
Adoptive Immunotherapy beyond CAR T-Cells


 and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
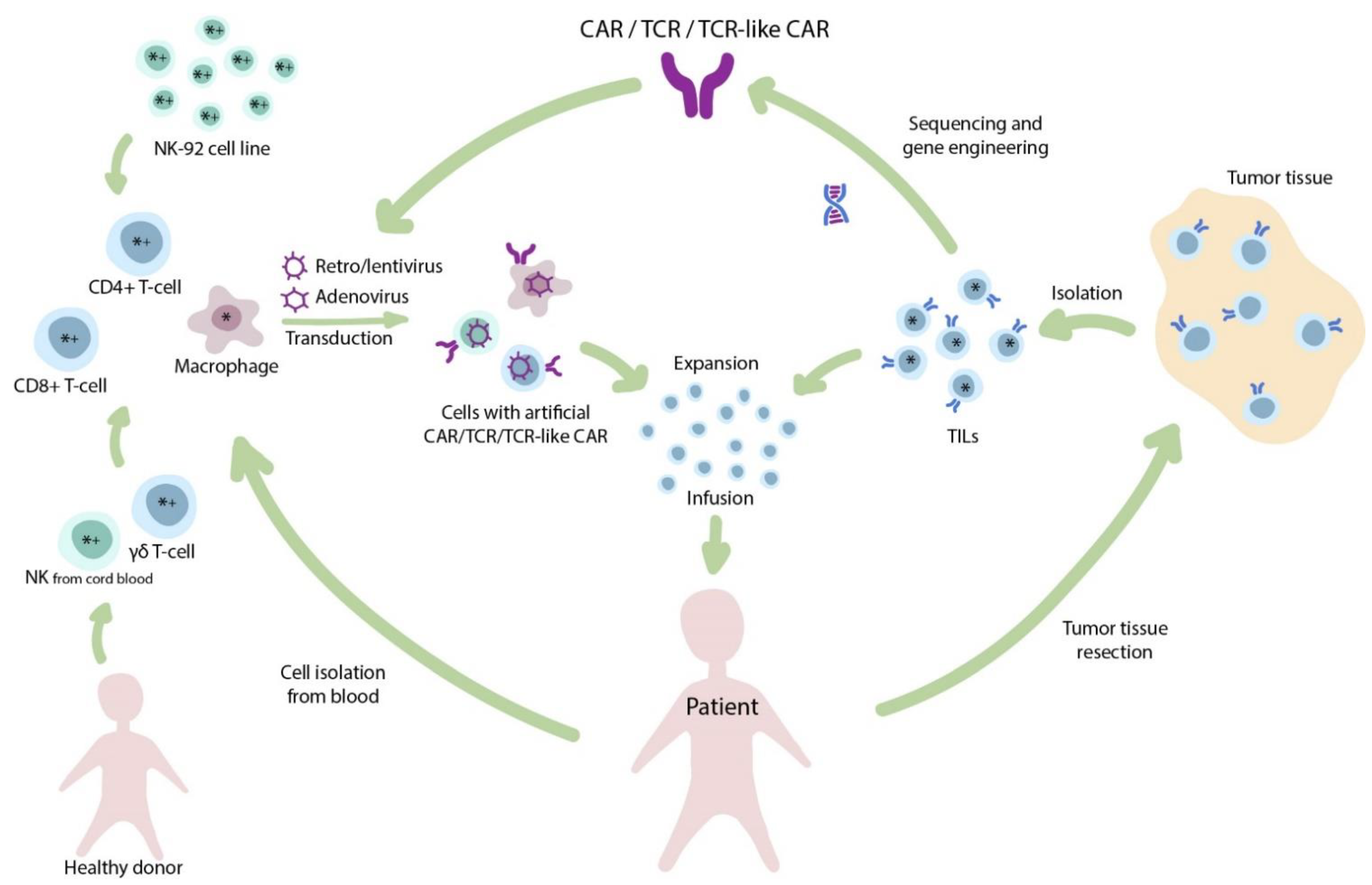
2. The Importance of Cell Source for Production of Conventional CAR T-Cells
3. CAR Cells, but Not CAR αβ T-Cells
3.1. γδ. T-Cells
3.2. NK-Cells
3.3. CAR M-Cells
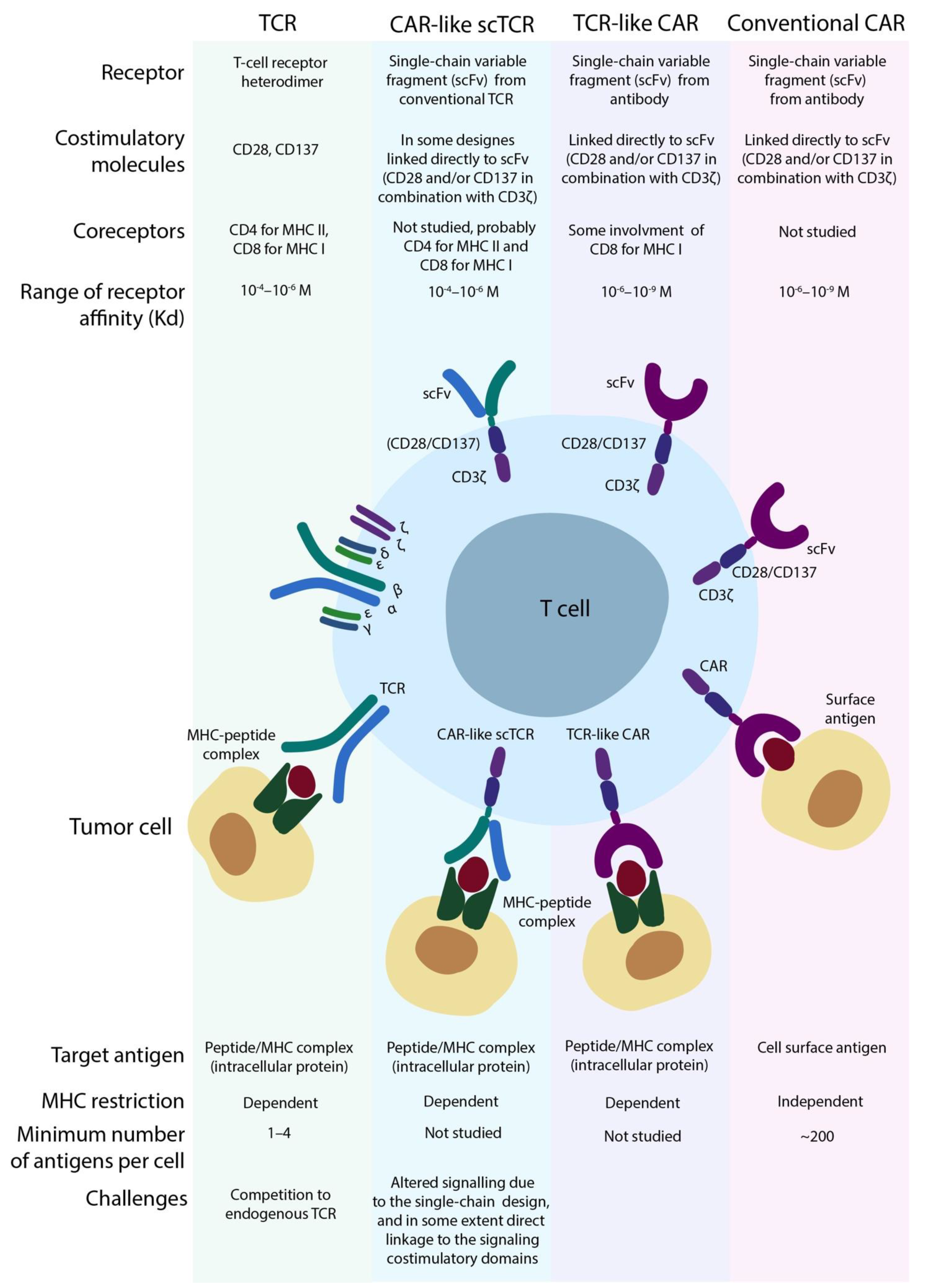
4. T-Cells, but Not CAR T-Cells. TCR-Based Therapies
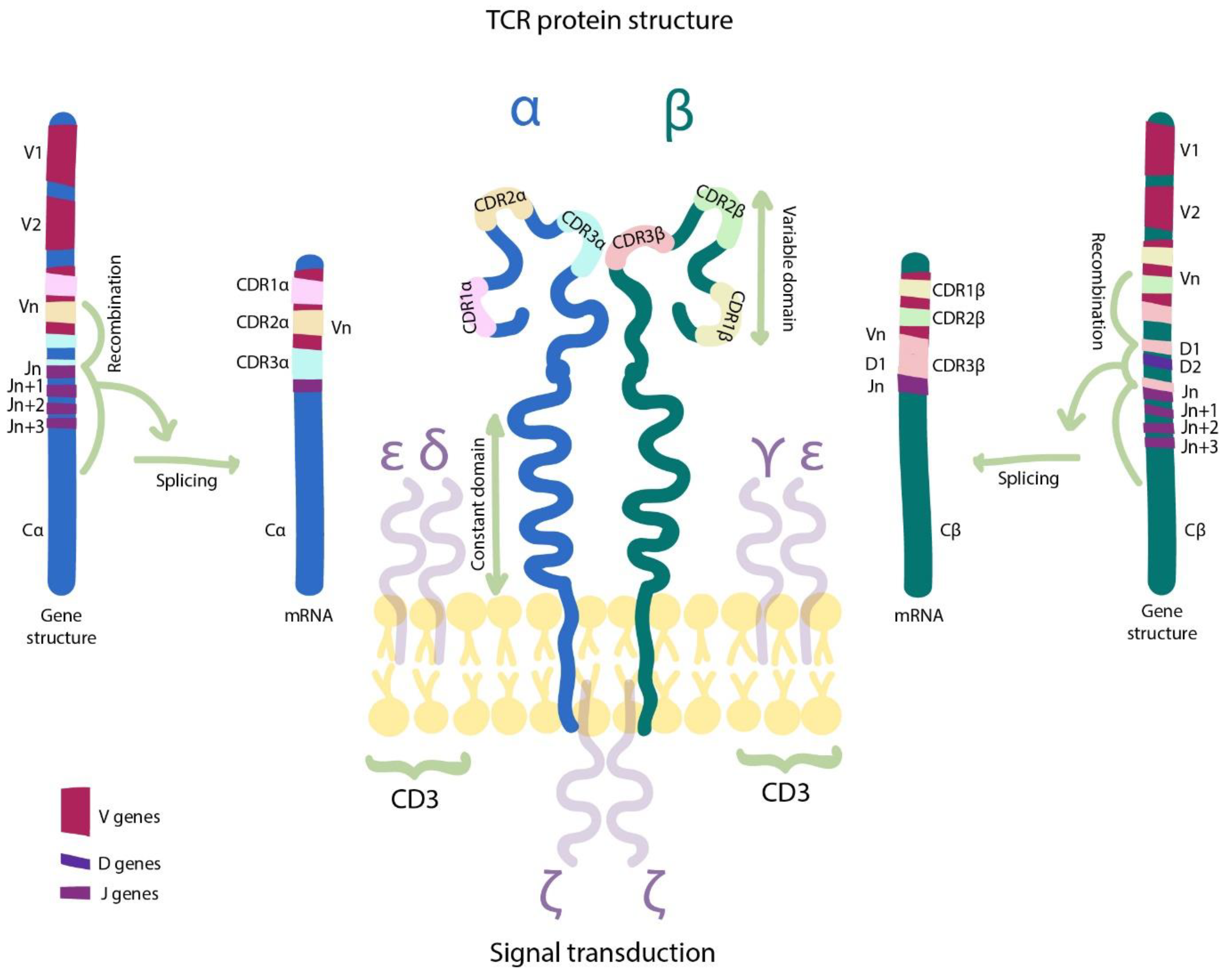
4.1. The Basis of the TCR Machinery
4.2. TILs—Not-Engineered T-Cells
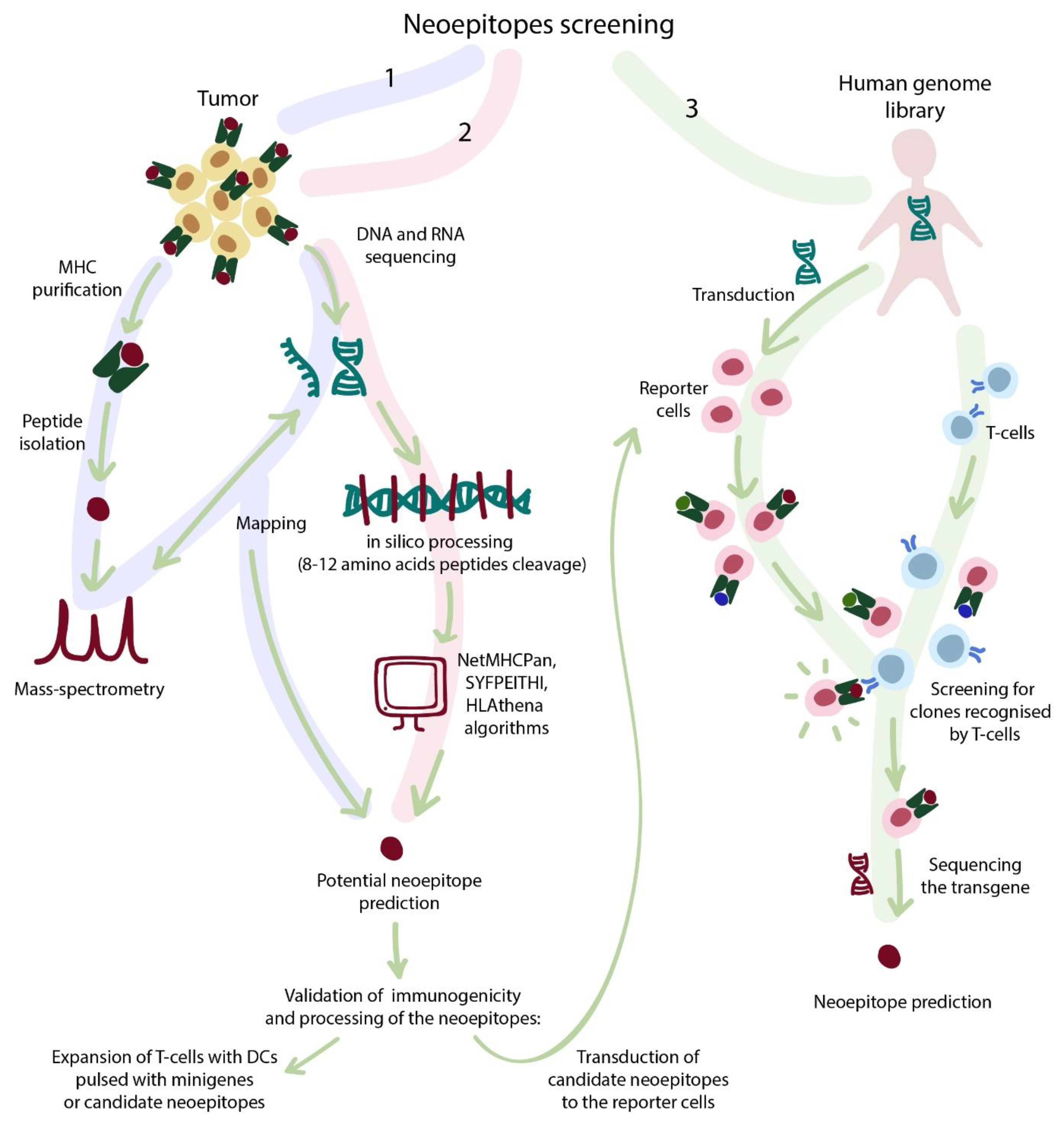
4.3. Strategies for Selection of TCR-Dependent Epitopes
4.3.1. Sequencing Strategies
4.3.2. Bioinformatic Selection
4.3.3. Library-Associated Epitope Screening
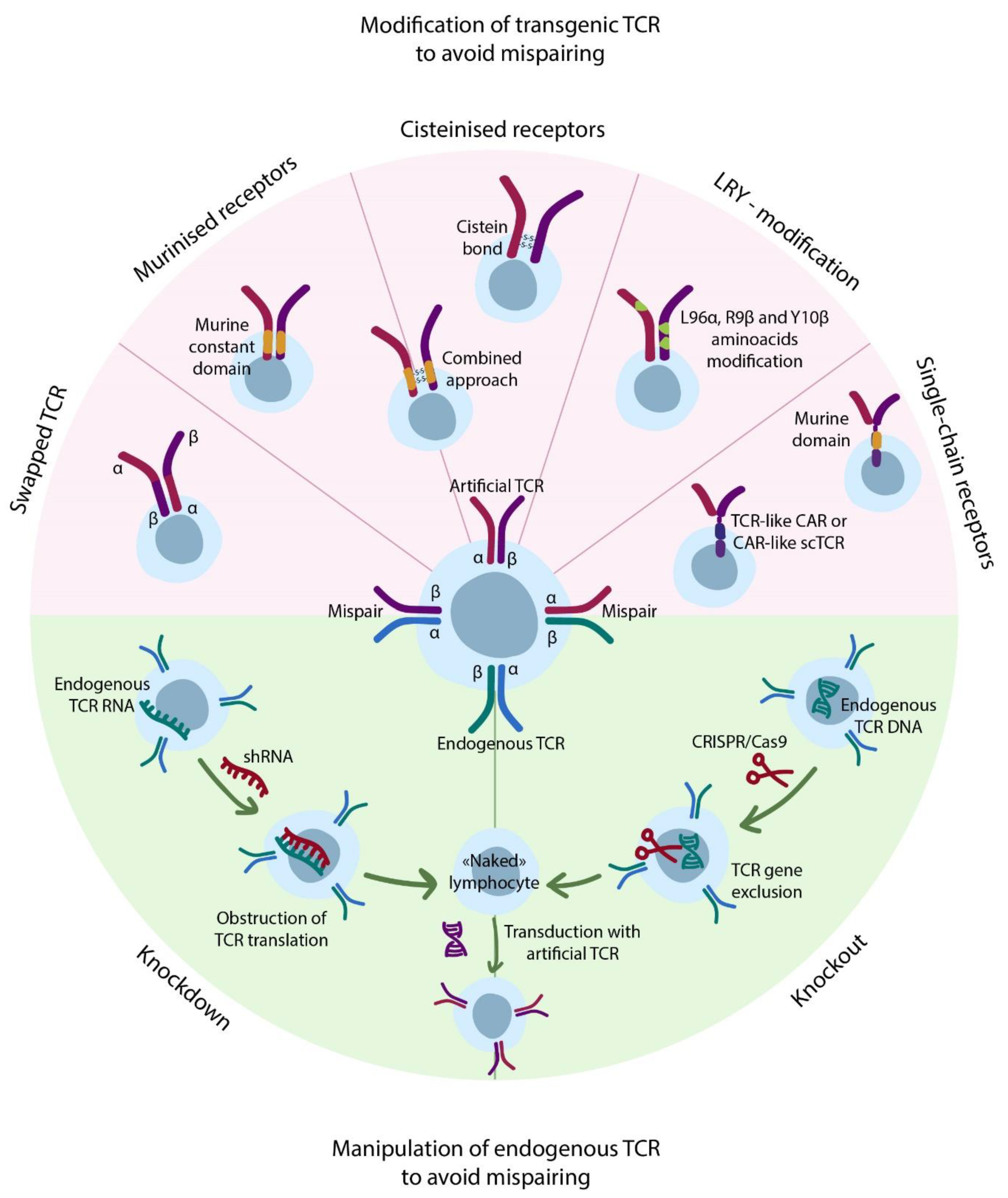
4.4. Transgenic TCRs
4.5. Exploiting Alternatives to Conventional TCRs
4.5.1. NK-Cell Receptors
4.5.2. Non-Conventional TCRs
5. Evolution of Manufacturing Therapeutic T-Cells
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACT | Adoptive Cell Transfer |
| APC | Antigen-Presenting Cell |
| BTLA | B- and T-lymphocyte attenuator |
| BTN2A1 | Butyrophilin-2A1 |
| CAR | Chimeric Antigen Receptor |
| CR | Complete Remission |
| DC | Dendritic Cell |
| FACS | Fluorescence-Activated Cell Sorting |
| GMP | Good Manufacturing Practice |
| GvHD | Graft versus Host Disease |
| HF | Hollow Fiber |
| HLA | Human Leukocyte Antigen |
| HSCT | Hematopoietic Stem Cell Transplantation |
| IL | Interleukin |
| Mel | Melanoma |
| KIR | Killer Cell Immunoglobulin-Like Receptor |
| MHC | Major Histocompatibility Complex |
| MRD | Minimal Residual Disease |
| MS | mass spectrometry |
| MSC | mesenchymal stem cell |
| MR1 | major histocompatibility complex class I-related gene protein |
| NK | Natural Killer |
| NOD/SCID | Nonobese Diabetic/Severe Combined Immunodeficiency |
| NR | No Response |
| ORF | Open Reading Frame |
| PBMC | Peripheral Blood Mononuclear Cell |
| pMHC | Peptide-loaded Major Histocompatibility Complex |
| PD | Progressive Disease |
| PoC | Point-of-Care |
| PR | Partial Remission |
| Pt/pts | patient/patients |
| RNA | Ribonucleic Acid |
| scTCR | single chain T-Cell Receptor |
| SD | Stable Disease |
| shRNA | short hairpin RNA |
| SCS | Synovial Cell Sarcoma |
| TCM | Central Memory T-cell |
| TIL | Tumor Infiltrating Lymphocyte |
| TM | TransMembrane |
| TMB | Tumor Mutation Burden |
| TME | Tumor Microenvironment |
| Treg | T regulatory cells |
| TSCM | Stem Memory T-cell |
| VGPR | Very Good Partial Remission |
| WIM | Wave-Induced Motion |
References
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Schmueck-Henneresse, M.; Omer, B.; Shum, T.; Tashiro, H.; Mamonkin, M.; Lapteva, N.; Sharma, S.; Rollins, L.; Dotti, G.; Reinke, P.; et al. Comprehensive Approach for Identifying the T Cell Subset Origin of CD3 and CD28 Antibody–Activated Chimeric Antigen Receptor–Modified T Cells. J. Immunol. 2017, 199, 348–362. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef]
- Rossi, J.; Paczkowski, P.; Shen, Y.-W.; Morse, K.; Flynn, B.; Kaiser, A.; Ng, C.; Gallatin, K.; Cain, T.; Fan, R.; et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 2018, 132, 804–814. [Google Scholar] [CrossRef] [Green Version]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell all patients. J. Clin. Invest. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramson, J.S.; Lia, P.M.; Gordon, L.I.; Lunning, M.A.; Arnason, J.E.; Wang, M. High Durable CR Rates in Relapsed/Refractory (R/R) Aggressive B-NHL Treated with the CD19-Directed CAR T Cell Product JCAR017 (TRANSCEND NHL 001): Defined Composition Allows for Dose-Finding and Definition of Pivotal Cohort. In Proceedings of the 2017 Annual American Society for Hematology Meeting, Atlanta, GA, USA, 9–12 December 2017. [Google Scholar]
- Sheih, A.; Voillet, V.; Hanafi, L.A.; DeBerg, H.A.; Yajima, M.; Hawkins, R.; Gersuk, V.; Riddell, S.R.; Maloney, D.G.; Wohlfahrt, M.E.; et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.; Petukhov, A.; Staliarova, A.; Motorin, D.; Bulatov, E.; Shuvalov, O.; Soond, S.M.; Piacentini, M.; Melino, G.; Zaritskey, A.; et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis. 2018, 9, 897. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titov, A.; Valiullina, A.; Zmievskaya, E.; Zaikova, E.; Petukhov, A.; Miftakhova, R.; Bulatov, E.; Rizvanov, A. Advancing CAR T-cell therapy for solid tumors: Lessons learned from lymphoma treatment. Cancers 2020, 12, 125. [Google Scholar] [CrossRef] [Green Version]
- Barral, D.C.; Brenner, M.B. CD1 antigen presentation: How it works. Nat. Rev. Immunol. 2007, 7, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, M.M.; Willcox, C.R.; Salim, M.; Paletta, D.; Fichtner, A.S.; Noll, A.; Starick, L.; Nöhren, A.; Begley, C.R.; Berwick, K.A.; et al. Butyrophilin-2A1 Directly Binds Germline-Encoded Regions of the Vγ9Vδ2 TCR and Is Essential for Phosphoantigen Sensing. Immunity 2020, 52, 487–498.e6. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, E.; Tsuboi, K.; Saijo, K.; Harada, H.; Takano, S.; Nose, T.; Ohno, T. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res. 2004, 24, 1861–1871. [Google Scholar] [PubMed]
- Yoshida, S.; Tanaka, R.; Takai, N.; Ono, K. Local Administration of Autologous Lymphokine-activated Killer Cells and Recombinant Interleukin 2 to Patients with Malignant Brain Tumors. Cancer Res. 1988, 48. [Google Scholar]
- Lupo, K.B.; Matosevic, S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers 2019, 11, 769. [Google Scholar] [CrossRef] [Green Version]
- Bertaina, A.; Zecca, M.; Buldini, B.; Sacchi, N.; Algeri, M.; Saglio, F.; Perotti, C.; Gallina, A.M.; Bertaina, V.; Lanino, E.; et al. Unrelated donor vs. HLA-haploidentical a/b T-cell– and B-cell–depleted HSCT in children with acute leukemia. Blood 2018, 132, 2594–2607. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.M.; Elfeky, R.; Nademi, Z.; Qasim, W.; Amrolia, P.; Chiesa, R.; Rao, K.; Lucchini, G.; Silva, J.M.F.; Worth, A.; et al. T-cell receptor αβ+ and CD19+ cell–depleted haploidentical and mismatched hematopoietic stem cell transplantation in primary immune deficiency. J. Allergy Clin. Immunol. 2018, 141, 1417–1426.e1. [Google Scholar] [CrossRef] [Green Version]
- Dovydenko, M.V.; Parovichnikova, E.N.; Kuzmina, L.A.; Vasilyeva, V.A.; Drokov, M.Y.; Koroleva, O.M.; Mikhaltsova, E.D.; Popova, N.N.; Konova, Z.V.; Dmitrova, A.A.; et al. Haploidentical Stem Cell Transplantation with TCR Alpha/Beta and CD19 Depletion in Adult Patients with Hematological Malignancies. Blood 2019, 134, 5648. [Google Scholar] [CrossRef]
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors into Precision Tools for Cancer Immunotherapy. Front. Immunol. 2019, 10, 2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Wels, W. CAR-NK cells as off-the-shelf therapeutics bridging innate and adaptive immunity. Presented at 2nd European CAR T-cell meeting, Barcelona, Spain, 30 January–1 February 2020. [Google Scholar]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-Man Clinical Trial of CAR NK-92 Cells: Safety Test of CD33-CAR NK-92 Cells in Patients with Relapsed and Refractory Acute Myeloid Leukemia; e-Century Publishing Corporation: Madison, WI, USA, 2018; Volume 8. [Google Scholar]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springuel, L.; Lonez, C.; Alexandre, B.; Van Cutsem, E.; Machiels, J.P.H.; Van Den Eynde, M.; Prenen, H.; Hendlisz, A.; Shaza, L.; Carrasco, J.; et al. Chimeric Antigen Receptor-T Cells for Targeting Solid Tumors: Current Challenges and Existing Strategies. BioDrugs 2019, 33, 515–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adusumilli, P.S.; Cherkassky, L.; Villena-Vargas, J.; Colovos, C.; Servais, E.; Plotkin, J.; Jones, D.R.; Sadelain, M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chulpanova, D.S.; Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Mouse Tumor Models for Advanced Cancer Immunotherapy. Int. J. Mol. Sci. 2020, 21, 4118. [Google Scholar] [CrossRef]
- Zhang, X.; Edwards, J.P.; Mosser, D.M. The expression of exogenous genes in macrophages: Obstacles and opportunities. Methods Mol. Biol. 2009, 531, 123–143. [Google Scholar] [CrossRef] [Green Version]
- Bobadilla, S.; Sunseri, N.; Landau, N.R. Efficient transduction of myeloid cells by an HIV-1-derived lentiviral vector that packages the Vpx accessory protein. Gene Ther. 2013, 20, 514–520. [Google Scholar] [CrossRef] [Green Version]
- Karponi, G.; Kritas, S.; Petridou, E.; Papanikolaou, E. Efficient transduction and expansion of ovine macrophages for gene therapy implementations. Vet. Sci. 2018, 5, 57. [Google Scholar] [CrossRef] [Green Version]
- Hajeri, P.B.; Sharma, N.S.; Yamamoto, M. Oncolytic Adenoviruses: Strategies for Improved Targeting and Specificity. Cancers 2020, 12, 1504. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.C.; Guan, X.H.; Li, Y.H.; Huang, J.Y.; Jiang, T.; Hou, L.H.; Li, J.X.; Yang, B.F.; Wang, L.; Wang, W.J.; et al. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2020, 396, 479–488. [Google Scholar] [CrossRef]
- Logunov, D.Y.; Dolzhikova, I.V.; Zubkova, O.V.; Tukhvatullin, A.I.; Shcheblyakov, D.V.; Dzharullaeva, A.S.; Grousova, D.M.; Erokhova, A.S.; Kovyrshina, A.V.; Botikov, A.G.; et al. Safety and immunogenicity of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine in two formulations: Two open, non-randomised phase 1/2 studies from Russia. Lancet 2020, 396, 887–897. [Google Scholar] [CrossRef]
- Wucherpfennig, K.W.; Gagnon, E.; Call, M.J.; Huseby, E.S.; Call, M.E. Structural Biology of the T-cell Receptor: Insights into Receptor Assembly, Ligand Recognition, and Initiation of Signaling. Cold Spring Harb. Perspect. Biol. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patten, P.A.; Rock, E.P.; Sonoda, T.; Fazekas de St Groth, B.; Jorgensen, J.L.; Davis, M.M. Transfer of putative complementarity-determining region loops of T cell receptor V domains confers toxin reactivity but not peptide/MHC specificity. J. Immunol. 1993, 150, 2281–2294. [Google Scholar] [PubMed]
- Smith, S.N.; Wang, Y.; Baylon, J.L.; Singh, N.K.; Baker, B.M.; Tajkhorshid, E.; Kranz, D.M. Changing the peptide specificity of a human T-cell receptor by directed evolution. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Piepenbrink, K.H.; Blevins, S.J.; Scott, D.R.; Baker, B.M. The basis for limited specificity and MHC restriction in a T cell receptor interface. Nat. Commun. 2013, 4, 1948. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, M.; Marits, P.; Dahl, K.; Dagöö, T.; Enerbäck, S.; Thörn, M.; Winqvist, O. Pilot study of sentinel-node-based adoptive immunotherapy in advanced colorectal cancer. Ann. Surg. Oncol. 2010, 17, 1747–1757. [Google Scholar] [CrossRef] [Green Version]
- Donia, M.; Larsen, S.M.; Met, Ö.; Svane, I.M. Simplified protocol for clinical-grade tumor-infiltrating lymphocyte manufacturing with use of the Wave bioreactor. Cytotherapy 2014, 16, 1117–1120. [Google Scholar] [CrossRef] [PubMed]
- Radvanyi, L.G.; Bernatchez, C.; Zhang, M.; Fox, P.S.; Miller, P.; Chacon, J.; Wu, R.; Lizee, G.; Mahoney, S.; Alvarado, G.; et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2012, 18, 6758–6770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, G.U.; Malekzadeh, P.; Shelton, T.; White, D.E.; Butman, J.A.; Yang, J.C.; Kammula, U.S.; Goff, S.L.; Rosenberg, S.A.; Sherry, R.M. Outcomes of Adoptive Cell Transfer with Tumor-infiltrating Lymphocytes for Metastatic Melanoma Patients with and Without Brain Metastases. J. Immunother. 2018, 41, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Goff, S.L.; Dudley, M.E.; Citrin, D.E.; Somerville, R.P.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J. Clin. Oncol. 2016, 34, 2389–2397. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Andersen, R.; Borch, T.H.; Draghi, A.; Gokuldass, A.; Rana, A.H.M.; Pedersen, M.; Nielsen, M.; Kongsted, P.; Kjeldsen, J.W.; Westergaard, C.W.M.; et al. T cells isolated from patients with checkpoint inhibitor-resistant melanoma are functional and can mediate tumor regression. Ann. Oncol. 2018, 29, 1575–1581. [Google Scholar] [CrossRef] [PubMed]
- Friese, C.; Harbst, K.; Borch, T.H.; Westergaard, M.C.W.; Pedersen, M.; Kverneland, A.; Jönsson, G.; Donia, M.; Svane, I.M.; Met, Ö. CTLA-4 blockade boosts the expansion of tumor-reactive CD8+ tumor-infiltrating lymphocytes in ovarian cancer. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodnala, S.K.; Eil, R.; Kishton, R.J.; Sukumar, M.; Yamamoto, T.N.; Ha, N.H.; Lee, P.H.; Shin, M.H.; Patel, S.J.; Yu, Z.; et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 2019, 363. [Google Scholar] [CrossRef]
- Hall, M.; Liu, H.; Malafa, M.; Centeno, B.; Hodul, P.J.; Pimiento, J.; Pilon-Thomas, S.; Sarnaik, A.A. Expansion of tumor-infiltrating lymphocytes (TIL) from human pancreatic tumors. J. Immunother. Cancer 2016, 4, 61. [Google Scholar] [CrossRef] [Green Version]
- Assarsson, E.; Sidney, J.; Oseroff, C.; Pasquetto, V.; Bui, H.-H.; Frahm, N.; Brander, C.; Peters, B.; Grey, H.; Sette, A. A Quantitative Analysis of the Variables Affecting the Repertoire of T Cell Specificities Recognized after Vaccinia Virus Infection. J. Immunol. 2007, 178, 7890–7901. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Sarkizova, S.; Klaeger, S.; Le, P.M.; Li, L.W.; Oliveira, G.; Keshishian, H.; Hartigan, C.R.; Zhang, W.; Braun, D.A.; Ligon, K.L.; et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020, 38, 199–209. [Google Scholar] [CrossRef]
- Laumont, C.M.; Perreault, C. Exploiting non-canonical translation to identify new targets for T cell-based cancer immunotherapy. Cell. Mol. Life Sci. 2018, 75, 607–621. [Google Scholar] [CrossRef]
- Ouspenskaia, T.; Law, T.; Clauser, K.; Klaeger, S.; Sarkizova, S.; Aguet, F.; Li, B.; Christian, E.; Knisbacher, B.; Le, P.; et al. Thousands of novel unannotated proteins expand the MHC I immunopeptidome in cancer. bioRxiv 2020. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingolia, N.T.; Brar, G.A.; Stern-Ginossar, N.; Harris, M.S.; Talhouarne, G.J.S.; Jackson, S.E.; Wills, M.R.; Weissman, J.S. Ribosome Profiling Reveals Pervasive Translation Outside of Annotated Protein-Coding Genes. Cell Rep. 2014, 8, 1365–1379. [Google Scholar] [CrossRef] [Green Version]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Stevanović, S.; Anna, P.; Gartner, J.J.; Tran, E.; Robbins, P.F.; Rosenberg, S.A.; Hinrichs, C.S. Adoptively transferred tumor-infiltrating T cells target somatic cancer mutations in a human papillomavirus+ cancer patient with complete tumor regression. J. Immunother. Cancer 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Parkhurst, M.; Gros, A.; Pasetto, A.; Prickett, T.; Crystal, J.S.; Robbins, P.; Rosenberg, S.A. Isolation of T-cell receptors specifically reactive with mutated tumor-associated antigens from tumor-infiltrating lymphocytes based on CD137 expression. Clin. Cancer Res. 2017, 23, 2491–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.M.; Nastke, M.D.; Stevanović, S. SYFPEITHI: Database for searching and T-cell epitope prediction. Methods Mol. Biol. 2007, 409, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Saxová, P.; Buus, S.; Brunak, S.; Keşmir, C. Predicting proteasomal cleavage sites: A comparison of available methods. Int. Immunol. 2003, 15, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lundegaard, C.; Lund, O.; Keşmir, C. The role of the proteasome in generating cytotoxic T-cell epitopes: Insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005, 57, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Perosanz, M.; Ras-Carmona, A.; Reche, P.A. Prediction of proteasomal cleavage sites using PCPS. In Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine, BIBM 2019, San Diego, CA, USA, 18–21 November 2019; pp. 2137–2140. [Google Scholar]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Yang, J.; Xu, Q.; Ding, N.; Yang, Y.; et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Invest. 2019, 129, 2056–2070. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Rive, C.M.; Holt, R.A. Rapid selection and identification of functional CD8+ T cell epitopes from large peptide-coding libraries. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kula, T.; Dezfulian, M.H.; Wang, C.I.; Abdelfattah, N.S.; Hartman, Z.C.; Wucherpfennig, K.W.; Lyerly, H.K.; Elledge, S.J. T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell 2019, 178, 1016–1028.e13. [Google Scholar] [CrossRef]
- Bunse, M.; Bendle, G.M.; Linnemann, C.; Bies, L.; Schulz, S.; Schumacher, T.N.; Uckert, W. RNAi-mediated TCR Knockdown Prevents Autoimmunity in Mice Caused by Mixed TCR Dimers Following TCR Gene Transfer. Mol. Ther. 2014, 22, 1983–1991. [Google Scholar] [CrossRef] [Green Version]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef]
- Okamoto, S.; Mineno, J.; Ikeda, H.; Fujiwara, H.; Yasukawa, M.; Shiku, H.; Kato, I. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res. 2009, 69, 9003–9011. [Google Scholar] [CrossRef] [Green Version]
- Cohen, C.J.; Li, Y.F.; El-Gamil, M.; Robbins, P.F.; Rosenberg, S.A.; Morgan, R.A. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007, 67, 3898–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, C.J.; Zhao, Y.; Zheng, Z.; Rosenberg, S.A.; Morgan, R.A. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006, 66, 8878–8886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, R.H.; Thomas, S.; Pfirschke, C.; Hauptrock, B.; Klobuch, S.; Kuball, J.; Grabowski, M.; Engel, R.; Guillaume, P.; Romero, P.; et al. Coexpression of the T-cell receptor constant α domain triggers tumor reactivity of single-chain TCR-transduced human T cells. Blood 2010, 115, 5154–5163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.; Mohammed, F.; Reijmers, R.M.; Woolston, A.; Stauss, T.; Kennedy, A.; Stirling, D.; Holler, A.; Green, L.; Jones, D.; et al. Framework engineering to produce dominant T cell receptors with enhanced antigen-specific function. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bethune, M.T.; Gee, M.H.; Bunse, M.; Lee, M.S.; Gschweng, E.H.; Pagadala, M.S.; Zhou, J.; Cheng, D.; Heath, J.R.; Kohn, D.B.; et al. Domain-swapped t cell receptors improve the safety of TCR gene therapy. Elife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Schaft, N.; Lankiewicz, B.; Drexhage, J.; Berrevoets, C.; Moss, D.J.; Levitsky, V.; Bonneville, M.; Lee, S.P.; McMichael, A.J.; Gratama, J.W.; et al. T cell re-targeting to EBV antigens following TCR gene transfer: CD28-containing receptors mediate enhanced antigen-specific IFNγ production. Int. Immunol. 2006, 18, 591–601. [Google Scholar] [CrossRef]
- Zhang, T.; He, X.; Tsang, T.C.; Harris, D.T. Transgenic TCR expression: Comparison of single chain with full-length receptor constructs for T-cell function. Cancer Gene Ther. 2004, 11, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, R.A.; Weijtens, M.E.M.; Ronteltap, C.; Eshhar, Z.; Gratama, J.W.; Chames, P.; Bolhuis, R.L.H. Grafting primary human T lymphocytes with cancer-specific chimeric single chain and two chain TCR. Gene Ther. 2000, 7, 1369–1377. [Google Scholar] [CrossRef]
- Aggen, D.H.; Chervin, A.S.; Schmitt, T.M.; Engels, B.; Stone, J.D.; Richman, S.A.; Piepenbrink, K.H.; Baker, B.M.; Greenberg, P.D.; Schreiber, H.; et al. Single-chain VαVβ T-cell receptors function without mispairing with endogenous TCR chains. Gene Ther. 2012, 19, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Harris, D.T.; Hager, M.V.; Smith, S.N.; Cai, Q.; Stone, J.D.; Kruger, P.; Lever, M.; Dushek, O.; Schmitt, T.M.; Greenberg, P.D.; et al. Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains. J. Immunol. 2018, 200, 1088–1100. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D.; June, C.H. Expanding the therapeutic window for CAR T cell therapy in solid tumors: The knowns and unknowns of CAR T cell biology. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Warshaviak, D.T.; Mkrtichyan, M.; Munguia, M.L.; Lin, A.; Chai, F.; Pigott, C.; Kang, J.; Gallo, M.; Kamb, A. Single variable domains from the T cell receptor β chain function as mono- and bifunctional CARs and TCRs. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Giordano-Attianese, G.; Gainza, P.; Gray-Gaillard, E.; Cribioli, E.; Shui, S.; Kim, S.; Kwak, M.J.; Vollers, S.; Corria Osorio, A.D.J.; Reichenbach, P.; et al. A computationally designed chimeric antigen receptor provides a small-molecule safety switch for T-cell therapy. Nat. Biotechnol. 2020, 38, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalían, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Celyad Presents Clinical Update for CYAD-01 at 24th Congress of the European Hematology Association. Available online: https://www.globenewswire.com/news-release/2019/06/17/1869905/0/en/Celyad-Presents-Clinical-Update-for-CYAD-01-at-24th-Congress-of-the-European-Hematology-Association.html (accessed on 1 December 2019).
- Celyad Highlights Safety and Clinical Activity of CYAD-101, a First-In-Class, Non-Gene Edited Allogeneic CAR-T Therapy for mCRC. Available online: https://www.bloomberg.com/press-releases/2019-11-11/celyad-highlights-safety-and-clinical-activity-of-cyad-101-a-first-in-class-non-gene-edited-allogeneic-car-t-therapy-for-mcrc-k2u1u867 (accessed on 1 December 2019).
- Crowther, M.D.; Dolton, G.; Legut, M.; Caillaud, M.E.; Lloyd, A.; Attaf, M.; Galloway, S.A.E.; Rius, C.; Farrell, C.P.; Szomolay, B.; et al. Genome-wide CRISPR–Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat. Immunol. 2020, 21, 178–185. [Google Scholar] [CrossRef]
- McWilliam, H.E.; Villadangos, J.A. MR1 antigen presentation to MAIT cells: New ligands, diverse pathways? Curr. Opin. Immunol. 2018, 52, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Vacchini, A.; Chancellor, A.; Spagnuolo, J.; Mori, L.; De Libero, G. MR1-Restricted T Cells Are Unprecedented Cancer Fighters. Front. Immunol. 2020, 11, 751. [Google Scholar] [CrossRef] [PubMed]
- McFarland, F. An Unconventional Weapon Against Cancer | University of Utah Health. Available online: https://uofuhealth.utah.edu/newsroom/news/2020/05/cancer-weapon.php (accessed on 2 November 2020).
- Bastien, J.P.; Fekete, N.; Beland, A.V.; Lachambre, M.P.; Laforte, V.; Juncker, D.; Dave, V.; Roy, D.C.; Hoesli, C.A. Closing the system: Production of viral antigen-presenting dendritic cells eliciting specific CD8+T cell activation in fluorinated ethylene propylene cell culture bags. J. Transl. Med. 2020, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, Y.R.; Tung, S.L.; Dudreuilh, C.; Lechler, R.I.; Fruhwirth, G.O.; Lombardi, G. The Future of Regulatory T Cell Therapy: Promises and Challenges of Implementing CAR Technology. Front. Immunol. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Hopewell, E.L.; Cox, C.; Pilon-Thomas, S.; Kelley, L.L. Tumor Infiltrating Lymphocytes Streamlining a Complex Manufacturing Process. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Chen, C.; Li, Z.; Zhu, S.; Tay, J.C.; Zhang, X.; Zha, S.; Zeng, J.; Tan, W.K.; Liu, X.; et al. Large-scale expansion of Vγ9Vδ2 T cells with engineered K562 feeder cells in G-Rex vessels and their use as chimeric antigen receptor–modified effector cells. Cytotherapy 2018, 20, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Bröker, K.; Sinelnikov, E.; Gustavus, D.; Schumacher, U.; Pörtner, R.; Hoffmeister, H.; Lüth, S.; Dammermann, W. Mass production of highly active NK cells for cancer immunotherapy in a GMP conform perfusion bioreactor. Front. Bioeng. Biotechnol. 2019, 7, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Hami, L.S.; Green, C.; Leshinsky, N.; Markham, E.; Miller, K.; Craig, S. GMP production and testing of Xcellerated T CellsTM for the treatment of patients with CLL. Cytotherapy 2004, 6, 554–562. [Google Scholar] [CrossRef]
- Meng, Y.; Sun, J.; Hu, T.; Ma, Y.; Du, T.; Kong, C.; Zhang, G.; Yu, T.; Piao, H. Rapid expansion in the WAVE bioreactor of clinical scale cells for tumor immunotherapy. Hum. Vaccines Immunother. 2018, 14, 2516–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, M.; Kempf, H.; Hetzel, M.; Hesse, C.; Hashtchin, A.R.; Brinkert, K.; Schott, J.W.; Haake, K.; Kühnel, M.P.; Glage, S.; et al. Bioreactor-based mass production of human iPSC-derived macrophages enables immunotherapies against bacterial airway infections. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nankervis, B.; Jones, M.; Vang, B.; Brent Rice, R.; Coeshott, C.; Beltzer, J. Optimizing T Cell Expansion in a Hollow-Fiber Bioreactor. Curr. Stem Cell Reports 2018, 4, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mock, U.; Nickolay, L.; Philip, B.; Cheung, G.W.K.; Zhan, H.; Johnston, I.C.D.; Kaiser, A.D.; Peggs, K.; Pule, M.; Thrasher, A.J.; et al. Automated manufacturing of chimeric antigen receptor T cells for adoptive immunotherapy using CliniMACS prodigy. Cytotherapy 2016, 18, 1002–1011. [Google Scholar] [CrossRef]
- Zmievskaya, E.; Valiullina, A.; Ganeeva, I.; Petukhov, A.; Rizvanov, A.; Bulatov, E. Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections. Biomedicines 2021, 9, 59. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | Commentary |
|---|---|
| May impact T-cell avidity and recognition of non-target cells with low target antigen levels |
| Prevents cross-reactivity by formation of hybrid TCRs with unknown specificity |
| Unknown additional specificity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Titov, A.; Zmievskaya, E.; Ganeeva, I.; Valiullina, A.; Petukhov, A.; Rakhmatullina, A.; Miftakhova, R.; Fainshtein, M.; Rizvanov, A.; Bulatov, E. Adoptive Immunotherapy beyond CAR T-Cells. Cancers 2021, 13, 743. https://doi.org/10.3390/cancers13040743
Titov A, Zmievskaya E, Ganeeva I, Valiullina A, Petukhov A, Rakhmatullina A, Miftakhova R, Fainshtein M, Rizvanov A, Bulatov E. Adoptive Immunotherapy beyond CAR T-Cells. Cancers. 2021; 13(4):743. https://doi.org/10.3390/cancers13040743
Chicago/Turabian StyleTitov, Aleksei, Ekaterina Zmievskaya, Irina Ganeeva, Aygul Valiullina, Alexey Petukhov, Aygul Rakhmatullina, Regina Miftakhova, Michael Fainshtein, Albert Rizvanov, and Emil Bulatov. 2021. "Adoptive Immunotherapy beyond CAR T-Cells" Cancers 13, no. 4: 743. https://doi.org/10.3390/cancers13040743
APA StyleTitov, A., Zmievskaya, E., Ganeeva, I., Valiullina, A., Petukhov, A., Rakhmatullina, A., Miftakhova, R., Fainshtein, M., Rizvanov, A., & Bulatov, E. (2021). Adoptive Immunotherapy beyond CAR T-Cells. Cancers, 13(4), 743. https://doi.org/10.3390/cancers13040743