
Novel Small Molecule Hsp90/Cdc37 Interface Inhibitors Indirectly Target K-Ras-Signaling


 , ,
, ,  and
and 
Abstract
:Simple Summary
Abstract
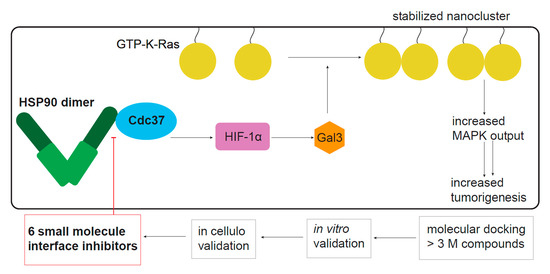
1. Introduction
2. Results
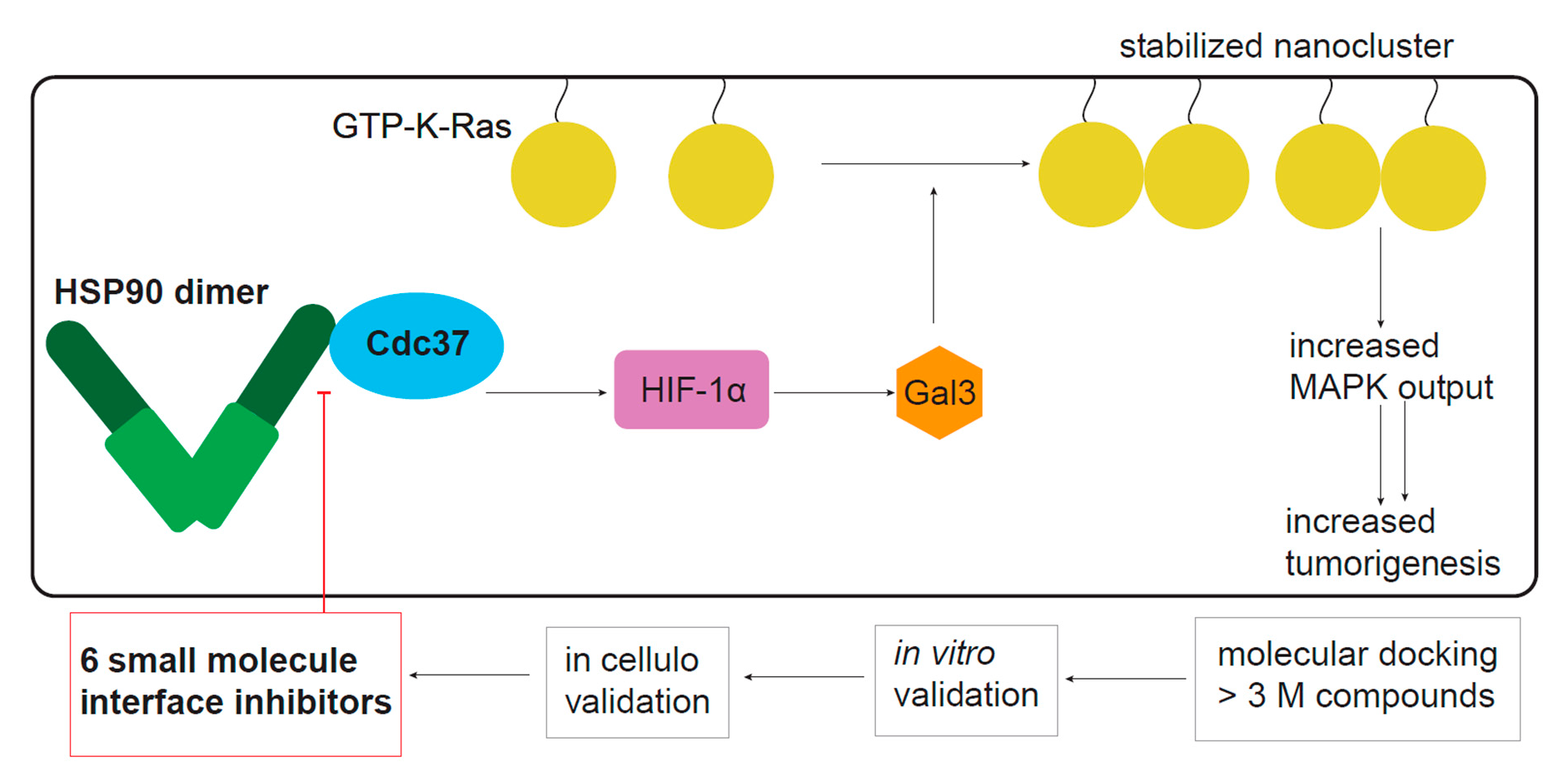
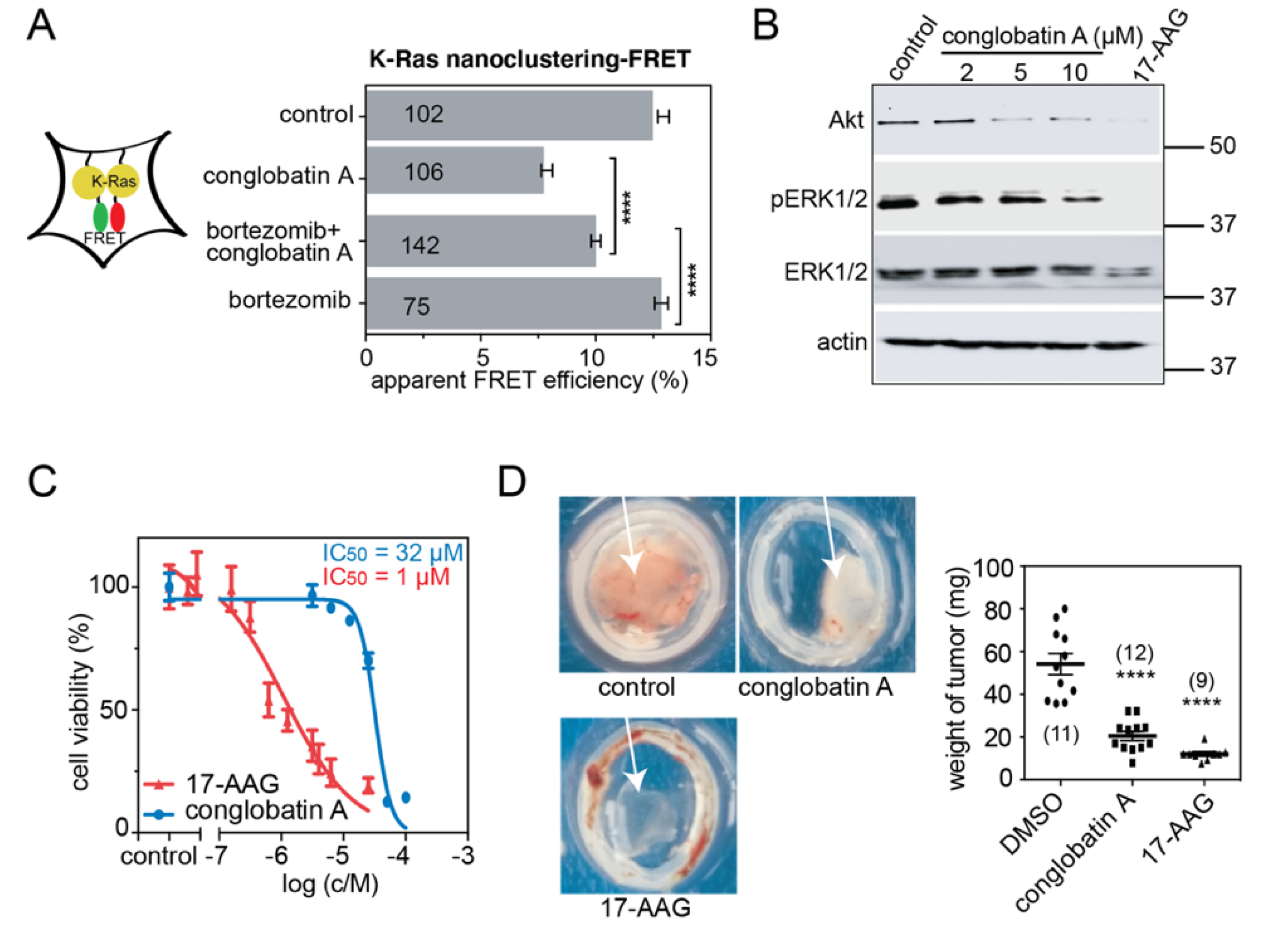
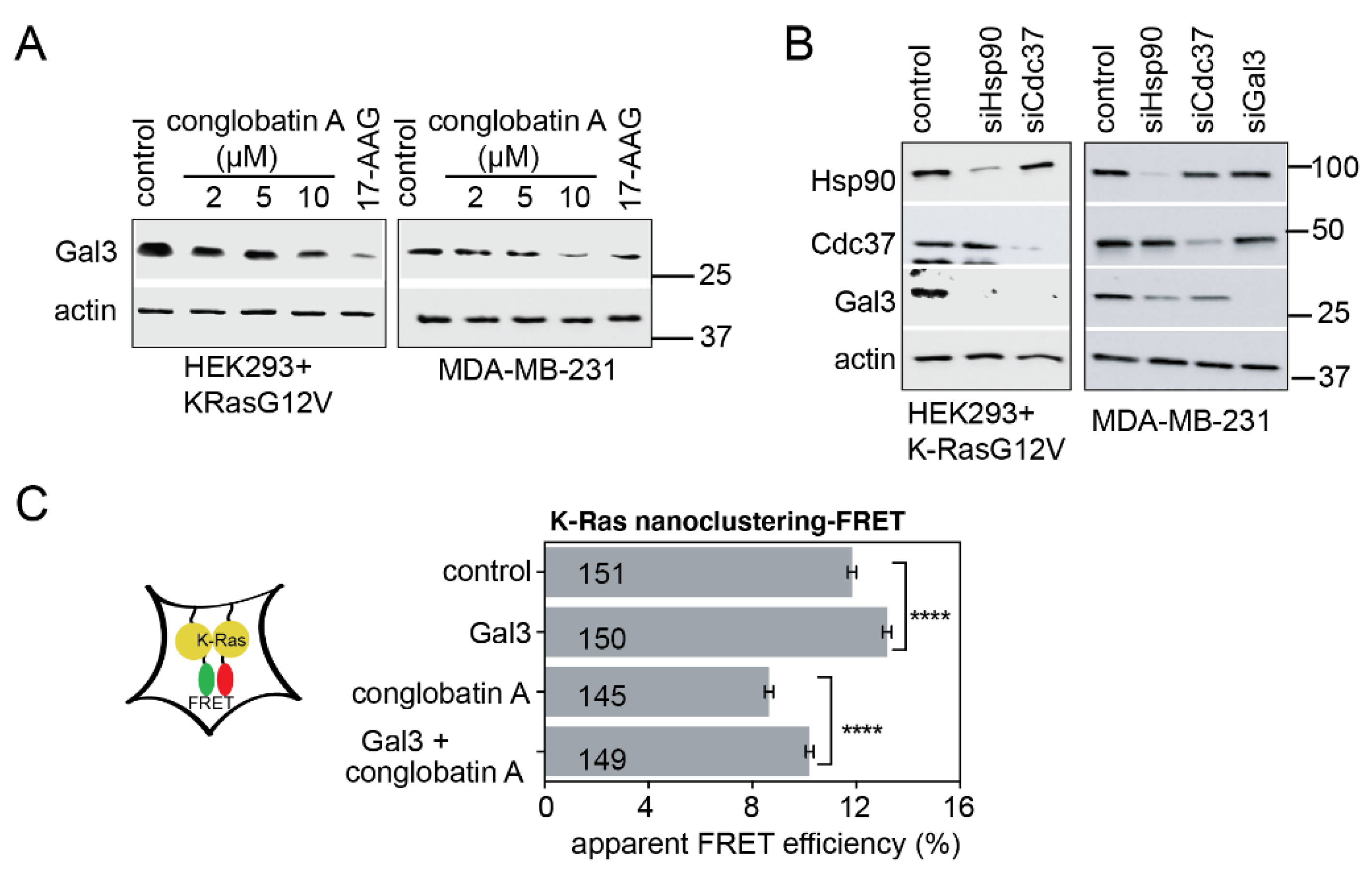
2.1. Conglobatin A Selectively Interferes with K-Ras Nanoclustering and Signaling by Inhibiting the Hsp90/Cdc37 Complex
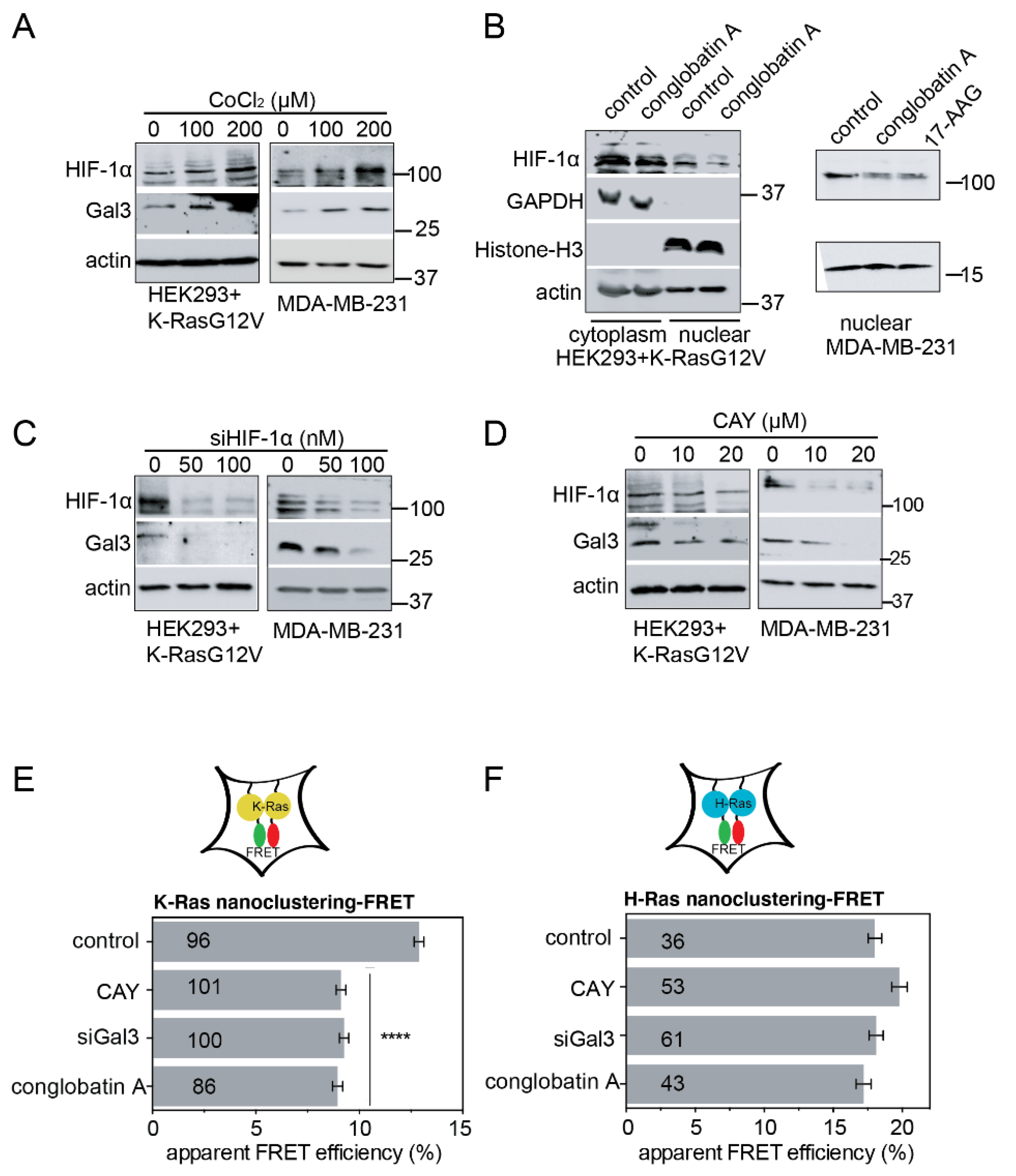
2.2. Hsp90 Inhibition Depletes the K-Ras Nanocluster Scaffold Galectin-3 Downstream of the Hsp90 Client HIF-1α
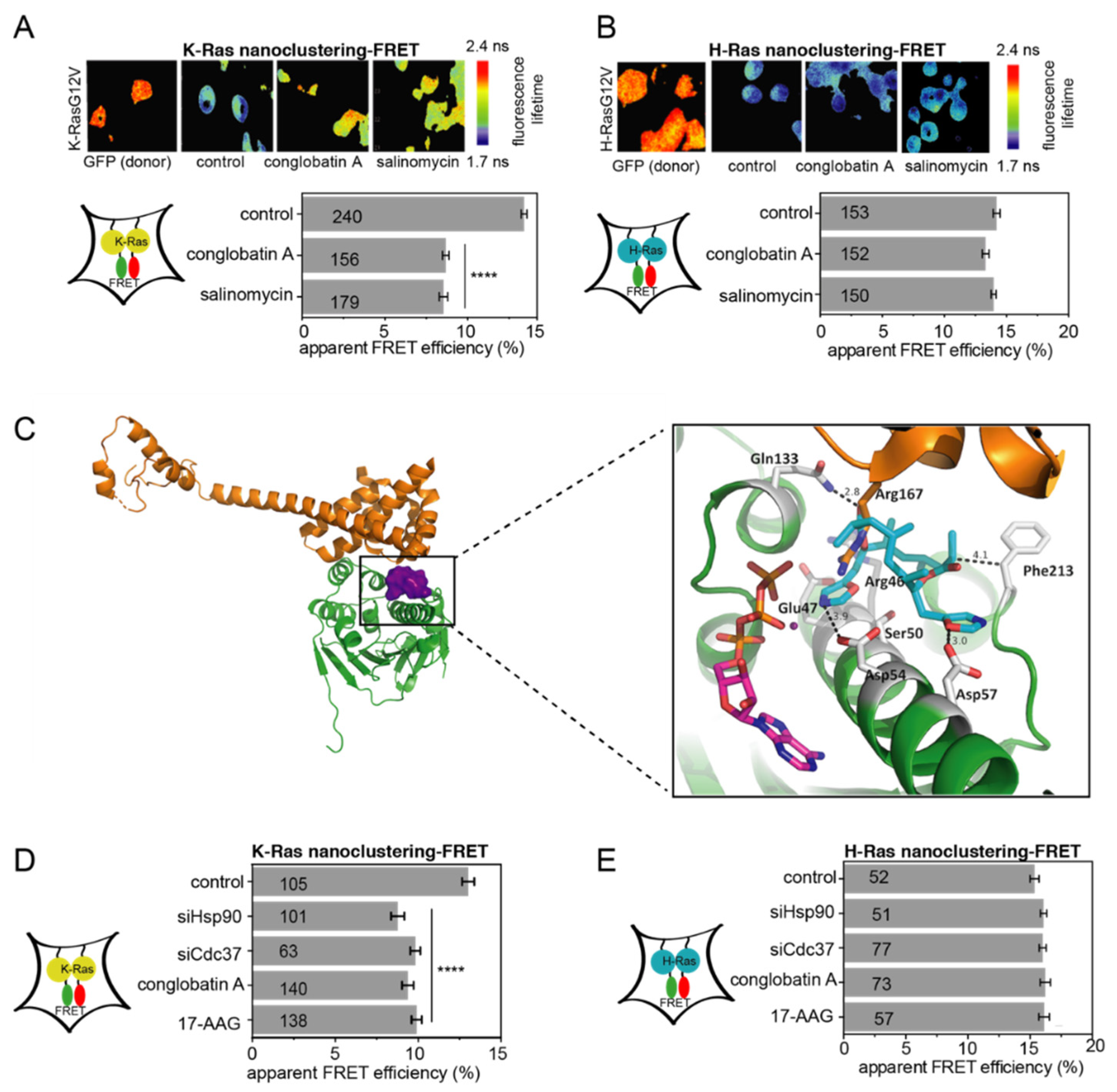
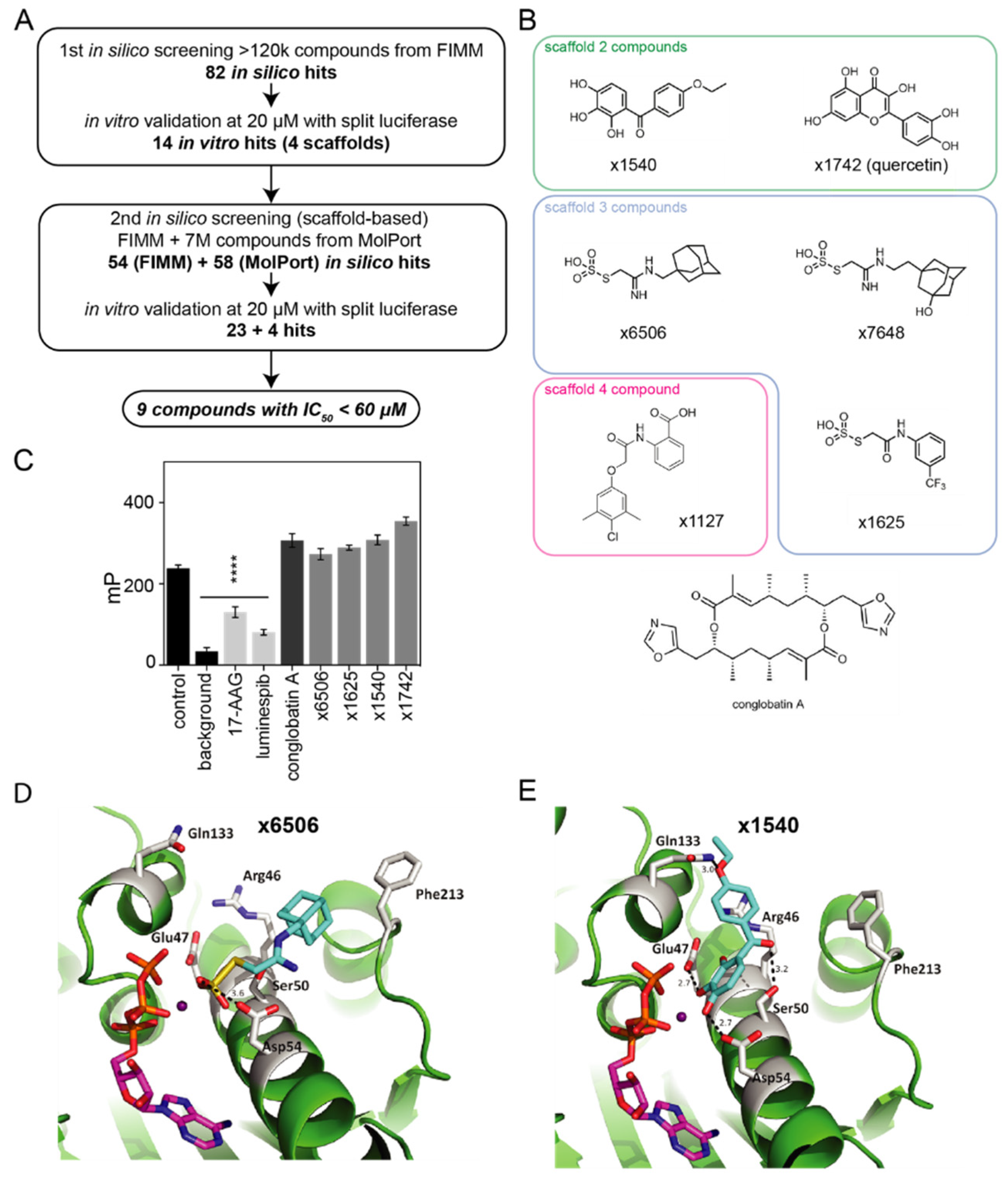
2.3. Computational and In Vitro Screening for Novel Hsp90/Cdc37 Interface Inhibitors
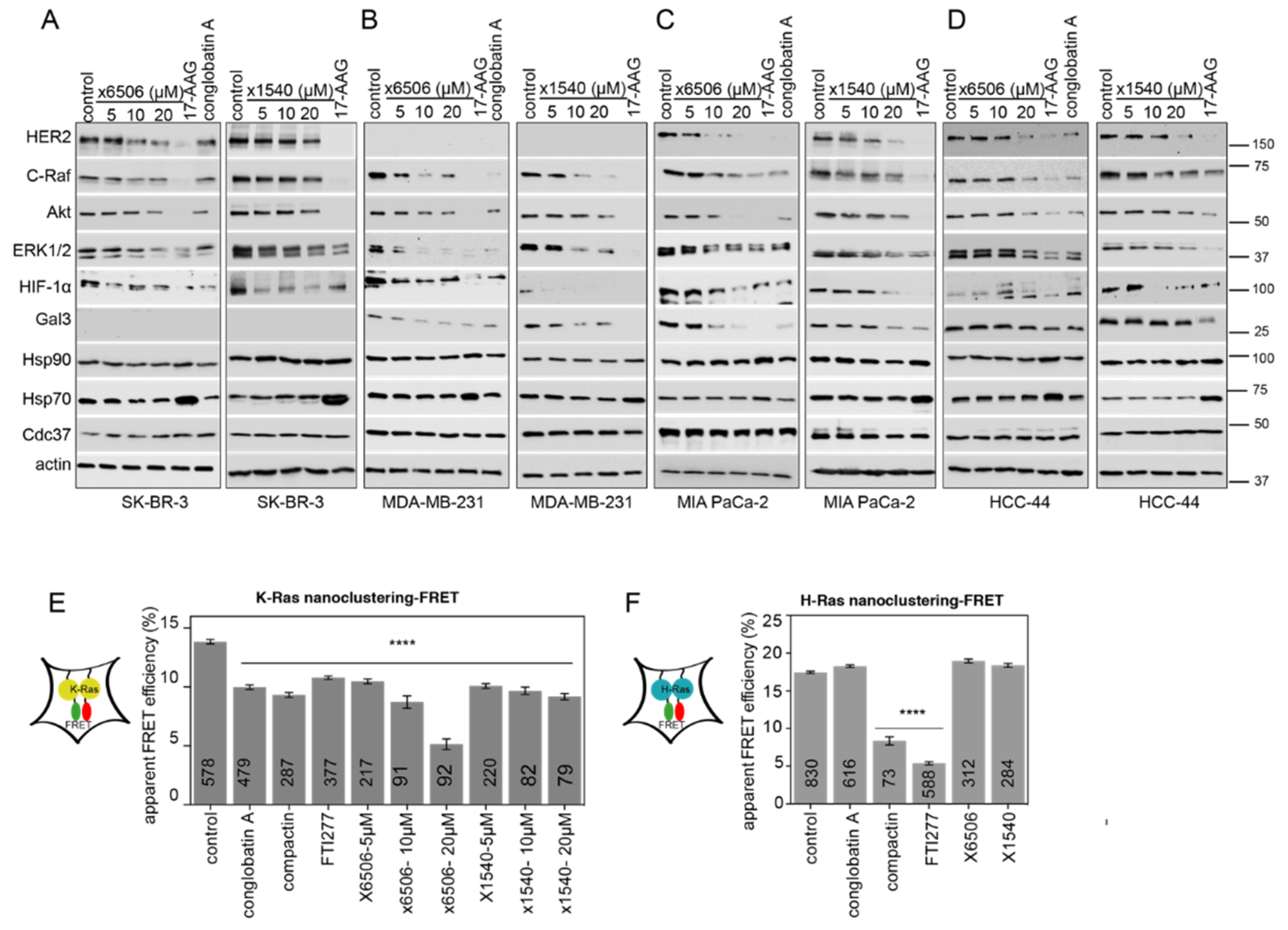
2.4. Both ×6506 and ×1540 Deplete HIF-1α and Gal3 to Selectively Affect K-Ras
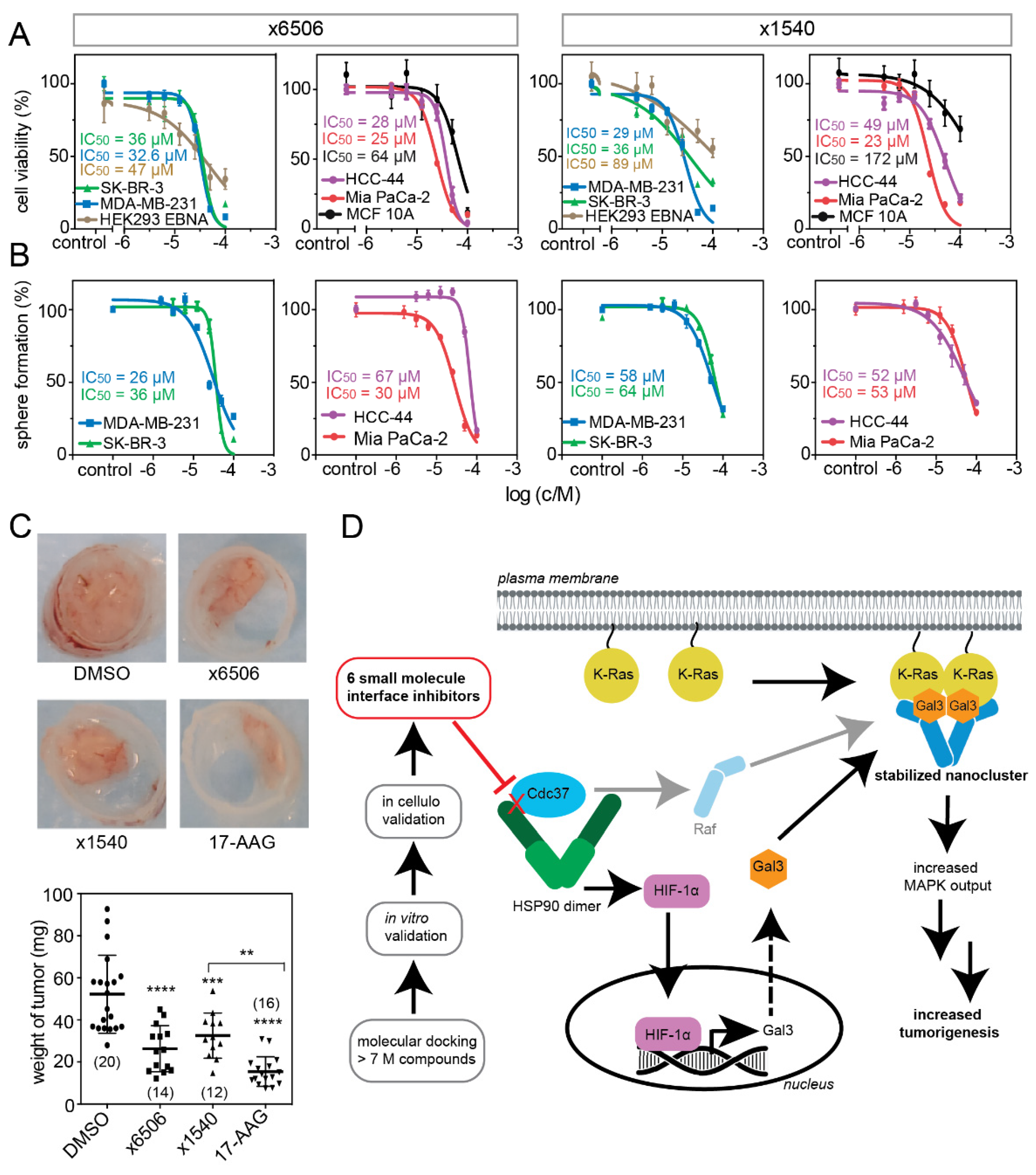
2.5. Compounds ×6506 and ×1540 Decrease Cancer Cell Proliferation, and Spheroid- and Microtumor-Formation
3. Discussion
4. Materials and Methods
4.1. DNA Constructs and siRNA
4.2. Chemical Compounds and Inhibitors
4.3. Cell Culture
4.4. Fluorescence Lifetime Imaging Microscopy (FLIM)-FRET
4.5. Computational Compound Screening and In Vitro Hit Validation Workflow
4.6. Hsp90/Cdc37 Split Renilla Luciferase Interaction Assays for Compound Screening
4.7. Hsp90 N-Terminal ATP-Binding Site Competition Assay
4.8. Western Blotting
4.9. Cytoplasmic and Nuclear Extracts
4.10. Tumorosphere Assay (Low Adhesion and Serum-Free 3D Spheroid Culture)
4.11. Cell Viability Assay (2D Cell Proliferation)
4.12. Chick Chorioallantoic Membrane (CAM) Assay
4.13. Biomarker Analysis in the TCGA PanCan Atlas Database
4.14. Survival Analysis in the Pan-Cancer Patient Tumor Cohorts
4.15. ATARiS Gene Dependency Data
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.L. Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2012, 1823, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbarao Sreedhar, A.; Kalmár, É.; Csermely, P.; Shen, Y.-F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Verba, K.A.; Wang, R.Y.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohl, A.; Rohrberg, J.; Buchner, J. The chaperone Hsp90: Changing partners for demanding clients. Trends Biochem. Sci. 2013, 38, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Keramisanou, D.; Aboalroub, A.; Zhang, Z.; Liu, W.; Marshall, D.; Diviney, A.; Larsen, R.W.; Landgraf, R.; Gelis, I. Molecular Mechanism of Protein Kinase Recognition and Sorting by the Hsp90 Kinome-Specific Cochaperone Cdc37. Mol. Cell 2016, 62, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, S.M.; Ali, M.M.; Meyer, P.; Vaughan, C.K.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. The Mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 2004, 116, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, C.K.; Gohlke, U.; Sobott, F.; Good, V.M.; Ali, M.M.; Prodromou, C.; Robinson, C.V.; Saibil, H.R.; Pearl, L.H. Structure of an Hsp90-Cdc37-Cdk4 complex. Mol. Cell 2006, 23, 697–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, L.M.; Ferraldeschi, R.; Armstrong, H.K.; Centenera, M.M.; Workman, P. Maximizing the Therapeutic Potential of HSP90 Inhibitors. Mol. Cancer Res. 2015, 13, 1445–1451. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, L.; You, Q.D.; Xu, X.L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. J. Med. Chem. 2020, 63, 1798–1822. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Kelland, L.R.; Sharp, S.Y.; Rogers, P.M.; Myers, T.G.; Workman, P. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J. Natl. Cancer Inst. 1999, 91, 1940–1949. [Google Scholar] [CrossRef] [Green Version]
- Travers, J.; Sharp, S.; Workman, P. HSP90 inhibition: Two-pronged exploitation of cancer dependencies. Drug Discov. Today 2012, 17, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Sidera, K.; Patsavoudi, E. HSP90 inhibitors: Current development and potential in cancer therapy. Recent Pat. Anticancer. Drug Discov. 2014, 9, 1–20. [Google Scholar] [CrossRef]
- Khandelwal, A.; Crowley, V.M.; Blagg, B.S.J. Resorcinol-Based Grp94-Selective Inhibitors. ACS Med. Chem. Lett. 2017, 8, 1013–1018. [Google Scholar] [CrossRef]
- Li, T.; Jiang, H.L.; Tong, Y.G.; Lu, J.J. Targeting the Hsp90-Cdc37-client protein interaction to disrupt Hsp90 chaperone machinery. J. Hematol. Oncol. 2018, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Bernard, D.; Yu, Y.; Xie, Y.; Zhang, T.; Li, Y.; Burnett, J.P.; Fu, X.; Wang, S.; Sun, D. Split Renilla luciferase protein fragment-assisted complementation (SRL-PFAC) to characterize Hsp90-Cdc37 complex and identify critical residues in protein/protein interactions. J. Biol. Chem. 2010, 285, 21023–21036. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Ye, M.; Zhang, L.R.; Wu, Q.D.; Zhang, M.; Xu, J.H.; Zheng, W. FW-04-806 inhibits proliferation and induces apoptosis in human breast cancer cells by binding to N-terminus of Hsp90 and disrupting Hsp90-Cdc37 complex formation. Mol. Cancer 2014, 13, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Wu, Q.D.; Zhang, M.; Kong, Y.L.; Cao, P.R.; Zheng, W.; Xu, J.H.; Ye, M. Novel Hsp90 inhibitor FW-04-806 displays potent antitumor effects in HER2-positive breast cancer cells as a single agent or in combination with lapatinib. Cancer Lett. 2015, 356, 862–871. [Google Scholar] [CrossRef]
- Li, D.; Li, C.; Li, L.; Chen, S.; Wang, L.; Li, Q.; Wang, X.; Lei, X.; Shen, Z. Natural Product Kongensin A is a Non-Canonical HSP90 Inhibitor that Blocks RIP3-dependent Necroptosis. Cell Chem. Biol. 2016, 23, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, T.; Jiang, Y.; Lee, H.F.; Schwartz, S.J.; Sun, D. (-)-Epigallocatechin-3-gallate inhibits Hsp90 function by impairing Hsp90 association with cochaperones in pancreatic cancer cell line Mia Paca-2. Mol. Pharm. 2009, 6, 1152–1159. [Google Scholar] [CrossRef] [Green Version]
- Grover, A.; Shandilya, A.; Agrawal, V.; Pratik, P.; Bhasme, D.; Bisaria, V.S.; Sundar, D. Hsp90/Cdc37 chaperone/co-chaperone complex, a novel junction anticancer target elucidated by the mode of action of herbal drug Withaferin A. BMC Bioinform. 2011, 12 (Suppl. 1), S30. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Hamza, A.; Cao, X.; Wang, B.; Yu, S.; Zhan, C.G.; Sun, D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer 2008, 7, 162–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, F.A.; Parkkola, H.; Manoharan, G.B.; Abankwa, D. Medium-Throughput Detection of Hsp90/Cdc37 Protein-Protein Interaction Inhibitors Using a Split Renilla Luciferase-Based Assay. SLAS Discov. 2020, 25, 195–206. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.; Li, L.; Jiang, J.; Zheng, Z.; Shang, J.; Wang, C.; Chen, W.; Bao, Q.; Xu, X.; et al. Small-molecule inhibitor targeting the Hsp90-Cdc37 protein-protein interaction in colorectal cancer. Sci. Adv. 2019, 5, eaax2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Jiang, J.; Zhang, L.; Zhang, Q.; Zhou, J.; Li, L.; Xu, X.-L.; You, Q.-D. Discovery and Optimization of Small Molecules Targeting the Protein-Protein Interaction of Heat Shock Protein 90 (Hsp90) and Cell Division Cycle 37 (Cdc37) As Orally Active Inhibitors for the treatment of colorectal cancer. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef]
- Stepanova, L.; Finegold, M.; DeMayo, F.; Schmidt, E.V.; Harper, J.W. The oncoprotein kinase chaperone CDC37 functions as an oncogene in mice and collaborates with both c-myc and cyclin D1 in transformation of multiple tissues. Mol. Cell Biol. 2000, 20, 4462–4473. [Google Scholar] [CrossRef] [Green Version]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D’Andrea, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. HSP90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldas-Lopes, E.; Cerchietti, L.; Ahn, J.H.; Clement, C.C.; Robles, A.I.; Rodina, A.; Moulick, K.; Taldone, T.; Gozman, A.; Guo, Y.; et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl. Acad. Sci. USA 2009, 106, 8368–8373. [Google Scholar] [CrossRef] [Green Version]
- Najumudeen, A.K.; Jaiswal, A.; Lectez, B.; Oetken-Lindholm, C.; Guzman, C.; Siljamaki, E.; Posada, I.M.; Lacey, E.; Aittokallio, T.; Abankwa, D. Cancer stem cell drugs target K-ras signaling in a stemness context. Oncogene 2016, 35, 5248–5262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazevits, O.; Mideksa, Y.G.; Solman, M.; Ligabue, A.; Ariotti, N.; Nakhaeizadeh, H.; Fansa, E.K.; Papageorgiou, A.C.; Wittinghofer, A.; Ahmadian, M.R.; et al. Galectin-1 dimers can scaffold Raf-effectors to increase H-ras nanoclustering. Sci. Rep. 2016, 6, 24165. [Google Scholar] [CrossRef] [Green Version]
- Rock, R.; Mayrhofer, J.E.; Torres-Quesada, O.; Enzler, F.; Raffeiner, A.; Raffeiner, P.; Feichtner, A.; Huber, R.G.; Koide, S.; Taylor, S.S.; et al. BRAF inhibitors promote intermediate BRAF(V600E) conformations and binary interactions with activated RAS. Sci. Adv. 2019, 5, eaav8463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, X.; Tamguney, T.M.; Collisson, E.A.; Lin, L.J.; Pitt, C.; Galeas, J.; Lewis, S.; Gray, J.W.; McCormick, F.; Chu, S. Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 7996–8001. [Google Scholar] [CrossRef] [Green Version]
- Mysore, V.P.; Zhou, Z.-W.; Ambrogio, C.; Li, L.; Kapp, J.N.; Lu, C.; Wang, Q.; Tucker, M.R.; Okoro, J.J.; Nagy-Davidescu, G.; et al. A structural model of a Ras-Raf signalosome. bioRxiv 2020, 263, 9853. [Google Scholar] [CrossRef]
- Tian, T.; Harding, A.; Inder, K.; Plowman, S.; Parton, R.G.; Hancock, J.F. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 2007, 9, 905–914. [Google Scholar] [CrossRef]
- Shalom-Feuerstein, R.; Plowman, S.J.; Rotblat, B.; Ariotti, N.; Tian, T.; Hancock, J.F.; Kloog, Y. K-ras nanoclustering is subverted by overexpression of the scaffold protein galectin-3. Cancer Res. 2008, 68, 6608–6616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abankwa, D.; Gorfe, A.A.; Hancock, J.F. Ras nanoclusters: Molecular structure and assembly. Semin. Cell Dev. Biol. 2007, 18, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Lokman, N.A.; Elder, A.S.; Ricciardelli, C.; Oehler, M.K. Chick chorioallantoic membrane (CAM) assay as an in vivo model to study the effect of newly identified molecules on ovarian cancer invasion and metastasis. Int. J. Mol. Sci. 2012, 13, 9959–9970. [Google Scholar] [CrossRef] [Green Version]
- Ikemori, R.Y.; Machado, C.M.; Furuzawa, K.M.; Nonogaki, S.; Osinaga, E.; Umezawa, K.; de Carvalho, M.A.; Verinaud, L.; Chammas, R. Galectin-3 up-regulation in hypoxic and nutrient deprived microenvironments promotes cell survival. PLoS ONE 2014, 9, e111592. [Google Scholar] [CrossRef] [Green Version]
- Minet, E.; Mottet, D.; Michel, G.; Roland, I.; Raes, M.; Remacle, J.; Michiels, C. Hypoxia-induced activation of HIF-1: Role of HIF-1alpha-Hsp90 interaction. FEBS Lett. 1999, 460, 251–256. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Jung, Y.J.; Mimnaugh, E.G.; Martinez, A.; Cuttitta, F.; Neckers, L.M. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J. Biol. Chem. 2002, 277, 29936–29944. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Danielson, K.G.; Albert, T.J.; Shapiro, I.M.; Risbud, M.V. HIF-1 alpha is a regulator of galectin-3 expression in the intervertebral disc. J. Bone Min. Res. 2007, 22, 1851–1861. [Google Scholar] [CrossRef]
- Yuan, Y.; Hilliard, G.; Ferguson, T.; Millhorn, D.E. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J. Biol. Chem. 2003, 278, 15911–15916. [Google Scholar] [CrossRef] [Green Version]
- Brahimi-Horn, C.; Mazure, N.; Pouyssegur, J. Signalling via the hypoxia-inducible factor-1alpha requires multiple posttranslational modifications. Cell Signal. 2005, 17, 1–9. [Google Scholar] [CrossRef]
- McDonald Iii, E.R.; de Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, , 577–586.e510. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, A.C.; Andrade, L.N.; Bustos, S.O.; Chammas, R. Galectin-3 Determines Tumor Cell Adaptive Strategies in Stressed Tumor Microenvironments. Front. Oncol. 2016, 6, 127. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell 2015, 163, 1237–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Zhang, Z.; Zhang, S.; Ren, J.; Zhang, R.; Zeng, H.; Li, Q.; Wu, G. Inhibitory action of Celastrol on hypoxia-mediated angiogenesis and metastasis via the HIF-1alpha pathway. Int. J. Mol. Med. 2011, 27, 407–415. [Google Scholar] [CrossRef]
- Guerra, B.; Rasmussen, T.D.; Schnitzler, A.; Jensen, H.H.; Boldyreff, B.S.; Miyata, Y.; Marcussen, N.; Niefind, K.; Issinger, O.G. Protein kinase CK2 inhibition is associated with the destabilization of HIF-1alpha in human cancer cells. Cancer Lett. 2015, 356, 751–761. [Google Scholar] [CrossRef]
- Whelan, K.A.; Schwab, L.P.; Karakashev, S.V.; Franchetti, L.; Johannes, G.J.; Seagroves, T.N.; Reginato, M.J. The oncogene HER2/neu (ERBB2) requires the hypoxia-inducible factor HIF-1 for mammary tumor growth and anoikis resistance. J. Biol. Chem. 2013, 288, 15865–15877. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Hamza, A.; Zhang, T.; Gu, M.; Zou, P.; Newman, B.; Li, Y.; Gunatilaka, A.A.; Zhan, C.G.; Sun, D. Withaferin A targets heat shock protein 90 in pancreatic cancer cells. Biochem. Pharm. 2010, 79, 542–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, A.D.; Goodwin, C.M.; Bryant, K.L.; Dagliyan, I.; George, S.D.; Lucas, K.E.; Gautam, P.; Wennerberg, K.; Der, C.J. Abstract SY20-02: Inhibitor combinations targeting KRAS effector signaling in KRAS-mutant pancreatic cancer. Cancer Res. 2018, 78. [Google Scholar] [CrossRef]
- Kennedy, S.A.; Jarboui, M.A.; Srihari, S.; Raso, C.; Bryan, K.; Dernayka, L.; Charitou, T.; Bernal-Llinares, M.; Herrera-Montavez, C.; Krstic, A.; et al. Extensive rewiring of the EGFR network in colorectal cancer cells expressing transforming levels of KRAS(G13D). Nat. Commun. 2020, 11, 499. [Google Scholar] [CrossRef]
- Kim, K.; Lee, H.W.; Lee, E.H.; Park, M.-I.; Lee, J.S.; Kim, M.-S.; Kim, K.; Roh, M.S.; Pak, M.G.; Oh, J.E.; et al. Differential expression of HSP90 isoforms and their correlations with clinicopathologic factors in patients with colorectal cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 978–986. [Google Scholar]
- Gray, P.J., Jr.; Stevenson, M.A.; Calderwood, S.K. Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res. 2007, 67, 11942–11950. [Google Scholar] [CrossRef] [Green Version]
- Abankwa, D.; Gorfe, A.A. Mechanisms of Ras Membrane Organization and Signaling: Ras Rocks Again. Biomolecules 2020, 10, 1522. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef] [Green Version]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef] [Green Version]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Smith, J.R.; de Billy, E.; Hobbs, S.; Powers, M.; Prodromou, C.; Pearl, L.; Clarke, P.A.; Workman, P. Restricting direct interaction of CDC37 with HSP90 does not compromise chaperoning of client proteins. Oncogene 2015, 34, 15–26. [Google Scholar] [CrossRef] [Green Version]
- D’Annessa, I.; Hurwitz, N.; Pirota, V.; Beretta, G.L.; Tinelli, S.; Woodford, M.; Freccero, M.; Mollapour, M.; Zaffaroni, N.; Wolfson, H.; et al. Design of Disruptors of the Hsp90-Cdc37 Interface. Molecules 2020, 25, 360. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.R.; Clarke, P.A.; de Billy, E.; Workman, P. Silencing the cochaperone CDC37 destabilizes kinase clients and sensitizes cancer cells to HSP90 inhibitors. Oncogene 2009, 28, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; He, Y.; Zhang, X.; Yuan, Y.; Pu, S.; Kong, Q.; Zheng, G.; Zhou, D. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene 2020, 39, 4909–4924. [Google Scholar] [CrossRef]
- Udell, C.M.; Rajakulendran, T.; Sicheri, F.; Therrien, M. Mechanistic principles of RAF kinase signaling. Cell. Mol. Life Sci. 2011, 68, 553–565. [Google Scholar] [CrossRef]
- Hombach, A.; Ommen, G.; Sattler, V.; Clos, J. Leishmania donovani P23 protects parasites against HSP90 inhibitor-mediated growth arrest. Cell Stress Chaperones 2015, 20, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Wang, R.E.; Hunt, C.R.; Chen, J.; Taylor, J.S. Biotinylated quercetin as an intrinsic photoaffinity proteomics probe for the identification of quercetin target proteins. Bioorg. Med. Chem. 2011, 19, 4710–4720. [Google Scholar] [CrossRef] [Green Version]
- Posada, I.M.D.; Lectez, B.; Siddiqui, F.A.; Oetken-Lindholm, C.; Sharma, M.; Abankwa, D. Opposite feedback from mTORC1 to H-ras and K-ras4B downstream of SREBP1. Sci. Rep. 2017, 7, 8944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, F.A.; Alam, C.; Rosenqvist, P.; Ora, M.; Sabt, A.; Manoharan, G.B.; Bindu, L.; Okutachi, S.; Catillon, M.; Taylor, T.; et al. PDE6D Inhibitors with a New Design Principle Selectively Block K-Ras Activity. ACS Omega 2020, 5, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Hogel, H.; Miikkulainen, P.; Bino, L.; Jaakkola, P.M. Hypoxia inducible prolyl hydroxylase PHD3 maintains carcinoma cell growth by decreasing the stability of p27. Mol. Cancer 2015, 14, 143. [Google Scholar] [CrossRef] [Green Version]
- Guzman, C.; Oetken-Lindholm, C.; Abankwa, D. Automated High-Throughput Fluorescence Lifetime Imaging Microscopy to Detect Protein-Protein Interactions. J. Lab. Autom. 2016, 21, 238–245. [Google Scholar] [CrossRef] [Green Version]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, L.; Xu, C.; Yu, F.; Zhou, H.; Zhao, Y.; Zhang, J.; Cai, J.; Mao, C.; Tang, L.; et al. Structure insights into mechanisms of ATP hydrolysis and the activation of human heat-shock protein 90. Acta Biochim. Biophys. Sin. 2012, 44, 300–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repecka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Hsp90/Cdc37 IC50 (µM) | N-Hsp90/Cdc37 IC50 (µM) |
|---|---|---|
| conglobatin A | 23 ± 6 | 4.5 ± 0.8 |
| ×6506 | 5 ± 1 | 4.1 ± 0.4 |
| ×7648 | 18 ± 2 | 14 ± 4 |
| ×1127 | 41 ± 10 | 11 ± 1 |
| ×1540 | 44 ± 18 | 44 ± 9 |
| ×1742 (quercetin) | 52 ± 6 | 42 ± 8 |
| ×1625 | 41 ± 8 | 94 ± 3 |
| Cell Line (Tissue, KRAS Mutation) | ×6506; IC50 (µM) | ×1540; IC50 (µM) |
|---|---|---|
| SK-BR-3 (breast, n.a.) | 36 ± 4 | 36 ± 3 |
| MDA-MB-231 (breast, G13D) | 32.6 ± 0.5 | 29 ± 3 |
| MIA PaCa-2 (pancreas, G12D) | 25 ± 2 | 23 ± 2 |
| HCC-44 (lung, G12C) | 28 ± 4 | 49 ± 6 |
| Cell Line | ×6506 IC50 (µM) | ×1540 IC50 (µM) |
|---|---|---|
| SK-BR-3 (breast, n.a.) | 36 ± 4 | 64 ± 5 |
| MDA-MB-231 (breast, G13D) | 26 ± 1 | 58 ± 5 |
| MIA PaCa-2 (pancreas, G12D) | 30 ± 3 | 53 ± 6 |
| HCC-44 (lung, G12C) | 67 ± 5 | 52 ± 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siddiqui, F.A.; Parkkola, H.; Vukic, V.; Oetken-Lindholm, C.; Jaiswal, A.; Kiriazis, A.; Pavic, K.; Aittokallio, T.; Salminen, T.A.; Abankwa, D. Novel Small Molecule Hsp90/Cdc37 Interface Inhibitors Indirectly Target K-Ras-Signaling. Cancers 2021, 13, 927. https://doi.org/10.3390/cancers13040927
Siddiqui FA, Parkkola H, Vukic V, Oetken-Lindholm C, Jaiswal A, Kiriazis A, Pavic K, Aittokallio T, Salminen TA, Abankwa D. Novel Small Molecule Hsp90/Cdc37 Interface Inhibitors Indirectly Target K-Ras-Signaling. Cancers. 2021; 13(4):927. https://doi.org/10.3390/cancers13040927
Chicago/Turabian StyleSiddiqui, Farid Ahmad, Hanna Parkkola, Vladimir Vukic, Christina Oetken-Lindholm, Alok Jaiswal, Alexandros Kiriazis, Karolina Pavic, Tero Aittokallio, Tiina A. Salminen, and Daniel Abankwa. 2021. "Novel Small Molecule Hsp90/Cdc37 Interface Inhibitors Indirectly Target K-Ras-Signaling" Cancers 13, no. 4: 927. https://doi.org/10.3390/cancers13040927
APA StyleSiddiqui, F. A., Parkkola, H., Vukic, V., Oetken-Lindholm, C., Jaiswal, A., Kiriazis, A., Pavic, K., Aittokallio, T., Salminen, T. A., & Abankwa, D. (2021). Novel Small Molecule Hsp90/Cdc37 Interface Inhibitors Indirectly Target K-Ras-Signaling. Cancers, 13(4), 927. https://doi.org/10.3390/cancers13040927