
Immunotherapy and Its Development for Gynecological (Ovarian, Endometrial and Cervical) Tumors: From Immune Checkpoint Inhibitors to Chimeric Antigen Receptor (CAR)-T Cell Therapy

 , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Immunotherapy in Gynecological Neoplasms: Rationale
2.1. Rationale for Immunotherapy in OC
2.2. Rationale for Immunotherapy in EC
2.3. Rationale for Immunotherapy in CC
3. Immunotherapy in Gynecological Neoplasms: Clinical Evidence
3.1. Immunotherapy in OC
- (1)
- Javelin 100 was a trial testing chemotherapy alone or in combination with anti-PD-L1 avelumab as first-line treatment. No differences between the two cohorts were reported in the interim analysis. The Javelin 200 trial tested the same combination in a second-line setting, reporting an objective response rate (ORR) of 9.6% and a median OS (mOS) of 11.2 months [6,32]. Wehnham et al. evaluated a second-line combination of weekly paclitaxel and anti-PD-1 pembrolizumab, observing an ORR of 51% and a 6-month progression-free survival (PFS) of 52% [33].
- (2)
- Zamarin et al. tested nivolumab alone or in combination with the anti-CTLA4 ipilimumab, reporting an overall response rate (ORR) of 12.2% (monotherapy cohort) vs. 31.4% (combo cohort), and an mOS of 21.8 months (monotherapy cohort) vs. 28.8 months (combo cohort) [34].
- (3)
- The TOPACIO-Keynote162 study investigated the combination of niraparib plus pembrolizumab, reporting an ORR of 25% [35]. The MEDIOLA trial, conducted on BRCA-mutated EC and testing the combination of olaparib plus anti-PD-L1 durvalumab, reported a disease control rate (DCR) of 81% [36]. The same combination was evaluated by Lee et al. in platinum-resistant OC, reporting a 37% DCR and an ORR of 14% [37].
- (4)
- Liu et al. tested the combination of nivolumab plus bevacizumab in recurrent EC, observing a median ORR (mORR) of 28.9% (ranging from 40% in platinum-sensitive EC to 16.7% in platinum-resistant disease) and a PFS of 8.1 months (ranging from 9.5 months in platinum-sensitive to 5.0 months in platinum-resistant disease) [38].
3.2. Immunotherapy in EC
3.3. Immunotherapy in CC
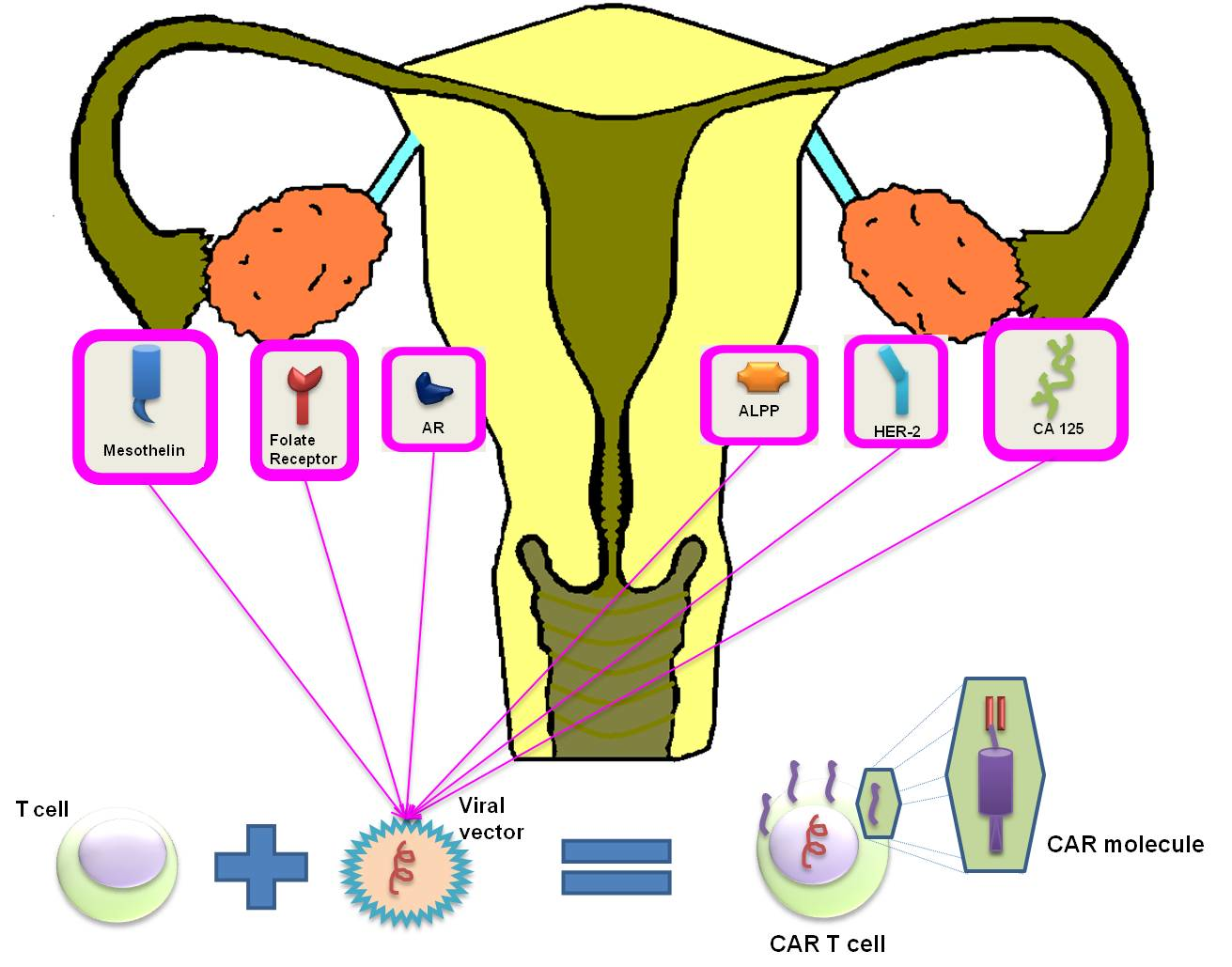
4. CAR-T: Structure, Function and Toxicities
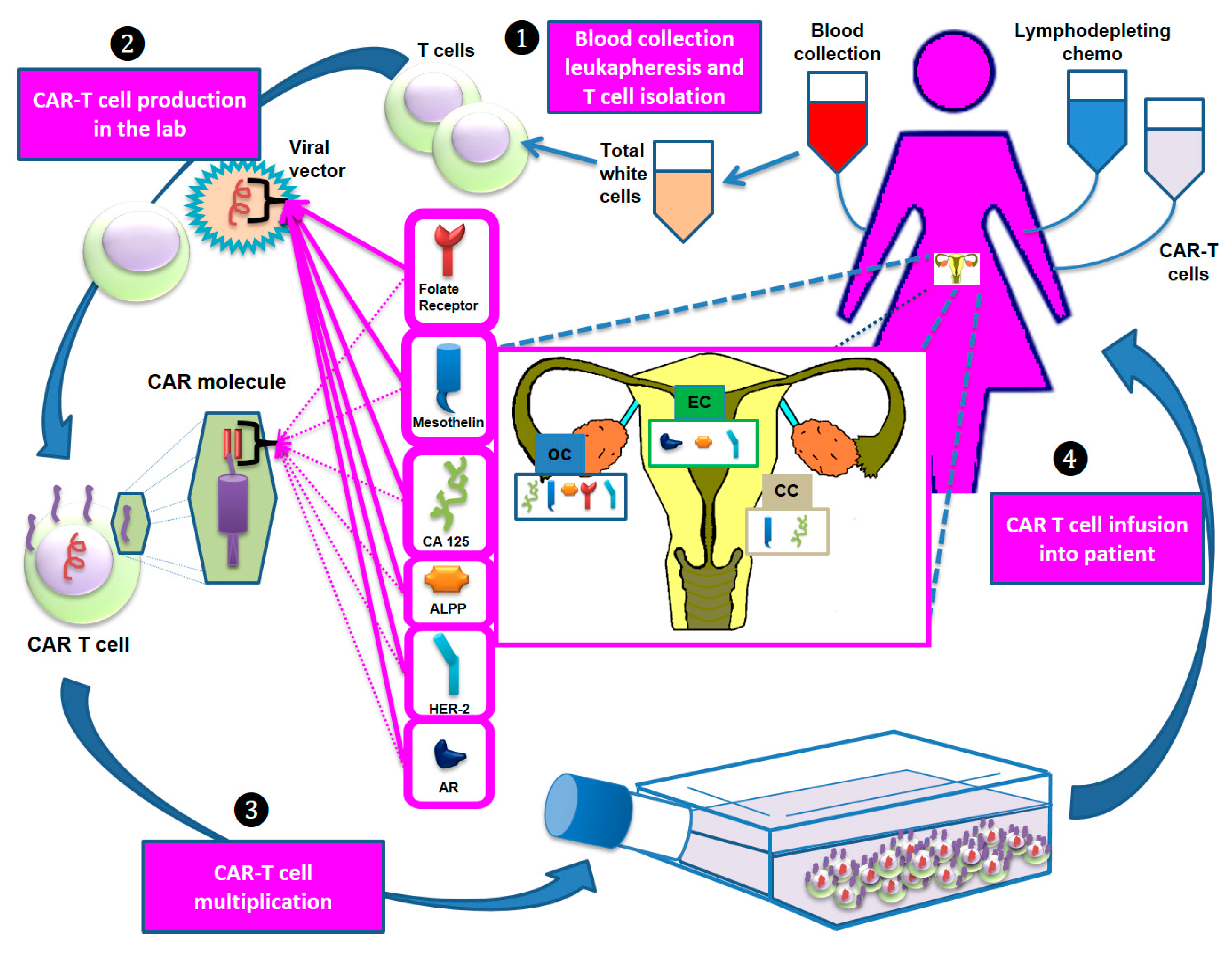
5. CAR-T Cell Therapy in Gynecological Tumors
5.1. CAR-T Cell Therapy in OC
5.2. CAR-T Cell Therapy in EC
5.3. CAR-T Cell Therapy in CC
6. Discussion
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Abiko, K.; Mandai, M.; Hamanishi, J.; Yoshioka, Y.; Matsumura, N.; Baba, T.; Yamaguchi, K.; Murakami, R.; Yamamoto, A.; Kharma, B.; et al. PD-L1 on Tumor Cells Is Induced in Ascites and Promotes Peritoneal Dissemination of Ovarian Cancer through CTL Dysfunction. Clin. Cancer Res. 2013, 19, 1363–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, A.L.; Roach, B.A.; Mays, M.P.; Chen, A.F.; Ginter, B.A.; Vierling, A.M.; Scoggins, C.R.; Martin, R.C.; Stromberg, A.J.; Hagendoorn, L.; et al. Prognostic Significance of Tumor Infiltrating Lymphocytes in Melanoma. Am. Surg. 2011, 77, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J.B.A.G. Adoptive cellular therapies: The current landscape. Virchows Arch. 2019, 474, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Levinson, K.; Dorigo, O.; Rubin, K.; Moore, K. Immunotherapy in Gynecologic Cancers: What We Know Now and Where We Are Headed. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, e126–e140. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [Green Version]
- Vanderstraeten, A.; Tuyaerts, S.; Amant, F. The immune system in the normal endometrium and implications for endometrial cancer development. J. Reprod. Immunol. 2015, 109, 7–16. [Google Scholar] [CrossRef]
- Longoria, T.C.; Eskander, R.N. Immunotherapy in endometrial cancer—An evolving therapeutic paradigm. Gynecol. Oncol. Res. Pract. 2015, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Giatromanolaki, A.; Bates, G.J.; Koukourakis, M.I.; Sivridis, E.; Gatter, K.C.; Harris, A.L.; Banham, A.H. The presence of tumor-infiltrating FOXP3+ lymphocytes correlates with intratumoral angiogenesis in endometrial cancer. Gynecol. Oncol. 2008, 110, 216–221. [Google Scholar] [CrossRef]
- Kübler, K.; Ayub, T.H.; Weber, S.K.; Zivanovic, O.; Abramian, A.; Keyver-Paik, M.-D.; Mallmann, M.R.; Kaiser, C.; Serçe, N.B.; Kuhn, W.; et al. Prognostic significance of tumor-associated macrophages in endometrial adenocarcinoma. Gynecol. Oncol. 2014, 135, 176–183. [Google Scholar] [CrossRef]
- Chang, W.-C.; Li, C.-H.; Huang, S.-C.; Chang, D.-Y.; Chou, L.-Y.; Sheu, B.-C. Clinical significance of regulatory T cells and CD8+ effector populations in patients with human endometrial carcinoma. Cancer 2010, 116, 5777–5788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, Z.; Liu, J.; Zhang, Q.; Chen, Z.; Mei, J.; Liu, L.; Yang, S.; Li, H.; Zhou, L.; You, Z. Expression of PD-1, PD-L1 and PD-L2 is associated with differentiation status and histological type of endometrial cancer. Oncol. Lett. 2016, 12, 944–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.A.; Howitt, B.E.; Wu, C.J.; Konstantinopoulos, P.A. Predicted neoantigen load in non-hypermutated endometrial cancers: Correlation with outcome and tumor-specific genomic alterations. Gynecol. Oncol. Rep. 2017, 19, 42–45. [Google Scholar] [CrossRef]
- Howitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.J.; Stover, E.H.; Strickland, K.C.; D’Andrea, A.D.; Wu, C.J.; et al. Association of Polymerase e–Mutated and Microsatellite-Instable Endometrial Cancers With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015, 1, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, I.C.; Eggink, F.A.; Freeman-Mills, L.; Stelloo, E.; Marchi, E.; De Bruyn, M.; Palles, C.; Nout, R.A.; De Kroon, C.D.; Osse, E.M.; et al. POLE Proofreading Mutations Elicit an Antitumor Immune Response in Endometrial Cancer. Clin. Cancer Res. 2015, 21, 3347–3355. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, C.O.; Vroobel, K.; Lalondrelle, S.; Taylor, A.; Nobbenhuis, M.; Attygalle, A.; Banerjee, S.; George, A. Prevalence and clinical implications of mismatch repair (MMR) deficiency in unselected endometrial cancer (EC) patients. Ann. Oncol. 2018, 29, viii342. [Google Scholar] [CrossRef]
- Deshpande, M.; Romanski, P.A.; Rosenwaks, Z.; Gerhardt, J. Gynecological Cancers Caused by Deficient Mismatch Repair and Microsatellite Instability. Cancers 2020, 12, 3319. [Google Scholar] [CrossRef]
- Makker, V.; Green, A.K.; Wenham, R.M.; Mutch, D.; Davidson, B.; Miller, D.S. New therapies for advanced, recurrent, and metastatic endometrial cancers. Gynecol. Oncol. Res. Pract. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einstein, M.H.; Schiller, J.T.; Viscidi, R.P.; Strickler, H.D.; Coursaget, P.; Tan, T.; Halsey, N.; Jenkins, D. Clinician’s guide to human papillomavirus immunology: Knowns and unknowns. Lancet Infect. Dis. 2009, 9, 347–356. [Google Scholar] [CrossRef]
- Arbyn, M.; Xu, L.; Simoens, C.; Martin-Hirsch, P.P.L. Prophylactic vaccination against human papillomaviruses to prevent cervical cancer and its precursors. Cochrane Database Syst. Rev. 2018, 9, CD009069. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.-B.; Lu, Y.; Huang, J.-L.; Long, Y.; Yao, D.-S. Prognostic genes in the tumor microenvironment in cervical squamous cell carcinoma. Aging 2019, 11, 10154–10166. [Google Scholar] [CrossRef]
- Hu, S.W.; Pu, D.; Xia, X.Y.; Guo, B.X.; Zhang, C.L. CTLA-4 rs5742909 polymorphism and cervical cancer risk: A meta-analysis. Medicine 2020, 99, e19433. [Google Scholar] [CrossRef] [PubMed]
- Karpathiou, G.; Chauleur, C.; Mobarki, M.; Peoc’H, M. The immune checkpoints CTLA-4 and PD-L1 in carcinomas of the uterine cervix. Pathol. - Res. Pract. 2020, 216, 216. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Liang, H.; Hu, J.; Liu, S.; Hao, X.; Wong, M.S.K.; Li, X.; Hu, L. PD-L1 Expression Correlates With Tumor Infiltrating Lymphocytes And Response To Neoadjuvant Chemotherapy In Cervical Cancer. J. Cancer 2018, 9, 2938–2945. [Google Scholar] [CrossRef]
- Burk, R.D.; Chen, Z.; Saller, C.; Tarvin, K.; Carvalho, A.L.; Scapulatempo-Neto, C.; Silveira, H.C.; Fregnani, J.H.; Creighton, C.J.; Anderson, M.L.; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar]
- Mezache, L.; Paniccia, B.; Nyinawabera, A.; Nuovo, G.J. Enhanced expression of PD L1 in cervical intraepithelial neoplasia and cervical cancers. Mod. Pathol. 2015, 28, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Song, Y.; Lu, Y.-L.; Sun, J.-Z.; Wang, H.-W. Increased expression of programmed death (PD)-1 and its ligand PD-L1 correlates with impaired cell-mediated immunity in high-risk human papillomavirus-related cervical intraepithelial neoplasia. Immunology 2013, 139, 513–522. [Google Scholar] [CrossRef]
- D’Alessandris, N.; Palaia, I.; Pernazza, A.; Tomao, F.; Di Pinto, A.; Musacchio, L.; Leopizzi, M.; Di Maio, V.; Pecorella, I.; Panici, P.B.; et al. PD-L1 expression is associated with tumor infiltrating lymphocytes that predict response to NACT in squamous cell cervical cancer. Virchows Arch. 2020, 1–9. [Google Scholar] [CrossRef]
- Odunsi, K. Immunotherapy in ovarian cancer. Ann. Oncol. 2017, 28, viii1–viii7. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Fujiwara, K.; Ledermann, J.; Oza, A.; Kristeleit, R.; Ray-Coquard, I.; Richardson, G.; Sessa, C.; Yonemori, K.; Banerjee, S.; et al. Avelumab alone or in combination with pegylated liposomal doxorubicin versus pegylated liposomal doxorubicin alone in platinum-resistant or refractory epithelial ovarian cancer: Primary and biomarker analysis of the phase III JAVELIN Ovarian 200 trial. Gynecol. Oncol. 2019, 154, 21–22. [Google Scholar] [CrossRef]
- Wenham, R.M.; Apte, S.M.; Shahzad, M.M.; Lee, J.K.; Dorman, D.; Chon, H.S. Phase II trial of dose dense (weekly) paclitaxel with pembrolizumab (MK-3475) in platinum-resistant recurrent ovarian cancer. J. Clin. Oncol. 2016, 34, TPS5612. [Google Scholar] [CrossRef]
- Zamarin, D.; Burger, R.A.; Sill, M.W.; Jr, D.J.P.; Lankes, H.A.; Feldman, M.D.; Zivanovic, O.; Gunderson, C.; Ko, E.; Mathews, C.; et al. Randomized Phase II Trial of Nivolumab Versus Nivolumab and Ipilimumab for Recurrent or Persistent Ovarian Cancer: An NRG Oncology Study. J. Clin. Oncol. 2020, 38, 1814–1823. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.E.; Vidal, G.A.; Mita, M.M.; Fleming, G.F.; Holloway, R.W.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. J. Clin. Oncol. 2018, 36, 106. [Google Scholar] [CrossRef]
- Drew, Y.; De Jonge, M.; Hong, S.; Park, Y.; Wolfer, A.; Brown, J.; Ferguson, M.; Gore, M.; Alvarez, R.; Gresty, C.; et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in germline BRCA-mutated (gBRCA m) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecol. Oncol. 2018, 149, 246–247. [Google Scholar] [CrossRef]
- Lee, J.-M.; Annunziata, C.; Houston, N.; Kohn, E.; Lipkowitz, S.; Minasian, L.; Nichols, E.; Trepel, J.; Trewhitt, K.; Zia, F.; et al. A phase II study of durvalumab, a PD-L1 inhibitor and olaparib in recurrent ovarian cancer (OvCa). Ann. Oncol. 2018, 29, viii334. [Google Scholar] [CrossRef]
- Liu, J.; Herold, C.; Luo, W.; Penson, R.; Horowitz, N.; Konstantinopoulos, P.; Castro, C.; Curtis, J.; Matulonis, U.; Cannistra, S.; et al. A phase II trial of combination nivolumab and bevacizumab in recurrent ovarian cancer. Ann. Oncol. 2018, 29, viii334–viii335. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fader, A.; Diaz, L.; Armstrong, D.; Tanner, E.; Uram, J.; Eyring, A.; Wang, H.; Fisher, G.; Greten, T.; Le, D. Preliminary results of a phase II study: PD-1 blockade in mismatch repair–deficient, recurrent or persistent endometrial cancer. Gynecol. Oncol. 2016, 141, 206–207. [Google Scholar] [CrossRef]
- Katsumata, N.; Tamura, K.; Hasegawa, K.; Matsumoto, K.; Mukai, H.; Takahashi, S.; Nomura, H.; Minami, H. Efficacy and safety of nivolumab in patients with uterine cervical cancer, uterine corpus cancer, or soft-tissue sarcoma. Ann. Oncol. 2018, 29, vii60. [Google Scholar] [CrossRef]
- Oaknin, A.; Duska, L.; Sullivan, R.; Pothuri, B.; Ellard, S.; Leath, C.; Moreno, V.; Kristeleit, R.; Guo, W.; Danaee, H.; et al. Preliminary safety, efficacy, and pharmacokinetic/pharmacodynamic characterization from GARNET, a phase I/II clinical trial of the anti–PD-1 monoclonal antibody, TSR-042, in patients with recurrent or advanced MSI-h and MSS endometrial cancer. Gynecol. Oncol. 2019, 154, 17. [Google Scholar] [CrossRef]
- Fleming, G.F.; Emens, L.A.; Eder, J.P.; Hamilton, E.P.; Liu, J.F.; Liu, B.; Molinero, L.; Fasso, M.; O’Hear, C.; Braiteh, F.S. Clinical activity, safety and biomarker results from a phase Ia study of atezolizumab (atezo) in advanced/recurrent endometrial cancer (rEC). J. Clin. Oncol. 2017, 35, 5585. [Google Scholar] [CrossRef]
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Di Simone, C.; Hyman, D.M.; E Stepan, D.; E Dutcus, C.; et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 711–718. [Google Scholar] [CrossRef]
- Rubinstein, M.M.; Caird, I.; Zhou, Q.; Iasonos, A.; Friedman, C.F.; Cadoo, K.A.; Konner, J.A.; O’Cearbhaill, R.E.; Tew, W.P.; Zamarin, D.; et al. A phase II trial of durvalumab with or without tremelimumab in patients with persistent or recurrent endometrial carcinoma and endometrial carcinosarcoma. J. Clin. Oncol. 2019, 37, 5582. [Google Scholar] [CrossRef]
- Ohno, S.; Kyo, S.; Myojo, S.; Dohi, S.; Ishizaki, J.; Miyamoto, K.I.; Morita, S.; Sakamoto, J.I.; Enomoto, T.; Kimura, T.; et al. Wilms’ tumor 1 (WTl) peptide immunotherapy for gynecological malignancy. Anticancer Res. 2009, 29, 4779–4784. [Google Scholar]
- Jackson, D.O.; Byrd, K.; Vreeland, T.J.; Hale, D.F.; Herbert, G.S.; Greene, J.M.; Schneble, E.J.; Berry, J.S.; Trappey, A.F.; Clifton, G.T.; et al. Interim analysis of a phase I/IIa trial assessing E39+GM-CSF, a folate binding protein vaccine, to prevent recurrence in ovarian and endometrial cancer patients. Oncotarget 2016, 8, 15912–15923. [Google Scholar] [CrossRef] [Green Version]
- Coosemans, A.; Vanderstraeten, A.; Tuyaerts, S.; Verschuere, T.; Moerman, P.; Berneman, Z.N.; Vergote, I.; Amant, F.; Van Gool, S.W. Wilms’ Tumor Gene 1 (WT1)--loaded dendritic cell immunotherapy in patients with uterine tumors: A phase I/II clinical trial. Anticancer. Res. 2013, 33, 5495–5500. [Google Scholar]
- Jäger, E.; Karbach, J.; Gnjatic, S.; Neumann, A.; Bender, A.; Valmori, D.; Ayyoub, M.; Ritter, E.; Ritter, G.; Jäger, D.; et al. Recombinant vaccinia/fowlpox NY-ESO-1 vaccines induce both humoral and cellular NY-ESO-1-specific immune responses in cancer patients. Proc. Natl. Acad. Sci. USA 2006, 103, 14453–14458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaumaya, P.T.; Foy, K.C.; Garrett, J.; Rawale, S.V.; Vicari, D.; Thurmond, J.M.; Lamb, T.; Mani, A.; Kane, Y.; Balint, C.R.; et al. Phase I Active Immunotherapy With Combination of Two Chimeric, Human Epidermal Growth Factor Receptor 2, B-Cell Epitopes Fused to a Promiscuous T-Cell Epitope in Patients With Metastatic and/or Recurrent Solid Tumors. J. Clin. Oncol. 2009, 27, 5270–5277. [Google Scholar] [CrossRef]
- Frenel, J.S.; Le Tourneau, C.; O’Neil, B.; Ott, P.A.; Piha-Paul, S.A.; Gomez-Roca, C.; Van Brummelen, E.M.J.; Rugo, H.S.; Thomas, S.; Saraf, S.; et al. Safety and efficacy of pembrolizumab in advanced, programmed death ligand 1-positive cervical cancer: Results from the phase IB KEYNOTE-028 trial. Proc. J. Clin. Oncol. 2017, 35, 4035–4041. [Google Scholar] [CrossRef]
- Chung, H.C.; Ros, W.; Delord, J.-P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef]
- Hollebecque, A.; Meyer, T.; Moore, K.N.; Machiels, J.-P.H.; De Greve, J.; López-Picazo, J.M.; Oaknin, A.; Kerger, J.N.; Boni, V.; Evans, T.R.J.; et al. An open-label, multicohort, phase I/II study of nivolumab in patients with virus-associated tumors (CheckMate 358): Efficacy and safety in recurrent or metastatic (R/M) cervical, vaginal, and vulvar cancers. J. Clin. Oncol. 2017, 35, 5504. [Google Scholar] [CrossRef]
- Santin, A.; Deng, W.; Frumovitz, M.M.; Huh, W.K.; Khleif, S.; Lankes, H.A.; Ratner, E.; O’Cearbhaill, R.; Jazaeri, A.A.; Birrer, M. A phase II evaluation of nivolumab, a fully human antibody against PD-1, in the treatment of persistent or recurrent cervical cancer. J. Clin. Oncol. 2018, 36, 5536. [Google Scholar] [CrossRef]
- Friedman, C.F.; Charen, A.S.; Zhou, Q.; A Carducci, M.; De Meritens, A.B.; Corr, B.R.; Fu, S.; Hollmann, T.J.; Iasonos, A.; A Konner, J.; et al. Phase II study of atezolizumab in combination with bevacizumab in patients with advanced cervical cancer. J. Immunother. Cancer 2020, 8, e001126. [Google Scholar] [CrossRef] [PubMed]
- Mayadev, J.; Brady, W.E.; Lin, Y.G.; Da Silva, D.M.; Lankes, H.A.; Fracasso, P.M.; Ghamande, S.A.; Moore, K.N.; Pham, H.Q.; Wilkinson, K.J.; et al. A phase I study of sequential ipilimumab in the definitive treatment of node positive cervical cancer: GOG 9929. J. Clin. Oncol. 2017, 35, 5526. [Google Scholar] [CrossRef]
- Lheureux, S.; Butler, M.O.; Clarke, B.; Cristea, M.C.; Martin, L.P.; Tonkin, K.; Fleming, G.F.; Tinker, A.V.; Hirte, H.W.; Tsoref, D.; et al. Association of ipilimumab with safety and antitumor activity inwomen with metastatic or recurrent human papillomavirus-related cervical carcinoma. JAMA Oncol. 2018, 4, e173776. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Mehta, A.; Jain, M.; Gupta, S.; Nagarkar, R.V.; John, S.; Petit, R. A Randomized Phase 2 Study of ADXS11-001 Listeria monocytogenes–Listeriolysin O Immunotherapy With or Without Cisplatin in Treatment of Advanced Cervical Cancer. Int. J. Gynecol. Cancer 2018, 28, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Huh, W.; Brady, W.; Dizon, D.; Powell, M.; Landrum, L.; Leath, C.; Tanner, E.; Higgins, R.; Ueda, S.; McHale, M.; et al. A prospective phase II trial of the listeria-based human papillomavirus immunotherpay axalimogene filolisbac in second- and third-line metastatic cervical cancer: A NRG oncology group trial. Gynecol. Oncol. 2017, 145, 220. [Google Scholar] [CrossRef]
- Cohen, E.E.; Moore, K.N.; Slomovitz, B.M.; Chung, C.H.; Anderson, M.L.; Morris, S.R.; Mauro, D.; Burtness, B. Phase I/II study of ADXS11-001 or MEDI4736 immunotherapies alone and in combination, in patients with recurrent/metastatic cervical or human papillomavirus (HPV)-positive head and neck cancer. J. Immunother. Cancer 2015, 3, P147. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.; Kemberling, H.; Eyring, A.; Skora, A.; Azad, N.S.; Laheru, D.A.; Donehower, R.C.; et al. PD-1 blockade in tumors with mismatch repair deficiency. J. Clin. Oncol. 2015, 33, LBA100. [Google Scholar] [CrossRef]
- Nakatsuka, S.-I.; Oji, Y.; Horiuchi, T.; Kanda, T.; Kitagawa, M.; Takeuchi, T.; Kawano, K.; Kuwae, Y.; Yamauchi, A.; Okumura, M.; et al. Immunohistochemical detection of WT1 protein in a variety of cancer cells. Mod. Pathol. 2006, 19, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Paraghamian, S.E.; Longoria, T.C.; Eskander, R.N. Metastatic small cell neuroendocrine carcinoma of the cervix treated with the PD-1 inhibitor, nivolumab: A case report. Gynecol. Oncol. Res. Pr. 2017, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharabi, A.; Kim, S.S.; Kato, S.; Sanders, P.D.; Patel, S.P.; Sanghvi, P.; Weihe, E.; Kurzrock, R. Exceptional Response to Nivolumab and Stereotactic Body Radiation Therapy (SBRT) in Neuroendocrine Cervical Carcinoma with High Tumor Mutational Burden: Management Considerations from the Center For Personalized Cancer Therapy at UC San Diego Moores Cancer. Oncologist 2017, 22, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Chitsike, L.; Duerksen-Hughes, P. The Potential of Immune Checkpoint Blockade in Cervical Cancer: Can Combinatorial Regimens Maximize Response? A Review of the Literature. Curr. Treat. Options Oncol. 2020, 21, 1–21. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the γ or ζ subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chailyan, A.; Marcatili, P.; Tramontano, A. The association of heavy and light chain variable domains in antibodies: Implications for antigen specificity. FEBS J. 2011, 278, 2858–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi, A.; Karulkar, A.; Ghosh, S.; Rafiq, A.; Purwar, R. Lymphocytes in cellular therapy: Functional regulation of CAR T cells. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Qin, L.; Lai, Y.; Zhao, R.; Wei, X.; Weng, J.; Lai, P.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; et al. Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells. J. Hematol. Oncol. 2017, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pan, J.; Guo, Z.; Yang, C.; Mao, L. CART cell therapy for prostate cancer: Status and promise. OncoTargets Ther. 2019, 12, 391–395. [Google Scholar] [CrossRef] [Green Version]
- Hombach, A.A.; Abken, H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int. J. Cancer 2011, 129, 2935–2944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cao, L.; Xie, J.; Shi, N.; Zhang, Z.; Luo, Z.; Yue, D.; Zhang, Z.; Wang, L.; Han, W.; et al. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: A meta-analysis. Oncotarget 2015, 6, 33961–33971. [Google Scholar] [CrossRef] [Green Version]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 Release by Engineered T Cells Expressing Chimeric Antigen Receptors Can Effectively Muster an Antigen-Independent Macrophage Response on Tumor Cells That Have Shut Down Tumor Antigen Expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Hillerdal, V.; Essand, M. Chimeric Antigen Receptor-Engineered T Cells for the Treatment of Metastatic Prostate Cancer. BioDrugs 2015, 29, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Petersen, C.T.; Krenciute, G. Next Generation CAR T Cells for the Immunotherapy of High-Grade Glioma. Front. Oncol. 2019, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.W.; Cho, J.-Y. Recent Advances in Allogeneic CAR-T Cells. Biomolecules 2020, 10, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [Green Version]
- Strohl, N. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Ahmad, A.; Uddin, S.; Steinhoff, M. CAR-T Cell Therapies: An Overview of Clinical Studies Supporting Their Approved Use against Acute Lymphoblastic Leukemia and Large B-Cell Lymphomas. Int. J. Mol. Sci. 2020, 21, 3906. [Google Scholar] [CrossRef]
- Schepisi, G.; Conteduca, V.; Casadei, C.; Gurioli, G.; Rossi, L.; Gallà, V.; Cursano, M.C.; Brighi, N.; Lolli, C.; Menna, C.; et al. Potential Application of Chimeric Antigen Receptor (CAR)-T Cell Therapy in Renal Cell Tumors. Front. Oncol. 2020, 10, 565857. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Pule, M.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Thistlethwaite, F.C.; Gilham, D.E.; Guest, R.D.; Rothwell, D.G.; Pillai, M.; Burt, D.J.; Byatte, A.J.; Kirillova, N.; Valle, J.W.; Sharma, S.K.; et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017, 66, 1425–1436. [Google Scholar] [CrossRef]
- Scarfò, I.; Maus, M.V. Current approaches to increase CAR T cell potency in solid tumors: Targeting the tumor microenvironment. J. Immunother. Cancer 2017, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- A Morgan, R.; Yang, J.C.; Kitano, M.; E Dudley, M.; Laurencot, C.M.; A Rosenberg, S. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of Metastatic Renal Cell Carcinoma With Autologous T-Lymphocytes Genetically Retargeted Against Carbonic Anhydrase IX: First Clinical Experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef] [PubMed]
- Arcangeli, S.; Magnani, C.F.; Tettamanti, S.; Biagi, E. “Switchable chimeric antigen receptor T cells: A novel universal chimeric antigen receptor platform for a safe control of T-cell activation”. Transl. Cancer Res. 2016, 5, S174–S177. [Google Scholar] [CrossRef]
- Zhang, E.; Xu, H. A new insight in chimeric antigen receptor-engineered T cells for cancer immunotherapy. J. Hematol. Oncol. 2017, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remmerie, M.; Janssens, V. PP2A: A Promising Biomarker and Therapeutic Target in Endometrial Cancer. Front. Oncol. 2019, 9, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, W.S.; Wang, H.; Maggio, D.; Kovach, J.S.; Zhang, Q.; Song, Q.; Marincola, F.M.; Heiss, J.D.; Gilbert, M.R.; Lu, R.; et al. Pharmacologic inhibition of protein phosphatase-2A achieves durable immune-mediated antitumor activity when combined with PD-1 blockade. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, N.L.; Xiu, J.; Chatterjee-Paer, S.; De Meritens, A.B.; Burke, W.M.; Tergas, A.I.; Wright, J.D.; Hou, J.Y. Distinct molecular landscapes between endometrioid and nonendometrioid uterine carcinomas. Int. J. Cancer 2017, 140, 1396–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, C.; Zanagnolo, V.; Ramirez, N.; Cohn, D.E.; Kelbick, N.; Copeland, L.; Maxwell, L.G.; Fowler, J.M. HER-2 Is an Independent Prognostic Factor in Endometrial Cancer: Association With Outcome in a Large Cohort of Surgically Staged Patients. J. Clin. Oncol. 2006, 24, 2376–2385. [Google Scholar] [CrossRef] [PubMed]
- Black, J.D.; Lopez, S.; Cocco, E.; Bellone, S.; Altwerger, G.; Schwab, C.L.; English, D.P.; Bonazzoli, E.; Predolini, F.; Ferrari, F.; et al. PIK3CA oncogenic mutations represent a major mechanism of resistance to trastuzumab in HER2/neu overexpressing uterine serous carcinomas. Br. J. Cancer 2015, 113, 1020–1026. [Google Scholar] [CrossRef]
- Zhao, S.; Choi, M.; Overton, J.D.; Bellone, S.; Roque, D.M.; Cocco, E.; Guzzo, F.; English, D.P.; Varughese, J.; Gasparrini, S.; et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 2916–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remmerie, M.; Janssens, V. Targeted Therapies in Type II Endometrial Cancers: Too Little, but Not Too Late. Int. J. Mol. Sci. 2018, 19, 2380. [Google Scholar] [CrossRef] [Green Version]
- Nicoletti, R.; Lopez, S.; Bellone, S.; Cocco, E.; Schwab, C.L.; Black, J.D.; Centritto, F.; Zhu, L.; Bonazzoli, E.; Buza, N.; et al. T-DM1, a novel antibody-drug conjugate, is highly effective against uterine and ovarian carcinosarcomas overexpressing HER2. Clin. Exp. Metastasis 2015, 32, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- English, D.P.; Bellone, S.; Schwab, C.L.; Bortolomai, I.; Bonazzoli, E.; Cocco, E.; Buza, N.; Hui, P.; Lopez, S.; Ratner, E.; et al. T-DM1, a novel antibody–drug conjugate, is highly effective against primary HER2 overexpressing uterine serous carcinoma in vitro and in vivo. Cancer Med. 2014, 3, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Tangen, I.L.; Onyango, T.B.; Kopperud, R.; Berg, A.; Halle, M.K.; Øyan, A.M.; Werner, H.M.; Trovik, J.; Kalland, K.H.; Salvesen, H.B.; et al. Androgen receptor as potential therapeutic target in metastatic endometrial cancer. Oncotarget 2016, 7, 49289–49298. [Google Scholar] [CrossRef] [Green Version]
- Barrett, D.M.; Grupp, S.A.; June, C.H. Chimeric Antigen Receptor– and TCR-Modified T Cells Enter Main Street and Wall Street. J. Immunol. 2015, 195, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.-F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Study | Phase | Treatment | Number of Patients | Patient Population | Results | TRAEs (%) |
|---|---|---|---|---|---|---|
| OC | ||||||
| Chemo-IMT combo | ||||||
| JAVELIN Ovarian 200 [32] | 3 | Arm 1: Avelumab alone Arm 2: PLD alone Arm 3: Avelumab + PLD | 566 | Platinum- resistant/ refractory OC | ORR 3.7% vs. 4.2% vs. 3.3% PFS 1.9 vs. 3.5 vs. 3.7 mos OS 11.8 vs. 13 vs. 15.7 mos | PPE syndrome (9.9) Rash (9.3) Neutropenia (9.3) Fatigue (7.1) Stomatitis (5.5) |
| Wenham et al. [33] | 2 | Weekly paclitaxel (80 mmg/m2) + pembrolizumab 200 mg q3 w | 37 | Recurrent platinum- resistant EO, more than 3 prior therapies | ORR 51.4% DCR 86.50% mPFS 7.6 mos mOS 13.4 mos | Neutropenia Nausea/vomiting Edema, diarrhea Dyspnea Neuropathy Abdominal pain |
| IMT combo | ||||||
| Zamarin et al. [34] | 2 | ARM 1 Nivolumab alone ARM 2 Nivolumab + Ipilimumab | 49 51 | Recurrent OC | ORR 31.4%, 28.1 mos 12.2%, 21 mos | Colitis/Diarrhea (16% vs. 4%) Anemia (16% vs. 4%) Rash (14% vs. 4%) |
| IMT-PARPi combo | ||||||
| TOPACIO [35] | 1/2 | Pembrolizumab 200 mg q3 w + Niraparib 200 mg q day | 60 | Recurrent OC | ORR 18% DCR 65% mPFS 3.4 mos, (6-mo 31% and 12-mo 12%). | Fatigue Anemia Nausea Constipation Myelosuppression |
| MEDIOLA [36] | 2 | Olaparib 300 mg BID × 4 w, then 300 mg BID + Durvalumab 1.5 g q4 w | 32 | gBRCAm platinum- sensitive relapsed OC | ORR 63% DCR 81% | Anemia, Neutropenia, Increased amylase/lipase |
| Lee et al. [37] | 2 | Durvalumab 1500 mg q4 w + Olaparib 300 mg BID | 35 | Platinum- resistant/ refractory OC | ORR 14.7% DCR 52.9% | Anemia, Lymphopenia Atrial fibrillation Nausea |
| IMT-VEGFi combo | ||||||
| Liu et al. [38] | 2 | Bevacizumab 10 mg/kg + Nivolumab 240 mg q 2 w until PD | 38 | Platinum sensitive/ resistant OC | ORR 28.9% DCR 34.2% mPFS 8.1 mos | Fatigue AST/ALT elevation Myalgia |
| EC | ||||||
| IMT | ||||||
| Le et al. [39] | 2 | Pembrolizumab 10 mg/kg q2 w | 15 | MMR- deficient EC with PD | ORR 53% DCR 73% | Colitis/Diarrhea Pancreatitis Hyperamylasemia |
| Fader et al. [40] | 2 | Pembrolizumab 10 mg/kg q2 w | 9 | Recurrent/ persistent MMR- deficient EC | ORR 56% DCR 88.9%, 1 y OS 89% | No grade 3 |
| Katsumata et al. [41] | 2 | Nivolumab 240 mg q2 w | 23 | Advanced/ recurrent EC | ORR: 23% PFS 3.4 mos 1 y OS 48.5% | Pruritus Increased lipase Diarrhea |
| GARNET [42] | 1/2 | Dostarlimab 500 mg q3 w × 4, then 1000 mg q6 w | 94 | Recurrent/ persistent EC | ORR 27% (50% MSI-H 19.1% MSS) DCR 48.90% | AST elevation |
| Fleming et al. [43] | 1 A | Atezolizumab 1200 mg q3 w | 15 | Advanced/ recurrent EC | ORR 13% DCR 26% mPFS 1.7 mos mOS 9.6 mos | Favorable safety profile |
| Makker et al. [44] | 2 | Pembrolizumab 200 mg q3 w + Lenvatinib 200 mg/die | 53 | Stage IV EC | ORR 39.6% DCR 86.8% PFS 7.4 mos | Hypertension Diarrhea Fatigue Hypothyroidism |
| Rubinstein et al. [45] | 2 | ARM 1 Durvalumab 1500 mg q4 w ARM 2 Durvalumab 1500 mg q4 w + Tremelimumab 75 mg q4 w | 56 | Recurrent/ persistent EC | ORR 14.8%, 6 mos PFS, 13.3 mos 11.1%, 18.5 mos | Fatigue Diarrhea Nausea/vomiting Pruritis |
| Vaccines | ||||||
| Ohno et al. [46] | 2 | HLA-A∗2402- restricted adjuvant WT1 peptide 3 mg 1/w × 12 w | 2 | HLA-A∗2402-positive EC resistant to standard therapy | ORR 0%, DCR 0% | Grade 1-2 erythema at injection site |
| Jackson et al. [47] | 1/2 A | HLA-A2 restricted FBP-derived peptide at different dosage: 100 mcg/0.5 mL 500 mcg/0.5 mL 1000 mcg/0.5 mL + 250 mcg/1.0 mL GM-CSF | 51 | HLA-2+ Gyn. cancers | 2-y DFS 43% for 1000 mcg dosage | Erythema at injection site Pruritus |
| Coosemans et al. [48] | 1/2 | Autologous DC electroporated with WT1 mRNA | 3 | Stage IV EC | ORR 0%, DCR 0% | Grade 1–2 erythema at injection site |
| Jager et al. [49] | 1 | rV-NY-ESO-1 3.1 × 107 PFU × 2, then rV-NY-ESO-1 7.41 × 107 PFU q4 w | 1 | Stage IV NY-ESO tumors | ORR = 0%, DCR = 0% | Grade 1-2 erythema at injection site |
| Kaumaya et al. [50] | 1 | Two HER2 B-cell epitopes combined with a T-cell epitope with n-MDP adjuvant 0.25 or 0.5 mg q3 w × 3 | 2 | Stage IV EC | ORR = 50% (1 PR) | Diarrhea Hyperglycemia |
| CC | ||||||
| IMT | ||||||
| KEYNOTE-028 [51] | 1 B | Pembrolizumab 10 mg/kg q2 w × 2 y | 24 | PD-L1+ previously treated CC | ORR 17% DCR 17% mPFS 2 mos mOS 11 mos | Rash, Fever, Proteinuria |
| KEYNOTE-158 [52] | 2 | Pembrolizumab 200 mg q2 w × 2 y | 98 | Pretreated CC patients | ORR 12.2% DCR 30.6% mPFS 2.1 mos mOS 9.4 mos | Hypothyroidism, Hyporexia, Fatigue |
| CheckMate 358 [53] | 1/2 | Nivolumab 240 mg q2 w | 19 | Pretreated CC patients | ORR 26.3% DCR 70.8% mPFS 5.5 mos | Diarrhea, Fatigue, Pneumonitis, Stomatitis Abdominal pain |
| Santin et al. [54] | 2 | Nivolumab 3 mg/kg q2 w | 25 | Persistent or recurrent CC | ORR 4% DCR 40% | Hepatotoxicity Type-1 diabetes |
| Friedman et al. [55] | 2 | Atezolizumab 1200 mg q3 w + Bevacizumab 15 mg/kg q3 w | 10 | Stage IV CC | DCR 50% mPFS 2.9 mos mOS 9 mos | Arachnoiditis, Hypoacusia, Weakness, Thrombosis |
| GOG208 [56] | 1 | Weekly Cisplatin (40 mg/m2) + extended field radiation, then Ipilimumab 3 mg/kg, 10 mg/kg, expansion cohort of 10 mg/kg | 19 | Stage IB2-IVA (n+) CC undergoing chemo- radiation | DCR 74% | Hyperlipasemia, Neutropenia, Rash |
| Lheureux et al. [57] | 1/2 | Ipilimumab 3 mg/kg q3 w × 4 cycles or 10 mg/kg q3 w × 4 cycles followed by maintenance q12 w) | 42 | Stage IV CC | ORR 2.9% DCR 32.4% mPFS 2.5 mos mOS 8.5 mos | Diarrhea |
| Vaccines | ||||||
| Basu et al. [58] | 2 | ADXS11–001 (3 times) 1 × 109 CFUs (80 mL infusion on day 1, 29, 57) Combo therapy ADX-011 (day 1) + weekly Cisplatin (40 mg/m2) post-vaccine 4 w × 5 w, then 1 cycle of ADS11–011 (3 times) | 69 | Stage IV CC | ORR: 17.1% mPFS 6.2 mos mOS 8.5 mos | Fever |
| Huh et al. [59] | 2 | ADXS11–001 (1 × 109 CFU) q3 w × 3 doses (stage 1) or 1 y (stage 2) | 50 | Stage IV CC | ORR 2% (1 CR) 1 y OS 38% | Fatigue Nausea, Anemia |
| Cohen et al. [60] | 1/2 | ARM 1 ADXS11–001 ARM 2 Pembrolizumab ARM 3 combo | 5 | Stage IV HPV+ CC | ORR 40% DCR 40% | Chills, Fever, Nausea, Hypotension, Diarrhea, Fatigue, Tachycardia, Headache |
| Drug [Reference] | Trade Name | Results | Approved For | Patient Population | Date of Approval |
|---|---|---|---|---|---|
| Tisagenlecleucel [80,81] | Kymriah | Complete Remission 90% | B-cell precursor acute lymphoblastic leukemia | Up to 25 years | 30 August 2017 |
| Complete Remission 38% ORR 52% | Large B-cell lymphoma | Adult | 1 May 2018 | ||
| Axicabtagene ciloleucel [82] | Yescarta | Complete Remission 51% ORR 82% | Large B-cell lymphoma | Adult | 18 October 2017 |
| Brexucabtagene autoleucel [83] | Tecartus | ORR 87% (Complete Response 62%) | Relapsed or refractory mantle cell lymphoma | Adult | 24 July 2020 |
| Study | Phase | Treatment | N | Patient Population | Endpoints | Locations | Current Status |
|---|---|---|---|---|---|---|---|
| MESO | |||||||
| NCT03916679 | 1/2 | anti-MESO CAR-T cells | 20 | MESO-positive OC patients | (1) Safety (2) ORR, PFS | Zhejiang University, Zhejiang, China | Recruiting |
| NCT04562298 | 1 | LCAR-M23, anti-MESO CAR-T | 34 | Relapsed and Refractory Epithelial OC | Dose-limiting toxicity and TRAEs | Shanghai East Hospital Shanghai, China | Recruiting |
| NCT03692637 | 1 | anti-MESO CAR-NK Cells | 30 | MESO-positive patients with stage II-IV epithelial OC | Occurrence of TRAEs | Allife Medical Science & Technology Co., Ltd. | Not yet recruiting |
| NCT04627740 | 1/2 | Retroviral vector- transduced autologous T cells to express anti-ALPP CARs | 20 | ALPP-Positive Metastatic OC and EC | (1) Safety (2) ORR, PFS | Xinqiao Hospital of Chongqing Chongqing, China | Not yet recruiting |
| NCT04503980 | 1 | MESO CAR-T Cells Secreting PD-1 Nanobodies | 10 | MESO-positive advanced solid tumors | (1) Dose-limiting toxicity (2) ORR, PFS, OS MTD | Shanghai Tenth people’s Hospital, Shanghai, China | Recruiting |
| NCT03814447 | 1 | anti- MESO CAR-T cells | 10 | Refractory/Relapsed OC | (1) TRAEs (2) ORR, PFS | Shanghai 6 th People’s Hospital, Shanghai, China | Recruiting |
| NCT03608618 | 1 | MCY-M11 | 27 | Platinum resistant high grade serous OC, Fallopian and peritoneal carcinoma | (1) TRAEs (2) RECIST and irRECIST | Multiple Institutions in the USA | Recruiting |
| NCT02159716 | 1 | anti-MESO CAR-T cells | 19 | MESO-positive advanced solid tumors | Occurrence of TRAEs | University of Pennsylvania, Philadelphia, USA | Completed |
| NCT03054298 | 1 | huCART-MESO cells +/− CTX and different administrations | 18 | MESO-positive advanced solid tumors | (1) TRAEs (2) RECIST PFS, OS | University of Pennsylvania, Philadelphia, USA | Recruiting |
| NCT01583686 | 1/2 | anti-MESO CAR-T cells | 15 | MESO-positive advanced solid tumors | ORR and TRAEs | National Cancer Institute, Bethesda, USA | Terminated due to slow/ insufficient accrual. |
| MUCINS (CA125) | |||||||
| NCT03907527 | 1 | PRGN-3005 UltraCAR-T cells | 71 | Advanced Stage Platinum Resistant OC | TRAE Incidence MTD | Fred Hutchinson Cancer Research Center, USA | Recruiting |
| NCT04025216 | 1 | TnMUC1- Targeted Genetically- modified CAR- T Cells | 112 | Advanced TnMUC1-Positive Solid Tumors and Multiple Myeloma | (1) Dose Identification and ORR (2) Safety and tolerability | Multiple Institutions in the USA | Recruiting |
| OTHER TARGETS | |||||||
| NCT04511871 | 1 | CCT303–406 CAR modified autologous T cells | 15 | Relapsed or refractory stage IV metastatic HER2-positive solid tumors | (1) MTD (2) ORR, DCR, DOR, PFS TRAEs | Fudan University, Shanghai, China | Recruiting |
| NCT03585764 | 1 | MOv19-BBz CAR-T cells | 18 | Alpha Folate Receptor -positive OC, Fallopian and peritoneal carcinoma | (1) TRAEs (2) ORR, PFS, OS | University of Pennsylvania, Philadelphia, USA | Recruiting |
| NCT02830724 | 1/2 | Anti-hCD70 CAR-T cells | 124 | CD70 Expressing cancers | (1) TRAEs (2) ORR | National Cancer Institute, Bethesda, USA | Recruiting |
| NCT03638206 | 1/2 | Multi-target Gene-modified CAR-T/TCR-T cell for Malignancies | 73 | Multi-target Gene-modified CAR-T/TCR-T Cell for Malignancies | (1) TRAEs (2) Clinical response | Zhengzhou University, Zhengzhou, China | Recruiting |
| Study | Phase | Treatment | No. | Patient Population | Endpoints | Locations |
|---|---|---|---|---|---|---|
| NCT01583686 | 1/2 | anti-MESO CAR-T cells | 15 | Mesothelin-positive CC patients | Safety | National Cancer Institute, Bethesda, USA |
| NCT04556669 | 1 | Autologous aPD-L1 armored anti-CD22 CAR-T cells | 30 | Refractory CC and other solid tumors | (1) Safety (2) ORR, PFS | 4 th Hospital of Hebei Medical University, Shijiazhuang, China |
| NCT03356795 | 1/2 | CC-specific CAR-T cells | 20 | GD2, PSMA, Muc1, Mesothelin or other markers positive CC | (1) Safety (2) ORR | Shenzhen Geno-immune Medical Institute, Shenzhen, China |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schepisi, G.; Casadei, C.; Toma, I.; Poti, G.; Iaia, M.L.; Farolfi, A.; Conteduca, V.; Lolli, C.; Ravaglia, G.; Brighi, N.; et al. Immunotherapy and Its Development for Gynecological (Ovarian, Endometrial and Cervical) Tumors: From Immune Checkpoint Inhibitors to Chimeric Antigen Receptor (CAR)-T Cell Therapy. Cancers 2021, 13, 840. https://doi.org/10.3390/cancers13040840
Schepisi G, Casadei C, Toma I, Poti G, Iaia ML, Farolfi A, Conteduca V, Lolli C, Ravaglia G, Brighi N, et al. Immunotherapy and Its Development for Gynecological (Ovarian, Endometrial and Cervical) Tumors: From Immune Checkpoint Inhibitors to Chimeric Antigen Receptor (CAR)-T Cell Therapy. Cancers. 2021; 13(4):840. https://doi.org/10.3390/cancers13040840
Chicago/Turabian StyleSchepisi, Giuseppe, Chiara Casadei, Ilaria Toma, Giulia Poti, Maria Laura Iaia, Alberto Farolfi, Vincenza Conteduca, Cristian Lolli, Giorgia Ravaglia, Nicole Brighi, and et al. 2021. "Immunotherapy and Its Development for Gynecological (Ovarian, Endometrial and Cervical) Tumors: From Immune Checkpoint Inhibitors to Chimeric Antigen Receptor (CAR)-T Cell Therapy" Cancers 13, no. 4: 840. https://doi.org/10.3390/cancers13040840
APA StyleSchepisi, G., Casadei, C., Toma, I., Poti, G., Iaia, M. L., Farolfi, A., Conteduca, V., Lolli, C., Ravaglia, G., Brighi, N., Altavilla, A., Martinelli, G., & De Giorgi, U. (2021). Immunotherapy and Its Development for Gynecological (Ovarian, Endometrial and Cervical) Tumors: From Immune Checkpoint Inhibitors to Chimeric Antigen Receptor (CAR)-T Cell Therapy. Cancers, 13(4), 840. https://doi.org/10.3390/cancers13040840