
Mechanisms of Resistance to Conventional Therapies for Osteosarcoma

 , and
, and 
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
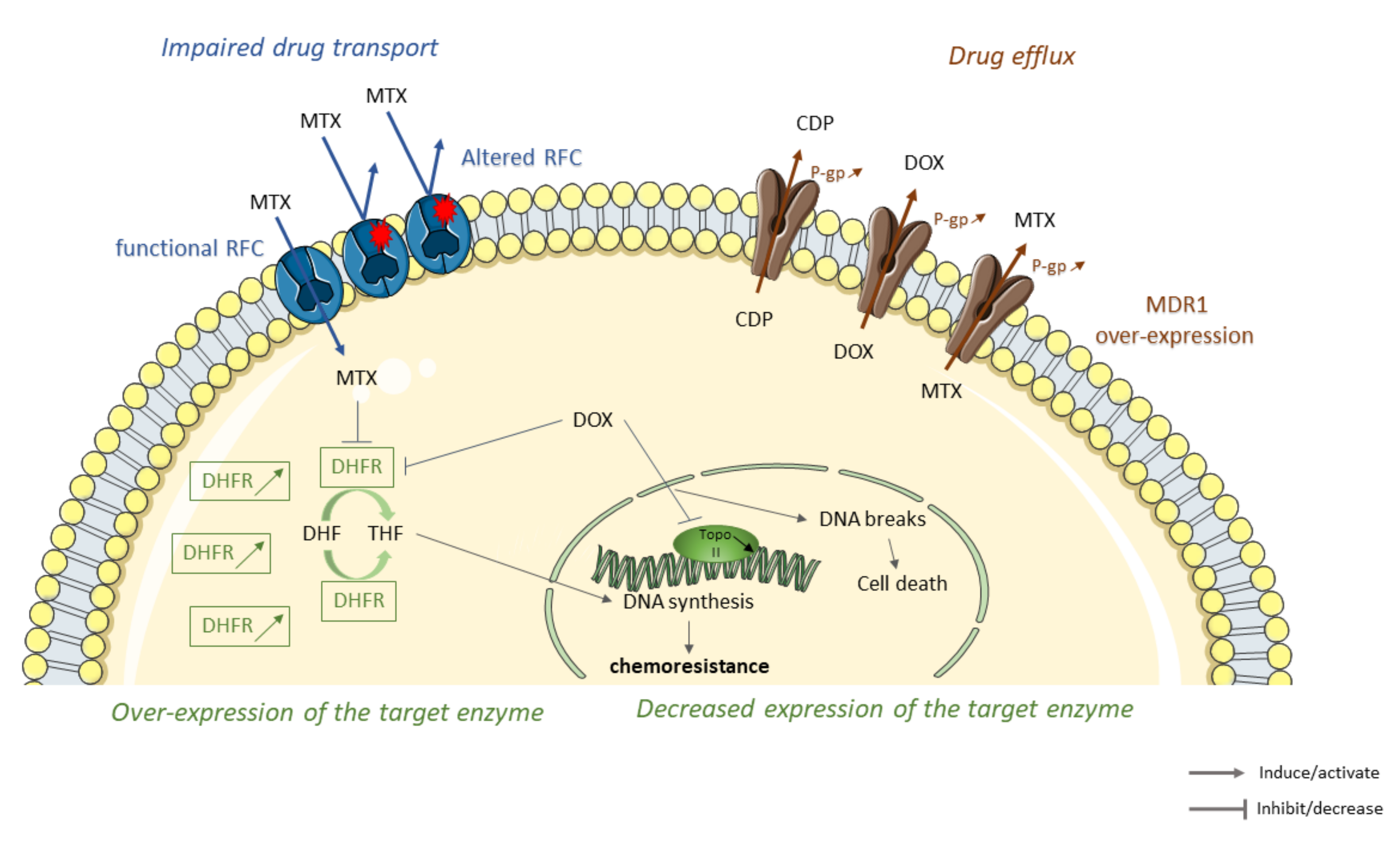
2. Decrease the Intracellular Accumulation of Drugs
2.1. Impaired Drug Transport
2.2. Drug Elimination
2.3. Alterations in the Structure or Expression of the Target Enzyme
3. Drug Detoxification/Inactivation
4. DNA Repair Improvement
5. Cell Cycle and Apoptosis Disruptions
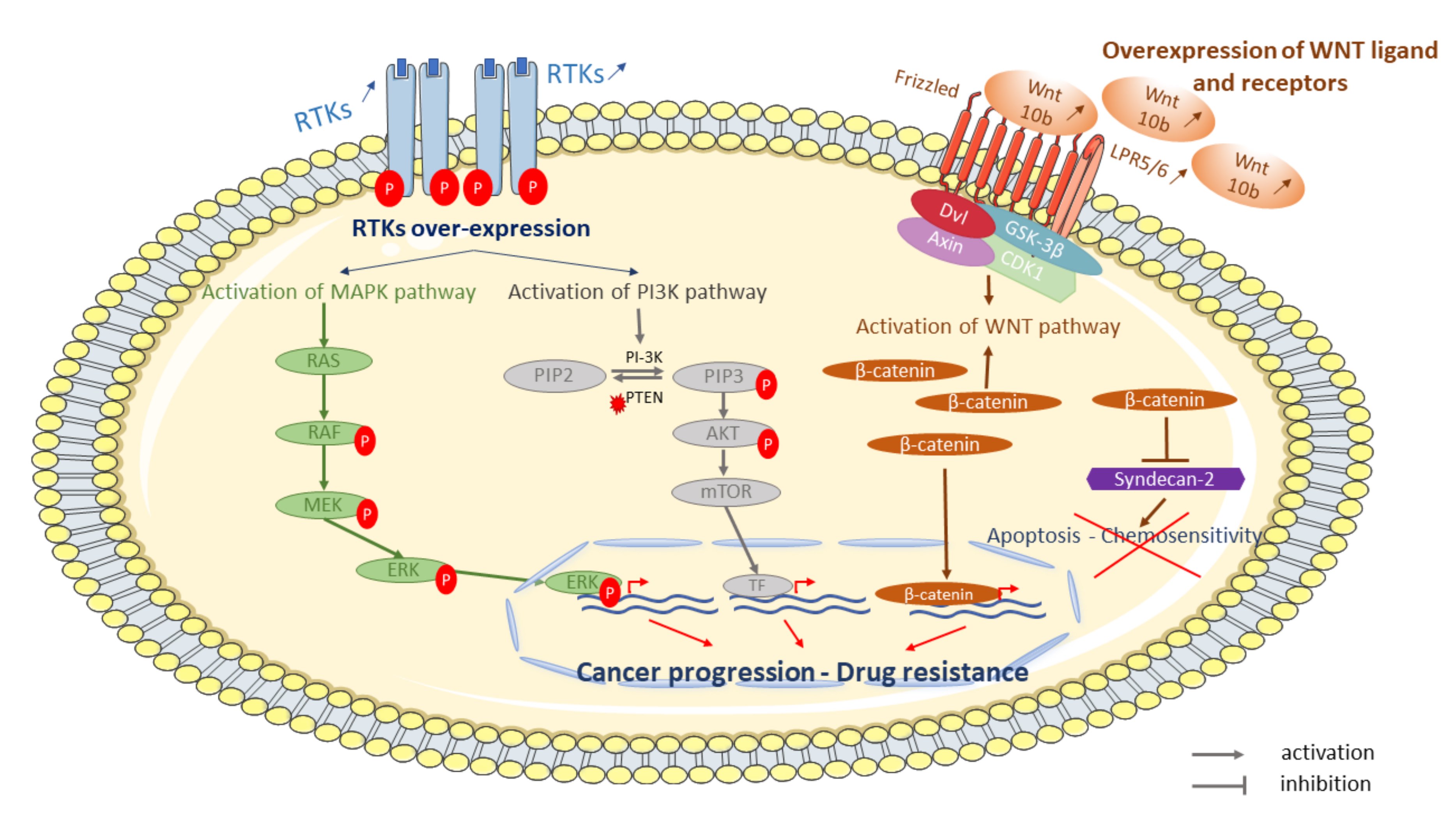
6. Involvement of Signaling and Signal Transduction Pathways
6.1. Cell-Surface Receptors: Her2, VEGF and IGF-1R
6.2. PI3K and MAPK Pathways
6.3. WNT Pathway
7. Autophagy Involvement
8. Cancer Stem Cells and Microenvironment
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lamoureux, F.; Trichet, V.; Chipoy, C.; Blanchard, F.; Gouin, F.; Redini, F. Recent advances in the management of osteosarcoma and forthcoming therapeutic strategies. Expert Rev. Anticancer Ther. 2007, 7, 169–181. [Google Scholar] [CrossRef]
- He, J.-P.; Hao, Y.; Wang, X.-L.; Yang, X.-J.; Shao, J.-F.; Guo, F.-J.; Feng, J.-X. Review of the Molecular Pathogenesis of Osteosarcoma. Asian Pac. J. Cancer Prev. 2014, 15, 5967–5976. [Google Scholar] [CrossRef] [Green Version]
- Marina, N.; Gebhardt, M.; Teot, L.; Gorlick, R. Biology and Therapeutic Advances for Pediatric Osteosarcoma. Oncologist 2004, 9, 422–441. [Google Scholar] [CrossRef]
- Sandberg, A.; Bridge, J.A. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: Osteosarcoma and related tumors. Cancer Genet. Cytogenet. 2003, 145, 1–30. [Google Scholar] [CrossRef]
- Klein, M.J.; Siegal, G.P. Osteosarcoma: Anatomic and histologic variants. Am. J. Clin. Pathol. 2006, 125, 555–581. [Google Scholar] [CrossRef]
- Isakoff, M.S.; Bielack, S.S.; Meltzer, P.S.; Gorlick, R. Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. J. Clin. Oncol. 2015, 33, 3029–3035. [Google Scholar] [CrossRef] [Green Version]
- Rejniak, K.A.; Lloyd, M.C.; Reed, D.; Bui, M.M. Diagnostic assessment of osteosarcoma chemoresistance based on Virtual Clinical Trials. Med. Hypotheses 2015, 85, 348–354. [Google Scholar] [CrossRef]
- ESMO/European Sarcoma Network Working Group. Bone sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. 3), iii113–iii123. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, J.; Zhang, N.; Yang, B.; He, Q.; Ying, M.; Ying, M. Advances in differentiation therapy for osteosarcoma. Drug Discov. Today 2020, 25, 497–504. [Google Scholar] [CrossRef]
- Anderson, P.M. Effectiveness of Radiotherapy for Osteosarcoma that Responds to Chemotherapy. Mayo Clin. Proc. 2003, 78, 145–146. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, R.; Bruland Øyvind, S.; Cassoni, A.; Schomberg, P.; Bielack, S. The Role of Radiotherapy in Oseosarcoma. Cancer Treat. Res. 2009, 152, 147–164. [Google Scholar] [CrossRef]
- Majó, J.; Cubedo, R.; Pardo, N. Treatment of Osteosarcoma. A Review. Rev. Esp. Cir. Ortop. Traumatol. 2010, 54, 329–336. [Google Scholar]
- Saraf, A.J.; Fenger, J.M.; Roberts, R.D. Osteosarcoma: Accelerating Progress Makes for a Hopeful Future. Front. Oncol. 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, S.; Palmerini, E. Adjuvant and neoadjuvant combination chemotherapy for osteogenic sarcoma. Curr. Opin. Oncol. 2007, 19, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Mialou, V.; Philip, T.; Kalifa, C.; Perol, D.; Gentet, J.-C.; Marec-Berard, P.; Pacquement, H.; Chastagner, P.; Defaschelles, A.; Hartmann, O. Metastatic osteosarcoma at diagnosis: Prognostic factors and long-term outcom—The French pediatric experience. Cancer 2005, 104, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, G.; Robert, R.S.; Huh, W.W.; Jaffe, N.; Ottaviani, G. Functional, Psychosocial and Professional Outcomes in Long-Term Survivors of Lower-Extremity Osteosarcomas: Amputation Versus Limb Salvage. Cancer Treat. Res. 2009, 152, 421–436. [Google Scholar] [CrossRef]
- Rosen, G.; Tan, C.; Sanmaneechai, A.; Beattie, E.J.; Marcove, R.; Murphy, M.L. The rationale for multiple drug chemotherapy in the treatment of osteogenic sarcoma. Cancer 1975, 35, 936–945. [Google Scholar] [CrossRef]
- Goorin, A.M.; Schwartzentruber, D.J.; Devidas, M.; Gebhardt, M.C.; Ayala, A.G.; Harris, M.B.; Helman, L.J.; Grier, H.E.; Link, M.P. Presurgical Chemotherapy Compared with Immediate Surgery and Adjuvant Chemotherapy for Nonmetastatic Osteosarcoma: Pediatric Oncology Group Study POG-8651. J. Clin. Oncol. 2003, 21, 1574–1580. [Google Scholar] [CrossRef]
- Wittig, J.C.; Bickels, J.; Priebat, D.; Jelinek, J.; Kellar-Graney, K.; Shmookler, B.; Malawer, M.M. Osteosarcoma: A multidisciplinary approach to diagnosis and treatment. Am. Fam. Physician 2002, 65, 1123–1132. [Google Scholar]
- Ando, K.; Heymann, M.-F.; Stresing, V.; Mori, K.; Redini, F.; Heymann, D. Current Therapeutic Strategies and Novel Approaches in Osteosarcoma. Cancers 2013, 5, 591–616. [Google Scholar] [CrossRef] [Green Version]
- Bielack, S.S.; Carrle, R.; Hardes, J.; Schuck, A.; Paulussen, M. Bone Tumors in Adolescents and Young Adults. Curr. Treat. Options Oncol. 2008, 9, 67–80. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Lippert, T.H.; Ruoff, H.-J.; Volm, M. Intrinsic and Acquired Drug Resistance in Malignant Tumors. The main reason for therapeutic failure. Arzneimittelforschung 2008, 58, 261–264. [Google Scholar] [CrossRef]
- Wang, X. Drug Resistance and Combating Drug Resistance in Cancer. Available online: https://cdrjournal.com/article/view/3039 (accessed on 7 May 2020).
- Chou, A.J.; Gorlick, R. Chemotherapy resistance in osteosarcoma: Current challenges and future directions. Expert Rev. Anticancer Ther. 2006, 6, 1075–1085. [Google Scholar] [CrossRef]
- Lewis, I.J.; Nooij, M.A.; Whelan, J.; Sydes, M.R.; Grimer, R.; Hogendoorn, P.C.W.; Memon, M.A.; Weeden, S.; Uscinska, B.M.; Van Glabbeke, M.; et al. Improvement in Histologic Response But Not Survival in Osteosarcoma Patients Treated With Intensified Chemotherapy: A Randomized Phase III Trial of the European Osteosarcoma Intergroup. J. Natl. Cancer Inst. 2007, 99, 112–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posthuma de Boer, J.; Royen, B.J.; Helder, M. Mechanisms of therapy resistance in osteosarcoma: A review. Oncol. Discov. 2013, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Fotoohi, A.K.; Albertioni, F. Mechanisms of antifolate resistance and methotrexate efficacy in leukemia cells. Leuk. Lymphoma 2008, 49, 410–426. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Ni, J.; Huang, J. Molecular mechanisms of chemoresistance in osteosarcoma (Review). Oncol. Lett. 2014, 7, 1352–1362. [Google Scholar] [CrossRef] [Green Version]
- Takemura, Y.; Kobayashi, H.; Miyachi, H. Cellular and molecular mechanisms of resistance to antifolate drugs: New analogues and approaches to overcome the resistance. Int. J. Hematol. 1997, 66, 459–477. [Google Scholar] [CrossRef]
- Jensen, D.E.; Black, A.R.; Swick, A.G.; Azizkhan, J.C. Distinct roles for Sp1 and E2F sites in the growth/cell cycle regulation of the DHFR promoter. J. Cell. Biochem. 1997, 67, 24–31. [Google Scholar] [CrossRef]
- Goldman, I.; Matherly, L.H. The cellular pharmacology of methotrexate. Pharmacol. Ther. 1985, 28, 77–102. [Google Scholar] [CrossRef]
- Bertino, J.R.; Göker, E.; Gorlick, R.; Li, W.W.; Banerjee, D. Resistance Mechanisms to Methotrexate in Tumors. Oncologist 1996, 1, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Moscow, J.A. Methotrexate transport and resistance. Leuk. Lymphoma 1998, 30, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Bertino, J.R. Karnofsky memorial lecture. Ode to methotrexate. J. Clin. Oncol. 1993, 11, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, G. Relationship between RFC gene expression and intracellular drug concentration in methotrexate-resistant osteosarcoma cells. Genet. Mol. Res. 2014, 13, 5313–5321. [Google Scholar] [CrossRef] [PubMed]
- Hattinger, C.M.; Reverter-Branchat, G.; Remondini, D.; Castellani, G.C.; Benini, S.; Pasello, M.; Manara, M.C.; Scotlandi, K.; Picci, P.; Serra, M. Genomic imbalances associated with methotrexate resistance in human osteosarcoma cell lines detected by comparative genomic hybridization-based techniques. Eur. J. Cell Biol. 2003, 82, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Flintoff, W.F.; Sadlish, H.; Gorlick, R.; Yang, R.; Williams, F.M. Functional analysis of altered reduced folate carrier sequence changes identified in osteosarcomas. Biochim. Biophys. Acta Mol. Basis Dis. 2004, 1690, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Ifergan, I.; Meller, I.; Issakov, J.; Assaraf, Y.G. Reduced folate carrier protein expression in osteosarcoma. Cancer 2003, 98, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Sowers, R.; Mazza, B.; Healey, J.H.; Huvos, A.; Grier, H.; Bernstein, M.; Beardsley, G.P.; Krailo, M.; Devidas, M.; et al. Sequence alterations in the reduced folate carrier are observed in osteosarcoma tumor samples. Clin. Cancer Res. 2003, 9, 837–844. [Google Scholar] [PubMed]
- Serra, M.; Reverter-Branchat, G.; Maurici, D.; Benini, S.; Shen, J.-N.; Chano, T.; Hattinger, C.-M.; Manara, M.-C.; Pasello, M.; Scotlandi, K.; et al. Analysis of dihydrofolate reductase and reduced folate carrier gene status in relation to methotrexate resistance in osteosarcoma cells. Ann. Oncol. 2004, 15, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Qin, J.; Hoang, B.H.; Healey, J.H.; Gorlick, R. Polymorphisms and Methylation of the Reduced Folate Carrier in Osteosarcoma. Clin. Orthop. Relat. Res. 2008, 466, 2046–2051. [Google Scholar] [CrossRef] [Green Version]
- Lilienthal, I.; Herold, N. Targeting Molecular Mechanisms Underlying Treatment Efficacy and Resistance in Osteosarcoma: A Review of Current and Future Strategies. Int. J. Mol. Sci. 2020, 21, 6885. [Google Scholar] [CrossRef]
- Memorial Sloan Kettering Cancer Center. Phase I Study of High Dose Methotrexate with Simultaneous Trimetrexate and Leucovorin in Patients with Recurrent Osteosarcoma. Report No.: NCT00119301. 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT00119301 (accessed on 15 December 2020).
- Sirotnak, F.M.; DeGraw, J.I.; Moccio, D.M.; Samuels, L.L.; Goutas, L.J. New folate analogs of the 10-deaza-aminopterin series Basis for structural design and biochemical and pharmacologic properties. Cancer Chemother. Pharmacol. 1984, 12, 18–25. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, O.A.; Pro, B.; Pinter-Brown, L.; Bartlett, N.; Popplewell, L.; Coiffier, B.; Lechowicz, M.J.; Savage, K.J.; Shustov, A.R.; Gisselbrecht, C.; et al. Pralatrexate in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results From the Pivotal PROPEL Study. J. Clin. Oncol. 2011, 29, 1182–1189. [Google Scholar] [CrossRef]
- Alberts, D.S.; Muggia, F.; Carmichael, J.; Winer, E.P.; Jahanzeb, M.; Venook, A.P.; Skubitz, K.M.; Rivera, E.; Sparano, J.A.; DiBella, N.J.; et al. Efficacy and safety of liposomal anthracyclines in Phase I/II clinical trials. Semin. Oncol. 2004, 31, 53–90. [Google Scholar] [CrossRef] [PubMed]
- O’Day, K.; Gorlick, R. Novel therapeutic agents for osteosarcoma. Expert Rev. Anticancer Ther. 2009, 9, 511–523. [Google Scholar] [CrossRef]
- Krishna, R.; Mayer, L.D. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci. 2000, 11, 265–283. [Google Scholar] [CrossRef]
- Choi, C.-H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef]
- Amawi, H.; Sim, H.-M.; Tiwari, A.K.; Ambudkar, S.V.; Shukla, S. ABC Transporter-Mediated Multidrug-Resistant Cancer. Adv. Exp. Med. Biol. 2019, 1141, 549–580. [Google Scholar] [CrossRef]
- Riordan, J.R.; Ling, V. Purification of P-glycoprotein from plasma membrane vesicles of Chinese hamster ovary cell mutants with reduced colchicine permeability. J. Biol. Chem. 1979, 254, 12701–12705. [Google Scholar] [CrossRef]
- Li, S.; Sun, W.; Wang, H.; Liao, Y.; Hua, Y.; Cai, Z. Research progress on the multidrug resistance mechanisms of osteosarcoma chemotherapy and reversal. Tumor Biol. 2015, 36, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of efflux pumps in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Kartner, N.; Riordan, J.R.; Ling, V. Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines. Science 1983, 221, 1285–1288. [Google Scholar] [CrossRef]
- Scotlandi, K.; Serra, M.; Manara, M.C.; Lollini, P.-L.; Maurici, D.; Del Bufalo, D.; Baldini, N. Pre-Treatment of human osteosarcoma cells with N-methylformamide enhances P-glycoprotein expression and resistance to doxorubicin. Int. J. Cancer 1994, 58, 95–101. [Google Scholar] [CrossRef]
- Gomes, C.; Van Paassen, H.; Romeo, S.; Welling, M.M.; Feitsma, R.; Abrunhosa, A.; Botelho, M.F.; Hogendoorn, P.C.; Pauwels, E.; Cleton-Jansen, A.M. Multidrug resistance mediated by ABC transporters in osteosarcoma cell lines: mRNA analysis and functional radiotracer studies. Nucl. Med. Biol. 2006, 33, 831–840. [Google Scholar] [CrossRef]
- Oda, Y.; Matsumoto, Y.; Harimaya, K.; Iwamoto, Y.; Tsuneyoshi, M. Establishment of new multidrug-resistant human osteosarcoma cell lines. Oncol. Rep. 2000, 7, 859–925. [Google Scholar] [CrossRef]
- Serra, M.; Pasello, M.; Manara, M.C.; Scotlandi, K.; Ferrari, S.; Bertoni, F.; Mercuri, M.; Alvegard, T.A.; Picci, P.; Bacci, G.; et al. May P-glycoprotein status be used to stratify high-grade osteosarcoma patients? Results from the Italian/Scandinavian Sarcoma Group 1 treatment protocol. Int. J. Oncol. 2006, 29, 1459–1468. [Google Scholar] [CrossRef]
- Hornicek, F.J.; Gebhardt, M.C.; Wolfe, M.W.; Kharrazi, F.D.; Takeshita, H.; Parekh, S.G.; Zurakowski, D.; Mankin, H.J. P-Glycoprotein Levels Predict Poor Outcome in Patients With Osteosarcoma. Clin. Orthop. Relat. Res. 2000, 373, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.S.L.; Haddad, G.; DeBoer, G.; Ling, V.; Grogan, T.M. P-glycoprotein Expression: Critical Determinant in the Response to Osteosarcoma Chemotherapy. J. Natl. Cancer Inst. 1997, 89, 1706–1715. [Google Scholar] [CrossRef] [Green Version]
- Baldini, N.; Scotlandi, K.; Barbanti-Bròdano, G.; Manara, M.C.; Maurici, D.; Bacci, G.; Bertoni, F.; Picci, P.; Sottili, S.; Campanacci, M.; et al. Expression of P-Glycoprotein in High-Grade Osteosarcomas in Relation to Clinical Outcome. N. Engl. J. Med. 1995, 333, 1380–1385. [Google Scholar] [CrossRef]
- Baldini, N.; Scotlandi, K.; Serra, M.; Picci, P.; Bacci, G.; Sottili, S.; Campanacci, M. P-glycoprotein expression in osteosarcoma: A basis for risk-adapted adjuvant chemotherapy. J. Orthop. Res. 1999, 17, 629–632. [Google Scholar] [CrossRef]
- Serra, M.; Scotlandi, K.; Reverter-Branchat, G.; Ferrari, S.; Manara, M.C.; Benini, S.; Incaprera, M.; Bertoni, F.; Mercuri, M.; Briccoli, A.; et al. Value of P-Glycoprotein and Clinicopathologic Factors as the Basis for New Treatment Strategies in High-Grade Osteosarcoma of the Extremities. J. Clin. Oncol. 2003, 21, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Caronia, D.; Patiño-Garcia, A.; Peréz-Martínez, A.; Pita, G.; Moreno, L.T.; Zalacain-Díez, M.; Molina, B.; Colmenero, I.; Sierrasesúmaga, L.; Benítez, J.F.; et al. Effect of ABCB1 and ABCC3 Polymorphisms on Osteosarcoma Survival after Chemotherapy: A Pharmacogenetic Study. PLoS ONE 2011, 6, e26091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Kim, H.; Oh, J.; Lee, S. The co-expression of p53 protein and P-glycoprotein is correlated to a poor prognosis in osteosarcoma. Int. Orthop. 2001, 24, 307–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, M.; Maurici, D.; Scotlandi, K.; Barbanti-Brodano, G.; Manara, M.C.; Benini, S.; Picci, P.; Bertoni, F.; Bacci, G.; Sottili, S.; et al. Relationship between P-glycoprotein expression and p53 status in high-grade osteosarcoma. Int. J. Oncol. 1999, 14, 301–307. [Google Scholar] [CrossRef]
- Sørensen, F.B.; Jensen, K.; Vaeth, M.; Hager, H.; Funder, A.M.D.; Safwat, A.; Keller, J.; Christensen, M. Immunohistochemical Estimates of Angiogenesis, Proliferative Activity, p53 Expression, and Multiple Drug Resistance Have No Prognostic Impact in Osteosarcoma: A Comparative Clinicopathological Investigation. Sarcoma 2008, 2008, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gorlick, R.; Huvos, A.G.; Heller, G.; Aledo, A.; Beardsley, G.P.; Healey, J.H.; Meyers, P.A. Expression of HER2/erbB-2 Correlates With Survival in Osteosarcoma. J. Clin. Oncol. 1999, 17, 2781. [Google Scholar] [CrossRef]
- Kumta, S.M.; Zhu, Q.S.; Lee, K.M.; Griffith, J.; Chow, L.T.; Leung, P.C. Clinical significance of P-glycoprotein immunohistochemistry and doxorubicin binding assay in patients with osteosarcoma. Int. Orthop. 2001, 25, 279–282. [Google Scholar] [CrossRef] [Green Version]
- Pakos, E.E.; Ioannidis, J.P.A. The association of P-glycoprotein with response to chemotherapy and clinical outcome in patients with osteosarcoma. A meta-analysis. Cancer 2003, 98, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-Z. Association between P-Glycoprotein expression and response to chemotherapy in patients with osteosarcoma: A systematic and meta-analysis. J. Cancer Res. Ther. 2014, 10, 206–209. [Google Scholar]
- Schwartz, C.L.; Gorlick, R.; Teot, L.; Krailo, M.; Chen, Z.; Goorin, A.; Grier, H.E.; Bernstein, M.L.; Meyers, P. Multiple Drug Resistance in Osteogenic Sarcoma: INT0133 from the Children’s Oncology Group. J. Clin. Oncol. 2007, 25, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, D.; Zamboni, S.; Federici, C.; Lugini, L.; Lozupone, F.; De Milito, A.; Cecchetti, S.; Cianfriglia, M.; Fais, S. P-glycoprotein binds to ezrin at amino acid residues 149–242 in the FERM domain and plays a key role in the multidrug resistance of human osteosarcoma. Int. J. Cancer 2011, 130, 2824–2834. [Google Scholar] [CrossRef]
- Khanna, C.; Wan, X.; Bose, S.M.; Cassaday, R.D.; Olomu, O.; Mendoza, A.; Yeung, C.; Gorlick, R.; Hewitt, S.M.; Helman, L.J. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat. Med. 2004, 10, 182–186. [Google Scholar] [CrossRef]
- Yang, X.; Yang, P.; Shen, J.; Osaka, E.; Choy, E.; Cote, G.; Harmon, D.; Zhang, Z.; Mankin, H.; Hornicek, F.J.; et al. Prevention of multidrug resistance (MDR) in osteosarcoma by NSC23925. Br. J. Cancer 2014, 110, 2896–2904. [Google Scholar] [CrossRef]
- Lu, Y.; Li, F.; Xu, T.; Sun, J. Tetrandrine prevents multidrug resistance in the osteosarcoma cell line, U-2OS, by preventing Pgp overexpression through the inhibition of NF-κB signaling. Int. J. Mol. Med. 2017, 39, 993–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Liu, L.; Chunyan, W.; Wang, C.; Wang, Y.; Han, K.; Lin, S.; Gan, Z.; Min, D. Pleiotrophin promotes chemoresistance to doxorubicin in osteosarcoma by upregulating P-glycoprotein. Oncotarget 2017, 8, 63857–63870. [Google Scholar] [CrossRef]
- Liu, T.; Li, Z.; Zhang, Q.; Bernstein, K.D.A.; Lozano-Calderon, S.; Choy, E.; Hornicek, F.J.; Duan, Z. Targeting ABCB1 (MDR1) in multi-drug resistant osteosarcoma cells using the CRISPR-Cas9 system to reverse drug resistance. Oncotarget 2016, 7, 83502–83513. [Google Scholar] [CrossRef] [Green Version]
- Fanelli, M.; Hattinger, C.M.; Vella, S.; Tavanti, E.; Michelacci, F.; Gudeman, B.; Barnett, D.; Picci, P.; Serra, M. Targeting ABCB1 and ABCC1 with their Specific Inhibitor CBT-1® can Overcome Drug Resistance in Osteosarcoma. Curr. Cancer Drug Targets 2016, 16, 261–274. [Google Scholar] [CrossRef]
- Jin, S.; Gorfajn, B.; Faircloth, G.; Scotto, K.W. Ecteinascidin 743, a transcription-targeted chemotherapeutic that inhibits MDR1 activation. Proc. Natl. Acad. Sci. USA 2000, 97, 6775–6779. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-H.; Yang, H.-W.; Yang, L.-C.; Lu, M.-Y.; Tsai, L.-L.; Yang, S.-F.; Huang, Y.-F.; Chou, M.-Y.; Yu, C.-C.; Hu, F.-W. DHFR and MDR1 upregulation is associated with chemoresistance in osteosarcoma stem-like cells. Oncol. Lett. 2017, 14, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques da Costa, M.E.; Marchais, A.; Gomez-Brouchet, A.; Job, B.; Assoun, N.; Daudigeos-Dubus, E.; Fromigué, O.; Santos, C.; Geoerger, B.; Gaspar, N. In-Vitro and In-Vivo Establishment and Characterization of Bioluminescent Orthotopic Chemotherapy-Resistant Human Osteosarcoma Models in NSG Mice. Cancers 2019, 11, 997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Fan, J.; Hochhauser, D.; Banerjee, D.; Zieliński, Z.; Almasan, A.; Yin, Y.; Kelly, R.; Wahl, G.M.; Bertino, J.R. Lack of functional retinoblastoma protein mediates increased resistance to antimetabolites in human sarcoma cell lines. Proc. Natl. Acad. Sci. USA 1995, 92, 10436–10440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowers, R.; Toguchida, J.; Qin, J.; Meyers, P.A.; Healey, J.H.; Huvos, A.; Banerjee, D.; Bertino, J.R.; Gorlick, R. mRNA expression levels of E2F transcription factors correlate with dihydrofolate reductase, reduced folate carrier, and thymidylate synthase mRNA expression in osteosarcoma. Mol. Cancer Ther. 2003, 2, 535–541. [Google Scholar]
- Hattinger, C.M.; Stoico, G.; Michelacci, F.; Pasello, M.; Scionti, I.; Remondini, D.; Castellani, G.C.; Fanelli, M.; Scotlandi, K.; Picci, P.; et al. Mechanisms of gene amplification and evidence of coamplification in drug-resistant human osteosarcoma cell lines. Genes Chromosom. Cancer 2008, 48, 289–309. [Google Scholar] [CrossRef]
- Song, B.; Wang, Y.A.; Titmus, M.; Botchkina, G.; Formentini, A.; Kornmann, M.; Ju, J. Molecular mechanism of chemoresistance by miR-215 in osteosarcoma and colon cancer cells. Mol. Cancer 2010, 9, 96. [Google Scholar] [CrossRef] [Green Version]
- Bodley, A.; Liu, L.F.; Israel, M.; Seshadri, R.; Koseki, Y.; Giuliani, F.C.; Kirschenbaum, S.; Silber, R.; Potmesil, M. DNA topoisomerase II-mediated interaction of doxorubicin and daunorubicin congeners with DNA. Cancer Res. 1989, 49, 5969–5978. [Google Scholar] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, T.; Yamuna, M. Multiple pathways are involved in drug resistance to doxorubicin in an osteosarcoma cell line. Anti Cancer Drugs 2008, 19, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Z.; Ma, S.-R.; Rong, X.-L.; Zhu, M.-J.; Ji, Q.-Y.; Meng, L.-J.; Gao, Y.-Y.; Yang, Y.-D.; Wang, Y. Characterization of multidrug-resistant osteosarcoma sublines and the molecular mechanisms of resistance. Mol. Med. Rep. 2016, 14, 3269–3276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conklin, K.A. Chemotherapy-Associated Oxidative Stress: Impact on Chemotherapeutic Effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [Green Version]
- Kelland, L.R. Preclinical perspectives on platinum resistance. Drugs 2000, 59 (Suppl. 4), 1–8. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Yue, L.; Yokoi, A.; Nishimura, R.; Uehara, T.; Koizumi, S.; Saikawa, Y. A novel single-nucleotide polymorphism in the 3′-untranslated region of the human dihydrofolate reductase gene with enhanced expression. Clin. Cancer Res. 2001, 7, 1952–1956. [Google Scholar]
- Tew, K.D.; Monks, A.; Barone, L.; Rosser, D.; Akerman, G.; Montali, J.A.; Wheatley, J.B.; Schmidt, D.E. Glutathione-associated enzymes in the human cell lines of the National Cancer Institute Drug Screening Program. Mol. Pharmacol. 1996, 50, 149–159. [Google Scholar] [PubMed]
- Tew, K.D. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994, 54, 4313–4320. [Google Scholar] [CrossRef] [Green Version]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [Green Version]
- Masanek, U.; Stammler, G.; Volm, M. Messenger RNA expression of resistance proteins and related factors in human ovarian carcinoma cell lines resistant to doxorubicin, taxol and cisplatin. Anti Cancer Drugs 1997, 8, 189–198. [Google Scholar] [CrossRef]
- Ban, N.; Takahashi, Y.; Takayama, T.; Kura, T.; Katahira, T.; Sakamaki, S.; Niitsu, Y. Transfection of glutathione S-transferase (GST)-pi antisense complementary DNA increases the sensitivity of a colon cancer cell line to adriamycin, cisplatin, melphalan, and etoposide. Cancer Res. 1996, 56, 3577–3582. [Google Scholar]
- Batist, G.; Tulpule, A.; Sinha, B.K.; Katki, A.G.; Myers, C.; Cowan, K.H. Overexpression of a novel anionic glutathione transferase in multidrug-resistant human breast cancer cells. J. Biol. Chem. 1986, 261, 15544–15549. [Google Scholar] [CrossRef]
- Bai, F.; Nakanishi, Y.; Kawasaki, M.; Takayama, K.; Yatsunami, J.; Pei, X.H.; Tsuruta, N.; Wakamatsu, K.; Hara, N. Immunohistochemical expression of glutathione S-transferase-Pi can predict chemotherapy response in patients with nonsmall cell lung carcinoma. Cancer 1996, 78, 416–421. [Google Scholar] [CrossRef]
- Pasello, M.; Michelacci, F.; Scionti, I.; Hattinger, C.M.; Zuntini, M.; Caccuri, A.M.; Scotlandi, K.; Picci, P.; Serra, M. Overcoming Glutathione S-Transferase P1–Related Cisplatin Resistance in Osteosarcoma. Cancer Res. 2008, 68, 6661–6668. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Song, X.; Wang, X.-W.; Li, M.; Uo, W.-S.Z. Expression of MDR1 and GST-pi in osteosarcoma and soft tissue sarcoma and their correlation with chemotherapy resistance. Zhonghua Zhong Liu Za Zhi Chin. J. Oncol. 2006, 28, 445–448. [Google Scholar]
- Huang, G.; Mills, L.; Worth, L.L. Expression of human glutathione S-transferase P1 mediates the chemosensitivity of osteosarcoma cells. Mol. Cancer Ther. 2007, 6, 1610–1619. [Google Scholar] [CrossRef] [Green Version]
- Komiya, S.; Gobhardt, M.C.; Mangham, D.C.; Inoue, A. Role of glutathione in cisplatin resistance in osteosarcoma cell lines. J. Orthop. Res. 1998, 16, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Shoieb, A.M.; Hahn, K.A.; Van Laack, R.L.; Barnhill, M.A. In vitro reversal of glutathione-S-transferase-mediated resistance in canine osteosarcoma (COS31) cells. In Vivo 1998, 12, 455–462. [Google Scholar] [PubMed]
- Shoieb, A.; Hahn, K. Detection and significance of glutathione-S-transferase pi in osteogenic tumors of dogs. Int. J. Oncol. 1997, 10, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Bruheim, S.; Bruland, O.S.; Breistol, K.; Maelandsmo, G.M.; Øystein, F. Human osteosarcoma xenografts and their sensitivity to chemotherapy. Pathol. Oncol. Res. 2004, 10, 133–141. [Google Scholar] [CrossRef]
- Uozaki, H.; Horiuchi, H.; Ishida, T.; Iijima, T.; Imamura, T.; Machinami, R. Overexpression of resistance-related proteins (metallothioneins, glutathione-S-transferase pi, heat shock protein 27, and lung resistance-related protein) in osteosarcoma. Relationship with poor prognosis. Cancer 1997, 79, 2336–2344. [Google Scholar] [CrossRef]
- Windsor, R.E.; Strauss, S.J.; Kallis, C.; Wood, N.E.; Whelan, J.S. Germline genetic polymorphisms may influence chemotherapy response and disease outcome in osteosarcoma. Cancer 2011, 118, 1856–1867. [Google Scholar] [CrossRef]
- Zhang, S.-L.; Mao, N.-F.; Sun, J.-Y.; Shi, Z.-C.; Wang, B.; Sun, Y.-J. Predictive Potential of Glutathione S-Transferase Polymorphisms for Prognosis of Osteosarcoma Patients on Chemotherapy. Asian Pac. J. Cancer Prev. 2012, 13, 2705–2709. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.-M.; Li, X.-H.; Bao, C.-F. Glutathione S-transferase P1 and DNA polymorphisms influence response to chemotherapy and prognosis of bone tumors. Asian Pac. J. Cancer Prev. 2012, 13, 5883–5886. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, H.; He, M.; Wu, H.; Zhu, Y.; Su, Z. The association of glutathione S-transferase polymorphisms in patients with osteosarcoma: Evidence from a meta-analysis. Eur. J. Cancer Care 2014, 24, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yi, Z.; Ling, M.; Shi, J.; Qiu, Y.; Yang, S. Predictive potential of ABCB1, ABCC3, and GSTP1 gene polymorphisms on osteosarcoma survival after chemotherapy. Tumor Biol. 2014, 35, 9897–9904. [Google Scholar] [CrossRef]
- Salinas-Souza, C.; Petrilli, A.S.; De Toledo, S.R. Glutathione S-transferase polymorphisms in osteosarcoma patients. Pharmacogenet. Genom. 2010, 20, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Ivanov, V.N.; Habelhah, H.; Tew, K.; Ronai, Z. Glutathione S-transferase p elicits protection against H2O2-induced cell death via coordinated regulation of stress kinases. Cancer Res. 2000, 60, 4053–4057. [Google Scholar] [PubMed]
- Adler, V.; Yin, Z.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.; et al. Regulation of JNK signaling by GSTp. EMBO J. 1999, 18, 1321–1334. [Google Scholar] [CrossRef]
- Lu, M.; Xia, L.; Luo, D.; Waxman, S.; Jing, Y. Dual effects of glutathione-S-transferase pi on As2O3 action in prostate cancer cells: Enhancement of growth inhibition and inhibition of apoptosis. Oncogene 2004, 23, 3945–3952. [Google Scholar] [CrossRef] [Green Version]
- Pasello, M.; Manara, M.C.; Michelacci, F.; Fanelli, M.; Hattinger, C.M.; Nicoletti, G.; Landuzzi, L.; Lollini, P.L.; Caccuri, A.; Picci, P.; et al. Targeting glutathione-S transferase enzymes in musculoskeletal sarcomas: A promising therapeutic strategy. Anal. Cell. Pathol. 2011, 34, 131–145. [Google Scholar] [CrossRef]
- Sau, A.; Filomeni, G.; Pezzola, S.; D’Aguanno, S.; Tregno, F.P.; Urbani, A.; Pasello, M.; Serra, M.; Picci, P.; Federici, G.; et al. Targeting GSTP1-1 induces JNK activation and leads to apoptosis in cisplatin-sensitive and -resistant human osteosarcoma cell lines. Mol. BioSyst. 2012, 8, 994–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Fanelli, M.; Tavanti, E.; Patrizio, M.P.; Vella, S.; Fernandez-Ramos, A.; Magagnoli, F.; Luppi, S.; Hattinger, C.M.; Serra, M. Cisplatin Resistance in Osteosarcoma: In vitro Validation of Candidate DNA Repair-Related Therapeutic Targets and Drugs for Tailored Treatments. Front. Oncol. 2020, 10, 331. [Google Scholar] [CrossRef] [Green Version]
- Fishel, M.L.; Kelley, M.R. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol. Asp. Med. 2007, 28, 375–395. [Google Scholar] [CrossRef]
- Wang, N.; Luo, M.; Kelley, M.R. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: Enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol. Cancer Ther. 2004, 3, 679–686. [Google Scholar] [PubMed]
- Yang, J.; Yang, D.; Cogdell, D.; Du, X.; Li, H.; Pang, Y.; Sun, Y.; Hu, L.; Sun, B.; Trent, J.; et al. APEX1 Gene Amplification and Its Protein Overexpression in Osteosarcoma: Correlation with Recurrence, Metastasis, and Survival. Technol. Cancer Res. Treat. 2010, 9, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Irani, K.; Heffron, S.E.; Jurnak, F.; Meyskens, F.L. Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE/Ref-1) in human melanoma and identification of the therapeutic potential of resveratrol as an APE/Ref-1 inhibitor. Mol. Cancer Ther. 2005, 4, 1923–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Kelley, M.R. Inhibition of the human apurinic/apyrimidinic endonuclease (APE1) repair activity and sensitization of breast cancer cells to DNA alkylating agents with lucanthone. Anticancer Res. 2004, 24, 2127–2134. [Google Scholar] [PubMed]
- Madhusudan, S.; Smart, F.; Shrimpton, P.; Parsons, J.L.; Gardiner, L.; Houlbrook, S.; Talbot, D.C.; Hammonds, T.; Freemont, P.A.; Sternberg, M.J.E.; et al. Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res. 2005, 33, 4711–4724. [Google Scholar] [CrossRef] [PubMed]
- Seiple, L.A.; Cardellina, J.H.; Akee, R.; Stivers, J.T. Potent Inhibition of Human Apurinic/Apyrimidinic Endonuclease 1 by Arylstibonic Acids. Mol. Pharmacol. 2007, 73, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Dai, N.; Qing, Y.; Cun, Y.; Zhong, Z.; Li, C.; Zhang, S.; Shan, J.; Yang, X.; Dai, X.; Cheng, Y.; et al. miR-513a-5p regulates radiosensitivity of osteosarcoma by targeting human apurinic/apyrimidinic endonuclease. Oncotarget 2016, 9, 25414–25426. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Li, C.; Li, M.; Wang, D.; Zhong, Z. MicroRNA-765 sensitizes osteosarcoma cells to cisplatin via downregulating APE1 expression. OncoTargets Ther. 2019, 12, 7203–7214. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhong, Z.-Y.; Li, M.-X.; Xiang, D.-B.; Li, Z.-P. Vector-based Ape1 small interfering RNA enhances the sensitivity of human osteosarcoma cells to endostatin in vivo. Cancer Sci. 2007, 98, 1993–2001. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Bae, J.S.; Kim, K.M.; Moon, Y.J.; Park, S.-H.; Ha, S.H.; Hussein, U.K.; Zhang, Z.; Park, H.S.; Park, B.-H.; et al. The PARP inhibitor olaparib potentiates the effect of the DNA damaging agent doxorubicin in osteosarcoma. J. Exp. Clin. Cancer Res. 2018, 37, 107. [Google Scholar] [CrossRef]
- National Cancer Institute (NCI). NCI-COG Pediatric MATCH (Molecular Analysis for Therapy Choice)-Phase 2 Subprotocol of Olaparib in Patients With Tumors Harboring Defects in DNA Damage Repair Genes. Report No.: NCT03233204. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03233204 (accessed on 20 December 2020).
- Janeway, K. Phase II Trial of Olaparib in Combination with Ceralasertib in Patients With Recurrent Osteosarcoma. Report No.: NCT04417062. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04417062 (accessed on 20 December 2020).
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Hattinger, C.M.; Michelacci, F.; Sella, F.; Magagnoli, G.; Benini, S.; Gambarotti, M.; Palmerini, E.; Picci, P.; Serra, M.; Ferrari, S. Excision repair cross-complementation group 1 protein expression predicts survival in patients with high-grade, non-metastatic osteosarcoma treated with neoadjuvant chemotherapy. Histopathology 2015, 67, 338–347. [Google Scholar] [CrossRef]
- Nathrath, M.; Kremer, M.; Letzel, H.; Remberger, K.; Höfler, H.; Ulle, T. Expression of genes of potential importance in the response to chemotherapy in osteosarcoma patients. Klin. Padiatr. 2002, 214, 230–235. [Google Scholar] [CrossRef]
- Li, X.; Guo, W.; Shen, D.-H.; Yang, R.-L.; Liu, J.; Zhao, H. Expressions of ERCC2 and ERCC4 genes in osteosarcoma and peripheral blood lymphocytes and their clinical significance. Beijing Da Xue Xue Bao 2007, 39, 467–471. [Google Scholar]
- Caronia, D.; Patinogarcia, A.; Milne, R.L.; Zalacaindiez, M.; Pita, G.; Alonso, M.R.; Moreno, L.T.; Sierrasesumaga-Ariznabarreta, L.; Benitez, J.; Gonzalezneira, A. Common variations in ERCC2 are associated with response to cisplatin chemotherapy and clinical outcome in osteosarcoma patients. Pharmacogenom. J. 2009, 9, 347–353. [Google Scholar] [CrossRef]
- Hao, T.; Feng, W.; Zhang, J.; Sun, Y.-J.; Wang, G. Association of four ERCC1 and ERCC2 SNPs with survival of bone tumour patients. Asian Pac. J. Cancer Prev. 2012, 13, 3821–3824. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Z.; Deng, C.; Tian, Y.; Ma, X. Meta-analysis showing that ERCC1 polymorphism is predictive of osteosarcoma prognosis. Oncotarget 2017, 8, 62769–62779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obiedat, H.; Alrabadi, N.; Sultan, I.; Al Shatti, M.; Zihlif, M. The effect of ERCC1 and ERCC2 gene polymorphysims on response to cisplatin based therapy in osteosarcoma patients. BMC Med. Genet. 2018, 19, 1–9. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6035436/ (accessed on 21 October 2020). [CrossRef]
- DeChant, M.J.; Ewerbeck, V.; Mau, H. Identification of drug-regulated genes in osteosarcoma cells. Int. J. Cancer 2003, 105, 636–643. [Google Scholar] [CrossRef]
- Liebermann, D.A.; Hoffman, B.; Steinman, R.A. Molecular controls of growth arrest and apoptosis: p53-dependent and independent pathways. Oncogene 1995, 11, 199–210. [Google Scholar]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Rao, P.H.; Favis, R.; Lu, X.-Y.; Elowitz, M.B.; Barany, F.; Ladanyi, M.; Gorlick, R.; Levine, A.J. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc. Natl. Acad. Sci. USA 2003, 100, 11547–11552. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.P.C.; Tsang, W.P.; Chau, P.Y.; Na Co, N.; Tsang, T.Y.; Kwok, T.T. p53-R273H gains new function in induction of drug resistance through down-regulation of procaspase-3. Mol. Cancer Ther. 2007, 6, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Sato, N.; Mizumoto, K.; Maehara, N.; Kusumoto, M.; Nishio, S.; Urashima, T.; Ogawa, T.; Tanaka, M. Enhancement of drug-induced apoptosis by antisense oligodeoxynucleotides targeted against Mdm2 and p21WAF1/CIP1. Anticancer Res. 2000, 20, 837–842. [Google Scholar]
- Asada, N.; Tsuchiya, H.; Tomita, K. De novo deletions of p53 gene and wild-type p53 correlate with acquired cisplatin-resistance in human osteosarcoma OST cell line. Anticancer Res. 2000, 19, 5131–5137. [Google Scholar]
- Tsuchiya, H.; Mori, Y.; Ueda, Y.; Okada, G.; Tomita, K. Sensitization and caffeine potentiation of cisplatin cytotoxicity resulting from introduction of wild-type p53 gene in human osteosarcoma. Anticancer Res. 2000, 20, 235–242. [Google Scholar]
- Fan, J.; Bertino, J.R. Modulation of Cisplatinum Cytotoxicity by p53: Effect of p53-Mediated Apoptosis and DNA Repair. Mol. Pharmacol. 1999, 56, 966–972. [Google Scholar] [CrossRef]
- Song, B.; Wang, Y.; Xi, Y.; Kudo, K.; Bruheim, S.; Botchkina, G.I.; Gavin, E.; Formentini, A.; Kornmann, M.; Fodstad, O.; et al. Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene 2009, 28, 4065–4074. [Google Scholar] [CrossRef] [Green Version]
- Goto, A.; Kanda, H.; Ishikawa, Y.; Matsumoto, S.; Kawaguchi, N.; Machinami, R.; Kato, Y.; Kitagawa, T. Association of Loss of Heterozygosity at the p53 Locus with Chemoresistance in Osteosarcomas. Jpn. J. Cancer Res. 1998, 89, 539–547. [Google Scholar] [CrossRef]
- Ferrari, S.; Bertoni, F.; Zanella, L.; Setola, E.; Bacchini, P.; Alberghini, M.; Versari, M.; Bacci, G. Evaluation of P-glycoprotein, HER-2/ErbB-2, p53, and Bcl-2 in primary tumor and metachronous lung metastases in patients with high-grade osteosarcoma. Cancer 2004, 100, 1936–1942. [Google Scholar] [CrossRef] [PubMed]
- Rossner, J.P.; Gammon, M.D.; Zhang, Y.-J.; Terry, M.B.; Hibshoosh, H.; Memeo, L.; Mansukhani, M.; Long, C.-M.; Garbowski, G.; Agrawal, M.; et al. Mutations in p53, p53 protein overexpression and breast cancer survival. J. Cell. Mol. Med. 2008, 13, 3847–3857. [Google Scholar] [CrossRef] [PubMed]
- Pakos, E.E.; Kyzas, P.A.; Ioannidis, J.P.A. Prognostic significance of TP53 tumor suppressor gene expression and mutations in human osteosarcoma: A meta-analysis. Clin. Cancer Res. 2004, 10 Pt 1, 6208–6214. [Google Scholar] [CrossRef] [Green Version]
- Wunder, J.S.; Gokgoz, N.; Parkes, R.; Bull, S.B.; Eskandarian, S.; Davis, A.M.; Beauchamp, C.P.; Conrad, E.U.; Grimer, R.J.; Healey, J.H.; et al. TP53 Mutations and Outcome in Osteosarcoma: A Prospective, Multicenter Study. J. Clin. Oncol. 2005, 23, 1483–1490. [Google Scholar] [CrossRef]
- Hata, A.N.; Engelman, J.A.; Faber, A.C. The BCL-2 family: Key mediators of the apoptotic response to targeted anti-cancer therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, J.C. Double identity for proteins of the Bcl-2 family. Nat. Cell Biol. 1997, 387, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, C.-L.; Zeng, B.-F.; Wu, X.-S.; Gao, T.-T.; Oda, Y. Enhanced chemosensitivity of drug-resistant osteosarcoma cells by lentivirus-mediated Bcl-2 silencing. Biochem. Biophys. Res. Commun. 2009, 390, 642–647. [Google Scholar] [CrossRef]
- Eliseev, R.A.; Dong, Y.-F.; Sampson, E.; Zuscik, M.J.; Schwarz, E.M.; O’Keefe, R.J.; Rosier, R.N.; Drissi, H. Runx2-mediated activation of the Bax gene increases osteosarcoma cell sensitivity to apoptosis. Oncogene 2008, 27, 3605–3614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zangemeister-Wittke, U. Antisense to apoptosis inhibitors facilitates chemotherapy and TRAIL-induced death signaling. Ann. N. Y. Acad. Sci. 2003, 1002, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, J.; Park, B.H.; Kinzler, K.W.; Vogelstein, B. Role of BAX in the Apoptotic Response to Anticancer Agents. Science 2000, 290, 989–992. [Google Scholar] [CrossRef]
- Wu, X.; Cai, Z.-D.; Lou, L.-M.; Zhu, Y.-B. Expressions of p53, c-MYC, BCL-2 and apoptotic index in human osteosarcoma and their correlations with prognosis of patients. Cancer Epidemiol. 2012, 36, 212–216. [Google Scholar] [CrossRef]
- Nedelcu, T.; Kubista, B.; Koller, A.; Sulzbacher, I.; Mosberger, I.; Arrich, F.; Trieb, K.; Kotz, R.; Toma, C.D. Livin and Bcl-2 expression in high-grade osteosarcoma. J. Cancer Res. Clin. Oncol. 2007, 134, 237–244. [Google Scholar] [CrossRef]
- Konstadoulakis, M.M.; Khaldi, L.; Gomatos, I.P.; Tzagarakis, G.P.; Alevizos, L.; Leandros, E.; Papagelopoulos, P.J.; Soucacos, P.N. Prognostic value of bax, bcl-2, and p53 staining in primary osteosarcoma. J. Surg. Oncol. 2008, 97, 259–266. [Google Scholar] [CrossRef]
- Patatsos, K.; Shekhar, T.M.; Hawkins, C.J. Pre-clinical evaluation of proteasome inhibitors for canine and human osteosarcoma. Vet. Comp. Oncol. 2018, 16, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignochino, Y.; Grignani, G.; Cavalloni, G.; Motta, M.; Tapparo, M.; Bruno, S.; Bottos, A.; Gammaitoni, L.; Migliardi, G.; Camussi, G.; et al. Sorafenib blocks tumour growth, angiogenesis and metastatic potential in preclinical models of osteosarcoma through a mechanism potentially involving the inhibition of ERK1/2, MCL-1 and ezrin pathways. Mol. Cancer 2009, 8, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, D.P.M.; Thomas, D.G.; Giordano, T.J.; Baker, L.H.; McDonagh, K.T. Cell Surface Expression of Epidermal Growth Factor Receptor and Her-2 with Nuclear Expression of Her-4 in Primary Osteosarcoma. Cancer Res. 2004, 64, 2047–2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Chen, S.; Deng, Z.; Long, H.; Zhang, S.; Chen, X. VEGF and EMMPRIN expression correlates with survival of patients with osteosarcoma. Surg. Oncol. 2011, 20, 13–19. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, J.; Dai, R.; Xu, J.; Feng, H. Transferrin receptor-1 and VEGF are prognostic factors for osteosarcoma. J. Orthop. Surg. Res. 2019, 14, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebb, D.H.; Meyers, P.; Grier, H.; Bernstein, M.; Gorlick, R.; Lipshultz, S.E.; Krailo, M.; Devidas, M.; Barkauskas, D.A.; Siegal, G.P.; et al. Phase II Trial of Trastuzumab in Combination With Cytotoxic Chemotherapy for Treatment of Metastatic Osteosarcoma With Human Epidermal Growth Factor Receptor 2 Overexpression: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 2545–2551. [Google Scholar] [CrossRef] [Green Version]
- Maris, J.M.; Courtright, J.; Houghton, P.J.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Lock, R.; Tajbakhsh, M.; Reynolds, C.P.; Keir, S.T.; et al. Initial testing of the VEGFR inhibitor AZD2171 by the pediatric preclinical testing program. Pediatr. Blood Cancer 2008, 50, 581–587. [Google Scholar] [CrossRef] [PubMed]
- MacEwen, E.G.; Pastor, J.; Kutzke, J.; Tsan, R.; Kurzman, I.D.; Thamm, D.H.; Wilson, M.; Radinsky, R. IGF-1 receptor contributes to the malignant phenotype in human and canine osteosarcoma. J. Cell. Biochem. 2004, 92, 77–91. [Google Scholar] [CrossRef]
- Hassan, S.E.; Ba, M.B.; Kim, M.Y.; Lin, J.; Bs, S.P.; Gorlick, R.; Geller, D.S. Cell surface receptor expression patterns in osteosarcoma. Cancer 2011, 118, 740–749. [Google Scholar] [CrossRef]
- Wang, Y.; Han, X.-D.; Qiu, Y.; Xiong, J.; Yu, Y.; Wang, B.; Zhu, Y.Q.Z.-Z.; Qian, B.-P.; Chen, Y.-X.; Wang, S.F.; et al. Increased expression of insulin-like growth factor-1 receptor is correlated with tumor metastasis and prognosis in patients with osteosarcoma. J. Surg. Oncol. 2011, 105, 235–243. [Google Scholar] [CrossRef]
- Luk, F.; Yu, Y.; Walsh, W.R.; Yang, J.-L. IGF1R-Targeted Therapy and Its Enhancement of Doxorubicin Chemosensitivity in Human Osteosarcoma Cell Lines. Cancer Investig. 2011, 29, 521–532. [Google Scholar] [CrossRef]
- Rettew, A.N.; Young, E.D.; Lev, D.; Kleinerman, E.S.; Abdulkarim, F.W.; Getty, P.J.; Greenfield, E.M. Multiple receptor tyrosine kinases promote the in vitro phenotype of metastatic human osteosarcoma cell lines. Oncogenesis 2012, 1, e34. [Google Scholar] [CrossRef]
- Anderson, P.M.; Bielack, S.S.; Gorlick, R.G.; Skubitz, K.; Daw, N.C.; Herzog, C.E.; Monge, O.R.; Lassaletta, A.; Boldrini, E.; Pápai, Z.; et al. A phase II study of clinical activity of SCH 717454 (robatumumab) in patients with relapsed osteosarcoma and Ewing sarcoma. Pediatr. Blood Cancer 2016, 63, 1761–1770. [Google Scholar] [CrossRef]
- Chitnis, M.M.; Yuen, J.S.; Protheroe, A.; Pollak, M.; Macaulay, V.M. The Type 1 Insulin-Like Growth Factor Receptor Pathway. Clin. Cancer Res. 2008, 14, 6364–6370. [Google Scholar] [CrossRef] [Green Version]
- Analysis of Aberrant Signal Transduction Pathways in Osteosarcoma Cell Lines. Cancer Research. Available online: https://cancerres.aacrjournals.org/content/65/9_Supplement/1075.2 (accessed on 8 November 2020).
- Adamopoulos, C.; Gargalionis, A.N.; Basdra, E.K.; Papavassiliou, A.G. Deciphering signaling networks in osteosarcoma pathobiology. Exp. Biol. Med. 2016, 241, 1296–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuijjer, M.L.; van den Akker, B.E.; Hilhorst, R.; Mommersteeg, M.; Buddingh, E.P.; Serra, M.; Buerger, H.; Hogendoorn, P.C.; Cleton-Jansen, A.-M. Kinome and mRNA expression profiling of high-grade osteosarcoma cell lines implies Akt signaling as possible target for therapy. BMC Med. Genom. 2014, 7, 4. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Gonzalez-Angulo, A.M. Targeting the mTOR Signaling Network for Cancer Therapy. J. Clin. Oncol. 2009, 27, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Gordon, I.K.; Ye, F.; Kent, M.S. Evaluation of the mammalian target of rapamycin pathway and the effect of rapamycin on target expression and cellular proliferation in osteosarcoma cells from dogs. Am. J. Vet. Res. 2008, 69, 1079–1084. [Google Scholar] [CrossRef]
- Gazitt, Y.; Kolaparthi, V.; Moncada, K.; Thomas, C.; Freeman, J. Targeted therapy of human osteosarcoma with 17AAG or rapamycin: Characterization of induced apoptosis and inhibition of mTOR and Akt/MAPK/Wnt pathways. Int. J. Oncol. 1992, 34, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Mendoza, A.; Khanna, C.; Helman, L.J. Rapamycin Inhibits Ezrin-Mediated Metastatic Behavior in a Murine Model of Osteosarcoma. Cancer Res. 2005, 65, 2406–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houghton, P.J.; Gorlick, R.; Kolb, E.A.; Lock, R.B.; Carol, H.; Bs, C.L.M.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Phelps, D.; et al. Initial testing (stage 1) of the mTOR kinase inhibitor AZD8055 by the pediatric preclinical testing program. Pediatr. Blood Cancer 2011, 58, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Chandhanayingyong, C.; Kim, Y.; Staples, J.R.; Hahn, C.; Lee, F.Y. MAPK/ERK Signaling in Osteosarcomas, Ewing Sarcomas and Chondrosarcomas: Therapeutic Implications and Future Directions. Sarcoma 2012, 2012, e404810. [Google Scholar] [CrossRef]
- Cai, Y.; Cai, T.; Chen, Y. Wnt Pathway in Osteosarcoma, from Oncogenic to Therapeutic. J. Cell. Biochem. 2014, 115, 625–631. [Google Scholar] [CrossRef]
- Vijayakumar, S.; Liu, G.; Rus, I.A.; Yao, S.; Chen, Y.; Akiri, G.; Grumolato, L.; Aaronson, S.A. High-Frequency Canonical Wnt Activation in Multiple Sarcoma Subtypes Drives Proliferation through a TCF/β-Catenin Target Gene, CDC25A. Cancer Cell 2011, 19, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Leow, P.-C.; Tian, Q.; Ong, Z.Y.; Yang, Z.; Ee, R.P.-L. Antitumor activity of natural compounds, curcumin and PKF118-310, as Wnt/β-catenin antagonists against human osteosarcoma cells. Investig. New Drugs 2009, 28, 766–782. [Google Scholar] [CrossRef]
- Guo, Y.; Rubin, E.M.; Xie, J.; Zi, X.; Hoang, B.H. Dominant Negative LRP5 Decreases Tumorigenicity and Metastasis of Osteosarcoma in an Animal Model. Clin. Orthop. Relat. Res. 2008, 466, 2039–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansara, M.; Tsang, M.; Kodjabachian, L.; Sims, N.A.; Trivett, M.K.; Ehrich, M.; Dobrovic, A.; Slavin, J.; Choong, P.F.; Simmons, P.J.; et al. Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J. Clin. Investig. 2009, 119, 837–851. [Google Scholar] [CrossRef] [Green Version]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef]
- Danieau, G.; Morice, S.; Redini, F.; Verrecchia, F.; Le Royer, B.B. New Insights about the Wnt/β-Catenin Signaling Pathway in Primary Bone Tumors and Their Microenvironment: A Promising Target to Develop Therapeutic Strategies? Int. J. Mol. Sci. 2019, 20, 3751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieudonné, F.-X.; Marion, A.; Marie, P.J.; Modrowski, D. Targeted inhibition of T-cell factor activity promotes syndecan-2 expression and sensitization to doxorubicin in osteosarcoma cells and bone tumors in mice. J. Bone Miner. Res. 2012, 27, 2118–2129. [Google Scholar] [CrossRef]
- Jiang, J.; Tian, S.; Yu, C.; Chen, M.; Sun, C. TRIM37 promoted the growth and migration of the pancreatic cancer cells. Tumor Biol. 2015, 37, 2629–2634. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Xin, M.; Cheng, H.; Huang, Z.; Huanchen, C.; Zhang, T.; Wang, J. TRIM37 promotes tumor cell proliferation and drug resistance in pediatric osteosarcoma. Oncol. Lett. 2017, 14, 6365–6372. [Google Scholar] [CrossRef]
- Fang, F.; VanCleave, A.; Helmuth, R.; Torres, H.; Rickel, K.; Wollenzien, H.; Sun, H.; Zeng, E.; Zhao, J.; Tao, J. Targeting the Wnt/β-catenin pathway in human osteosarcoma cells. Oncotarget 2018, 9, 36780–36792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Harb, J.; Lin, P.-J.; Hao, J. Recent Development of Wnt Signaling Pathway Inhibitors for Cancer Therapeutics. Curr. Oncol. Rep. 2019, 21, 12. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiuri, M.; Zalckvar, E.; Kimchi, A.; Kroemer, G.; Maiuri, M.C.; Zalckvar, E.; Kimchi, A. Kroemer GSelf-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, S.G.; Kim, Y.J.; Park, J.-E.; Lee, K.Y.; Yoo, Y.H.; Kim, J.-M. Cytoprotective role of autophagy during paclitaxel-induced apoptosis in Saos-2 osteosarcoma cells. Int. J. Oncol. 2013, 42, 1985–1992. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Shao, Z.; Xiong, L.; Che, B.; Deng, C.; Xu, W. Expression of Beclin1 in osteosarcoma and the effects of down-regulation of autophagy on the chemotherapeutic sensitivity. J. Huazhong Univ. Sci. Technol. Med. Sci. 2009, 29, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Coupienne, I.; Fettweis, G.; Piette, J. RIP3 expression induces a death profile change in U2OS osteosarcoma cells after 5-ALA-PDT. Lasers Surg. Med. 2011, 43, 557–564. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Cheh, C.-W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.A.; Tracey, K.J.; et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.J.; Cao, L.; Tang, D.; Ni, J. Targeting HMGB1-mediated autophagy as a novel therapeutic strategy for osteosarcoma. Autophagy 2012, 8, 275–277. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Ni, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.; Cao, L.; Tang, D. HMGB1 Promotes Drug Resistance in Osteosarcoma. Cancer Res. 2012, 72, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Gao, R.; Wang, J.; Yuan, W.; Wang, C.; Zhou, X. High-mobility group nucleosome-binding domain 5 increases drug resistance in osteosarcoma through upregulating autophagy. Tumor Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 6357–6363. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, W.; Li, Y.; Yang, D.; Li, X.; Shen, C.; Liu, Y.; Ke, X.; Guo, S.; Guo, Z. HSP90AA1-mediated autophagy promotes drug resistance in osteosarcoma. J. Exp. Clin. Cancer Res. 2018, 37, 201. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Carew, J.S.; Medina, E.C.; Ii, J.A.E.; Mahalingam, D.; Swords, R.; Kelly, K.; Zhang, H.; Huang, P.; Mita, A.C.; Mita, M.M.; et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell. Mol. Med. 2009, 14, 2448–2459. [Google Scholar] [CrossRef]
- Shen, C.; Wang, W.; Tao, L.; Liu, B.; Yang, Z.; Tao, H. Chloroquine blocks the autophagic process in cisplatin-resistant osteosarcoma cells by regulating the expression of p62/SQSTM1. Int. J. Mol. Med. 2013, 32, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.D.; Sun, W.; Hua, Y.Q.; Wang, S.G.; Cai, Z.D. Effect of rapamycin and chloroquine on osteosarcoma. Zhonghua Yi Xue Za Zhi 2017, 97, 1510–1514. [Google Scholar] [PubMed]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Al Mazeedi, M.A.M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.; Wei, X. Cancer stem cells and drug resistance: The potential of nanomedicine. Nanomedicine 2012, 7, 597–615. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Ma, W.; Jha, R.K.; Gurung, K. Cancer stem cells in osteosarcoma: Recent progress and perspective. Acta Oncol. 2011, 50, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, H.; Zheng, J.; Yu, P.; Xu, L.; Jiang, P.; Gao, J.; Wang, H.; Zhang, Y. Transforming Growth Factor β1 Signal is Crucial for Dedifferentiation of Cancer Cells to Cancer Stem Cells in Osteosarcoma. Stem Cells 2013, 31, 433–446. [Google Scholar] [CrossRef]
- Gibbs, C.P.; Kukekov, V.G.; Reith, J.D.; Tchigrinova, O.; Suslov, O.N.; Scott, E.W.; Ghivizzani, S.C.; Ignatova, T.N.; Steindler, D.A. Stem-Like Cells in Bone Sarcomas: Implications for Tumorigenesis. Neoplasia 2005, 7, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Tirino, V.; Desiderio, V.; d’Aquino, R.; De Francesco, F.; Pirozzi, G.; Graziano, A.; Galderisi, U.; Cavaliere, C.; de Rosa, A.; Papaccio, G. Detection and characterization of CD133+ cancer stem cells in human solid tumours. PLoS ONE 2008, 3, e3469. [Google Scholar] [CrossRef]
- Di Fiore, R.; Santulli, A.; Ferrante, R.D.; Giuliano, M.; De Blasio, A.; Messina, C.; Pirozzi, G.; Tirino, V.; Tesoriere, G.; Vento, R. Identification and expansion of human osteosarcoma-cancer-stem cells by long-term 3-aminobenzamide treatment. J. Cell. Physiol. 2009, 219, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Martins-Neves, S.R.; Lopes, Á.O.; do Carmo, A.; Paiva, A.A.; Simões, P.C.; Abrunhosa, A.J.; Gomes, C.M.F. Therapeutic implications of an enriched cancer stem-like cell population in a human osteosarcoma cell line. BMC Cancer 2012, 12, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, H.; Honoki, K.; Tsujiuchi, T.; Kido, A.; Yoshitani, K.; Takakura, Y. Sphere-forming stem-like cell populations with drug resistance in human sarcoma cell lines. Int. J. Oncol. 2009, 34, 1381–1386. [Google Scholar] [PubMed]
- Honoki, K.; Fujii, H.; Kubo, A.; Kido, A.; Mori, T.; Tanaka, Y.; Tsujiuchi, T. Possible involvement of stem-like populations with elevated ALDH1 in sarcomas for chemotherapeutic drug resistance. Oncol. Rep. 2010, 24, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-H.; Morrow, J.K.; Li, C.-F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.E.; Cai, Z.; Xu, D.; et al. Pharmacological Inactivation of Skp2 SCF Ubiquitin Ligase Restricts Cancer Stem Cell Traits and Cancer Progression. Cell 2013, 154, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Wang, C.; Cui, Y.; Han, X.; Zhou, Y.; Bai, J.; Li, R. S-phase kinase-associated protein 2 is involved in epithelial-mesenchymal transition in methotrexate-resistant osteosarcoma cells. Int. J. Oncol. 2018, 52, 1841–1852. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Guo, Y.; Yang, Y.; Kan, J.; Dai, S.; Helian, M.; Li, B.; Xu, J.; Liu, C. Nitidine chloride suppresses epithelial-to-mesenchymal transition in osteosarcoma cell migration and invasion through Akt/GSK-3β/Snail signaling pathway. Oncol. Rep. 2016, 36, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Laverdière, C.; Kolb, E.A.; Supko, J.G.; Gorlick, R.; Meyers, P.A.; Maki, R.G.; Wexler, L.; Demetri, G.D.; Healey, J.H.; Huvos, A.G.; et al. Phase II study of ecteinascidin 743 in heavily pretreated patients with recurrent osteosarcoma. Cancer 2003, 98, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Ratti, C.; Botti, L.; Cancila, V.; Galvan, S.; Torselli, I.; Garofalo, C.; Manara, M.C.; Bongiovanni, L.; Valenti, C.F.; Burocchi, A.; et al. Trabectedin Overrides Osteosarcoma Differentiative Block and Reprograms the Tumor Immune Environment Enabling Effective Combination with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 5149–5161. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchandet, L.; Lallier, M.; Charrier, C.; Baud’huin, M.; Ory, B.; Lamoureux, F. Mechanisms of Resistance to Conventional Therapies for Osteosarcoma. Cancers 2021, 13, 683. https://doi.org/10.3390/cancers13040683
Marchandet L, Lallier M, Charrier C, Baud’huin M, Ory B, Lamoureux F. Mechanisms of Resistance to Conventional Therapies for Osteosarcoma. Cancers. 2021; 13(4):683. https://doi.org/10.3390/cancers13040683
Chicago/Turabian StyleMarchandet, Louise, Morgane Lallier, Céline Charrier, Marc Baud’huin, Benjamin Ory, and François Lamoureux. 2021. "Mechanisms of Resistance to Conventional Therapies for Osteosarcoma" Cancers 13, no. 4: 683. https://doi.org/10.3390/cancers13040683
APA StyleMarchandet, L., Lallier, M., Charrier, C., Baud’huin, M., Ory, B., & Lamoureux, F. (2021). Mechanisms of Resistance to Conventional Therapies for Osteosarcoma. Cancers, 13(4), 683. https://doi.org/10.3390/cancers13040683