Gemcitabine in Women with Recurrent, Platinum-Sensitive Epithelial Ovarian, Fallopian Tube, or Peritoneal Cancer. Cancer Chemother. Pharmacol. 2015, 76, 417–423. [Google Scholar] [CrossRef]
- Blumenschein, G.R.; Kies, M.S.; Papadimitrakopoulou, V.A.; Lu, C.; Kumar, A.J.; Ricker, J.L.; Chiao, J.H.; Chen, C.; Frankel, S.R. Phase II Trial of the Histone Deacetylase Inhibitor Vorinostat (ZolinzaTM, Suberoylanilide Hydroxamic Acid, SAHA) in Patients with Recurrent and/or Metastatic Head and Neck Cancer. Investig. New Drugs 2008, 26, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Krug, L.M.; Kindler, H.L.; Calvert, H.; Manegold, C.; Tsao, A.S.; Fennell, D.; Öhman, R.; Plummer, R.; Eberhardt, W.E.E.; Fukuoka, K.; et al. Vorinostat in Patients with Advanced Malignant Pleural Mesothelioma Who Have Progressed on Previous Chemotherapy (VANTAGE-014): A Phase 3, Double-Blind, Randomised, Placebo-Controlled Trial. Lancet Oncol. 2015, 16, 447–456. [Google Scholar] [CrossRef]
- Kelly, W.K.; O’Connor, O.A.; Krug, L.M.; Chiao, J.H.; Heaney, M.; Curley, T.; MacGregore-Cortelli, B.; Tong, W.; Secrist, J.P.; Schwartz, L.; et al. Phase I Study of an Oral Histone Deacetylase Inhibitor, Suberoylanilide Hydroxamic Acid, in Patients with Advanced Cancer. J. Clin. Oncol. 2005, 23, 3923–3931. [Google Scholar] [CrossRef]
- Hoang, T.; Campbell, T.C.; Zhang, C.; Kim, K.M.; Kolesar, J.M.; Oettel, K.R.; Blank, J.H.; Robinson, E.G.; Ahuja, H.G.; Kirschling, R.J.; et al. Vorinostat and Bortezomib as Third-Line Therapy in Patients with Advanced Non-Small Cell Lung Cancer: A Wisconsin Oncology Network Phase II Study. Investig. New Drugs 2014, 32, 195–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, N.B.; Quirt, I.; Hotte, S.; McWhirter, E.; Polintan, R.; Litwin, S.; Adams, P.D.; McBryan, T.; Wang, L.; Martin, L.P.; et al. Phase II Trial of Vorinostat in Advanced Melanoma. Investig. New Drugs 2014, 32, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.W.; McGuire, W.P.; Shafer, D.A.; Sterling, R.K.; Lee, H.M.; Matherly, S.C.; Roberts, J.D.; Bose, P.; Tombes, M.B.; Shrader, E.E.; et al. Phase i Study of Sorafenib and Vorinostat in Advanced Hepatocellular Carcinoma. Am. J. Clin. Oncol. Cancer Clin. Trials 2019, 42, 649–654. [Google Scholar] [CrossRef]
- Goncalves, P.H.; Heilbrun, L.K.; Barrett, M.T.; Kummar, S.; Hansen, A.R.; Siu, L.L.; Piekarz, R.L.; Sukari, A.W.; Chao, J.; Pilat, M.J.; et al. A Phase 2 Study of Vorinostat in Locally Advanced, Recurrent, or Metastatic Adenoid Cystic Carcinoma. Oncotarget 2017, 8, 32918–32929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanis, E.; Keith Anderson, S.; Miller, C.R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F.; Nebozhyn, M.; et al. Phase I/II Trial of Vorinostat Combined with Temozolomide and Radiation Therapy for Newly Diagnosed Glioblastoma: Results of Alliance N0874/ABTC 02. Neuro-Oncol. 2018, 20, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Galanis, E.; Jaeckle, K.A.; Maurer, M.J.; Reid, J.M.; Ames, M.M.; Hardwick, J.S.; Reilly, J.F.; Loboda, A.; Nebozhyn, M.; Fantin, V.R.; et al. Phase II Trial of Vorinostat in Recurrent Glioblastoma Multiforme: A North Central Cancer Treatment Group Study. J. Clin. Oncol. 2009, 27, 2052–2058. [Google Scholar] [CrossRef] [Green Version]
- Fakih, M.G.; Fetterly, G.; Egorin, M.J.; Muindi, J.R.; Espinoza-Delgado, I.; Zwiebel, J.A.; Litwin, A.; Holleran, J.L.; Wang, K.; Diasio, R.B. A Phase I, Pharmacokinetic, and Pharmacodynamic Study of Two Schedules of Vorinostat in Combination with 5-Fluorouracil and Leucovorin in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2010, 16, 3786–3794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakih, M.G.; Pendyala, L.; Fetterly, G.; Toth, K.; Zwiebel, J.A.; Espinoza-Delgado, I.; Litwin, A.; Rustum, Y.M.; Ross, M.E.; Holleran, J.L.; et al. A Phase I, Pharmacokinetic and Pharmacodynamic Study on Vorinostat in Combinationwith 5-Fluorouracil, Leucovorin, and Oxaliplatin in Patients with Refractory Colorectal Cancer. Clin. Cancer Res. 2009, 15, 3189–3195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakih, M.G.; Groman, A.; McMahon, J.; Wilding, G.; Muindi, J.R. A Randomized Phase II Study of Two Doses of Vorinostat in Combination with 5-FU/LV in Patients with Refractory Colorectal Cancer. Cancer Chemother. Pharmacol. 2012, 69, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Hamaguchi, T.; Shirao, K.; Chin, K.; Hatake, K.; Noguchi, K.; Otsuki, T.; Mehta, A.; Ohtsu, A. Evaluation of Safety, Pharmacokinetics, and Efficacy of Vorinostat, a Histone Deacetylase Inhibitor, in the Treatment of Gastrointestinal (GI) Cancer in a Phase I Clinical Trial. Int. J. Clin. Oncol. 2013, 18, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Arlinghaus, L.R.; Cardin, D.B.; Goff, L.; Berlin, J.D.; Parikh, A.; Abramson, R.G.; Yankeelov, T.E.; Hiebert, S.; Merchant, N.; et al. Phase I Trial of Vorinostat Added to Chemoradiation with Capecitabine in Pancreatic Cancer. Radiother. Oncol. 2016, 119, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jenke, R.; Reßing, N.; Hansen, F.K.; Aigner, A.; Büch, T. Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers 2021, 13, 634. https://doi.org/10.3390/cancers13040634
Jenke R, Reßing N, Hansen FK, Aigner A, Büch T. Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers. 2021; 13(4):634. https://doi.org/10.3390/cancers13040634
Chicago/Turabian StyleJenke, Robert, Nina Reßing, Finn K. Hansen, Achim Aigner, and Thomas Büch. 2021. "Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives" Cancers 13, no. 4: 634. https://doi.org/10.3390/cancers13040634
APA StyleJenke, R., Reßing, N., Hansen, F. K., Aigner, A., & Büch, T. (2021). Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers, 13(4), 634. https://doi.org/10.3390/cancers13040634