
The Revolutionary Roads to Study Cell–Cell Interactions in 3D In Vitro Pancreatic Cancer Models

 , ,
, ,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
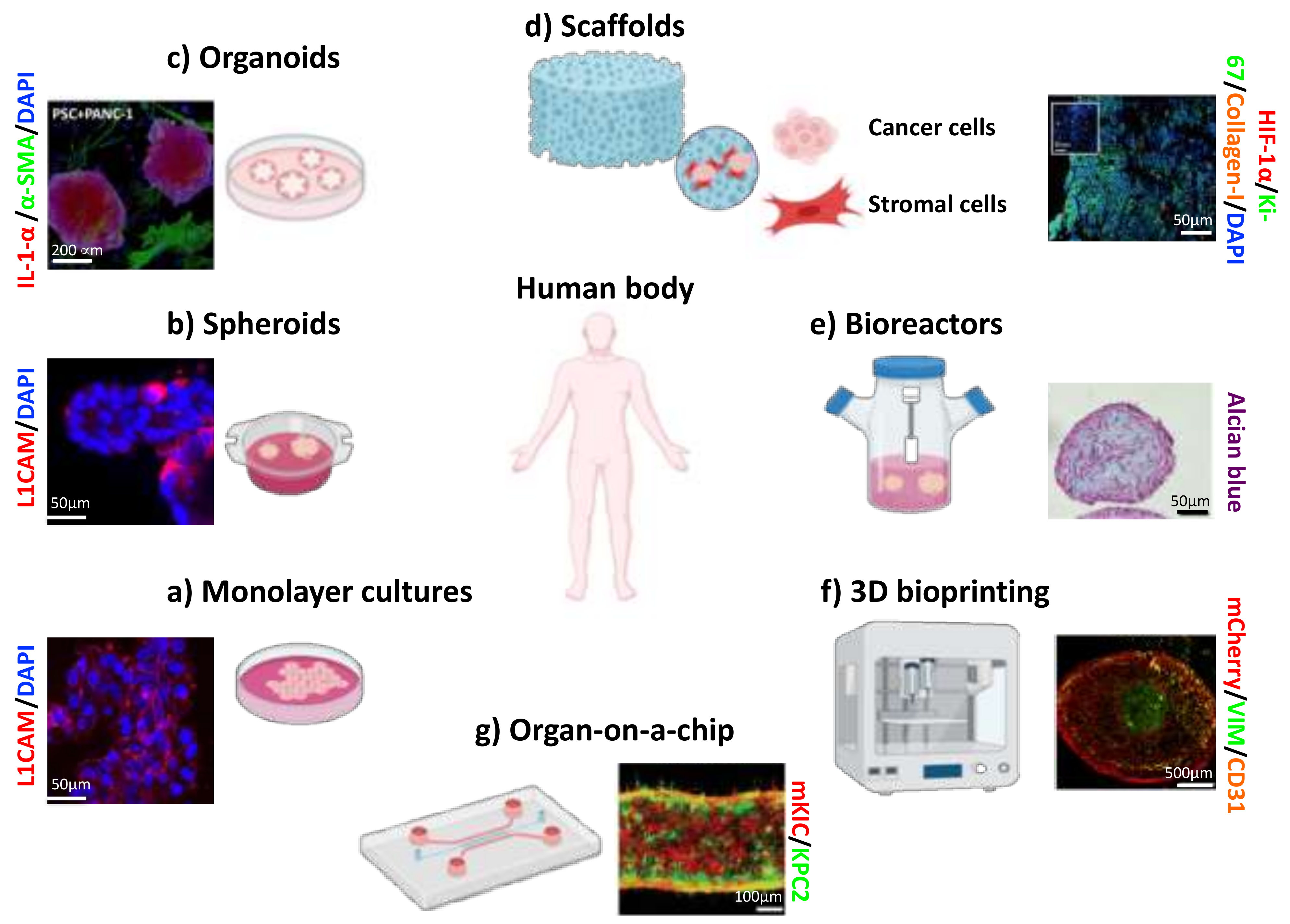
2. 3D In Vitro Models
2.1. Spheroid
2.2. Patient-Derived Organoids (PDOs)
3. Reconstitution of Tumor Cell Heterogeneity and Complexity in 3D In Vitro Models
3.1. Scaffolds
3.2. 3D Cell/TME
3.3. 3D suspension Bioreactors
4. Investigating Cell–Cell Interactions in In Vitro 3D Models
4.1. 3D Bioprinting
4.2. Organ-on-A-Chip
5. The End of Perpetual Chemotherapy: Nanoparticles (NPs) for Cell Targeting
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fitzmaurice, C. Global Burden of Disease Cancer Collaboration. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 2006 to 2016: A systematic analysis for the Global Burden of Disease study. J. Clin. Oncol. 2018, 36, 1568. [Google Scholar] [CrossRef] [Green Version]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestris, N.; Brunetti, O.; Bittoni, A.; Cataldo, I.; Corsi, D.; Crippa, S.; D’Onofrio, M.; Fiore, M.; Giommoni, E.; Milella, M.; et al. Clinical Practice Guidelines for Diagnosis, Treatment and Follow-up of Exocrine Pancreatic Ductal Adenocarcinoma: Evidence Evaluation and Recommendations by the Italian Association of Medical Oncology (AIOM). Cancers 2020, 12, 1681. [Google Scholar] [CrossRef]
- Swayden, M.; Soubeyran, P.; Iovanna, J. Upcoming Revolutionary Paths in Preclinical Modeling of Pancreatic Adenocarcinoma. Front. Oncol. 2020, 9, 1443. [Google Scholar] [CrossRef]
- Michalski, C.W.; Rosendahl, J.; Michl, P.; Kleeff, J. Translational Pancreatic Cancer Research: From Understanding of Mechanisms to Novel Clinical Trials. In Molecular and Translational Medicine; Springer International Publishing: Cham, Switzerland, 2020; ISBN 978-3-030-49475-9. [Google Scholar]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-Derived Xenografts Undergo Murine-Specific Tumor Evolution. Nat. Genet. 2017, 49, 1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, A.S.; Costa, E.C.; Nunes, A.S.; De Melo-Diogo, D.; Correia, I.J. Comparative study of the therapeutic effect of Doxorubicin and Resveratrol combination on 2D and 3D (spheroids) cell culture models. Int. J. Pharm. 2018, 551, 76–83. [Google Scholar] [CrossRef]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef]
- Dougan, S.K. The Pancreatic Cancer Microenvironment. Cancer J. 2017, 23, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Westphalen, C.B.; Olive, K.P. Genetically Engineered Mouse Models of Pancreatic Cancer. Cancer J. 2012, 18, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Gopinathan, A.; Morton, J.P.; Jodrell, D.I.; Sansom, O.J. GEMMs as preclinical models for testing pancreatic cancer therapies. Dis. Model. Mech. 2015, 8, 1185–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomás-Bort, E.; Kieler, M.; Sharma, S.; Candido, J.B.; Loessner, D. 3D approaches to model the tumor microenvironment of pancreatic cancer. Theranostics 2020, 10, 5074–5089. [Google Scholar] [CrossRef]
- Ehlen, L.; Arndt, J.; Treue, D.; Bischoff, P.; Loch, F.N.; Hahn, E.M.; Kotsch, K.; Klauschen, F.; Beyer, K.; Margonis, G.A.; et al. Novel methods for in vitro modeling of pancreatic cancer reveal important aspects for successful primary cell culture. BMC Cancer 2020, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cavo, M.; Serio, F.; Kale, N.R.; D’Amone, E.; Gigli, G.; Del Mercato, L.L. Electrospun nanofibers in cancer research: From engineering of in vitro 3D cancer models to therapy. Biomater. Sci. 2020, 8, 4887–4905. [Google Scholar] [CrossRef] [PubMed]
- Polini, A.; Del Mercato, L.L.; Barra, A.; Zhang, Y.S.; Calabi, F.; Gigli, G. Towards the development of human immune-system-on-a-chip platforms. Drug Discov. Today 2019, 24, 517–525. [Google Scholar] [CrossRef]
- Turetta, M.; Del Ben, F.; Brisotto, G.; Biscontin, E.; Bulfoni, M.; Cesselli, D.; Colombatti, A.; Scoles, G.; Gigli, G.; Del Mercato, L.L. Emerging Technologies for Cancer Research: Towards Personalized Medicine with Microfluidic Platforms and 3D Tumor Models. Curr. Med. Chem. 2018, 25, 4616–4637. [Google Scholar] [CrossRef]
- Kimlin, L.; Kassis, J.; Virador, V. 3Din vitrotissue models and their potential for drug screening. Expert Opin. Drug Discov. 2013, 8, 1455–1466. [Google Scholar] [CrossRef]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.-R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef] [Green Version]
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Cavo, M.; Cave, D.D.; D’Amone, E.; Gigli, G.; Lonardo, E.; Del Mercato, L.L. A synergic approach to enhance long-term culture and manipulation of MiaPaCa-2 pancreatic cancer spheroids. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Zeeberg, K.; Cardone, R.A.; Greco, M.R.; Saccomano, M.; Nøhr-Nielsen, A.; Alves, F.; Pedersen, S.F.; Reshkin, S.J. Assessment of different 3D culture systems to study tumor phenotype and chemosensitivity in pancreatic ductal adenocarcinoma. Int. J. Oncol. 2016, 49, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, C.-I.; Boj, S.F.; Clevers, H.; Tuveson, D.A. Preclinical models of pancreatic ductal adenocarcinoma. J. Pathol. 2016, 238, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Moreira, L.; Bakir, B.; Chatterji, P.; Dantes, Z.; Reichert, M.; Rustgi, A.K. Pancreas 3D Organoids: Current and Future Aspects as a Research Platform for Personalized Medicine in Pancreatic Cancer. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, M.; Clevers, H.; Sato, T. Modeling Human Digestive Diseases with CRISPR-Cas9–Modified Organoids. Gastroenterology 2019, 156, 562–576. [Google Scholar] [CrossRef] [Green Version]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [Green Version]
- Driehuis, E.; Van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467. [Google Scholar] [CrossRef] [Green Version]
- Frappart, P.; Walter, K.; Gout, J.; Beutel, A.K.; Morawe, M.; Arnold, F.; Breunig, M.; Barth, T.F.; Marienfeld, R.; Schulte, L.; et al. Pancreatic cancer-derived organoids—A disease modeling tool to predict drug response. United Eur. Gastroenterol. J. 2020, 8, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.R.; Zhang, C.; Roche, S.; O’Neill, F.; Swan, N.; Luo, Y.; Larkin, A.; Crown, J.; Walsh, N. Modelling of pancreatic cancer biology: Transcriptomic signature for 3D PDX-derived organoids and primary cell line organoid development. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Schuster, B.; Junkin, M.; Kashaf, S.S.; Romero-Calvo, I.; Kirby, K.; Matthews, J.; Weber, C.R.; Rzhetsky, A.; White, K.P.; Tay, S. Automated microfluidic platform for dynamic and combinatorial drug screening of tumor organoids. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Cave, D.D.; Di Guida, M.; Costa, V.; Sevillano, M.; Ferrante, L.; Heeschen, C.; Corona, M.; Cucciardi, A.; Lonardo, E. TGF-β1 secreted by pancreatic stellate cells promotes stemness and tumourigenicity in pancreatic cancer cells through L1CAM downregulation. Oncogene 2020, 39, 4271–4285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [Green Version]
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puls, T.J.; Tan, X.; Whittington, C.F.; Voytik-Harbin, S.L. 3D collagen fibrillar microstructure guides pancreatic cancer cell phenotype and serves as a critical design parameter for phenotypic models of EMT. PLoS ONE 2017, 12, e0188870. [Google Scholar] [CrossRef] [Green Version]
- Chiellini, F.; Puppi, D.; Piras, A.M.; Morelli, A.; Bartoli, C.; Migone, C. Modelling of pancreatic ductal adenocarcinoma in vitro with three-dimensional microstructured hydrogels. RSC Adv. 2016, 6, 54226–54235. [Google Scholar] [CrossRef]
- Ricci, C.; Mota, C.; Moscato, S.; D’Alessandro, D.; Ugel, S.; Sartoris, S.; Bronte, V.; Boggi, U.; Campani, D.; Funel, N.; et al. Interfacing polymeric scaffolds with primary pancreatic ductal adenocarcinoma cells to develop 3D cancer models. Biomatter 2014, 4, e955386. [Google Scholar] [CrossRef] [Green Version]
- Bregenzer, M.E.; Horst, E.N.; Mehta, P.; Novak, C.M.; Raghavan, S.; Snyder, C.S.; Mehta, G. Integrated cancer tissue engineering models for precision medicine. PLoS ONE 2019, 14, e0216564. [Google Scholar] [CrossRef] [Green Version]
- Burdett, E.; Kasper, F.K.; Mikos, A.G.; Ludwig, J.A. Engineering Tumors: A Tissue Engineering Perspective in Cancer Biology. Tissue Eng. Part B Rev. 2010, 16, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Totti, S.; Allenby, M.C.; Dos Santos, S.B.; Mantalaris, A.; Velliou, E.G. A 3D bioinspired highly porous polymeric scaffolding system forin vitrosimulation of pancreatic ductal adenocarcinoma. RSC Adv. 2018, 8, 20928–20940. [Google Scholar] [CrossRef] [Green Version]
- Baker, L.A.; Tiriac, H.; Clevers, H.; Tuveson, D.A. Modeling Pancreatic Cancer with Organoids. Trends Cancer 2016, 2, 176–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillaume, L.; Rigal, L.; Fehrenbach, J.; Severac, C.; Ducommun, B.; Lobjois, V. Characterization of the physical properties of tumor-derived spheroids reveals critical insights for pre-clinical studies. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Prakash, J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer 2019, 5, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Birbrair, A. Tumor Microenvironment: Non-Hematopoietic Cells. In Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020; Volume 1234, ISBN 978-3-030-37183-8. [Google Scholar]
- Lonardo, E.; Frias-Aldeguer, J.; Hermann, P.C.; Heeschen, C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 2012, 11, 1282–1290. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norberg, K.J.; Liu, X.; Moro, C.F.; Strell, C.; Nania, S.; Blümel, M.; Balboni, A.; Bozóky, B.; Heuchel, R.L.; Löhr, J.M. A novel pancreatic tumour and stellate cell 3D co-culture spheroid model. BMC Cancer 2020, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Jang, S.D.; Kim, H.; Chung, S.; Park, J.K.; Kuh, H.-J. Phenotypic Heterogeneity and Plasticity of Cancer Cell Migration in a Pancreatic Tumor Three-Dimensional Culture Model. Cancers 2020, 12, 1305. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Anbil, S.; Bulin, A.-L.; Obaid, G.; Mai, Z.; Baglo, Y.; Rizvi, I.; Hasan, T. Modulation of redox metabolism negates cancer-associated fibroblasts-induced treatment resistance in a heterotypic 3D culture platform of pancreatic cancer. Biomaterials 2019, 222, 119421. [Google Scholar] [CrossRef]
- Karimnia, V.; Rizvi, I.; Slack, F.J.; Celli, J.P. Photodestruction of Stromal Fibroblasts Enhances Tumor Response to PDT in 3D Pancreatic Cancer Coculture Models. Photochem. Photobiol. 2020, 13339. [Google Scholar] [CrossRef] [PubMed]
- Gioeli, D.; Snow, C.J.; Simmers, M.B.; Hoang, S.A.; Figler, R.A.; Allende, J.A.; Roller, D.G.; Parsons, J.T.; Wulfkuhle, J.D.; Petricoin, E.F.; et al. Development of a multicellular pancreatic tumor microenvironment system using patient-derived tumor cells. Lab Chip 2019, 19, 1193–1204. [Google Scholar] [CrossRef] [Green Version]
- Di Maggio, F.; Arumugam, P.; Delvecchio, F.R.; Batista, S.; Lechertier, T.; Hodivala-Dilke, K.; Kocher, H.M. Pancreatic stellate cells regulate blood vessel density in the stroma of pancreatic ductal adenocarcinoma. Pancreatology 2016, 16, 995–1004. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Pérez-Mancera, P.A.; Kocher, H.; Nisbet, A.; Schettino, G.; Velliou, E.G. A Novel Scaffold-Based Hybrid Multicellular Model for Pancreatic Ductal Adenocarcinoma—Toward a Better Mimicry of the in vivo Tumor Microenvironment. Front. Bioeng. Biotechnol. 2020, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Lai, B.F.L.; Lu, R.X.Z.; Hu, Y.; Huyer, L.D.; Dou, W.; Wang, E.Y.; Radulovich, N.; Tsao, M.S.; Sun, Y.; Radisic, M. Recapitulating Pancreatic Tumor Microenvironment through Synergistic Use of Patient Organoids and Organ-on-a-Chip Vasculature. Adv. Funct. Mater. 2020. [Google Scholar] [CrossRef]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kuen, J.; Darowski, D.; Kluge, T.; Majety, M. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PLoS ONE 2017, 12, e0182039. [Google Scholar] [CrossRef]
- Tang, J.; Cui, J.; Chen, R.; Guo, K.; Kang, X.; Li, Y.; Gao, D.; Sun, L.; Xu, C.; Chen, J.; et al. A three-dimensional cell biology model of human hepatocellular carcinoma in vitro. Tumor Biol. 2010, 32, 469–479. [Google Scholar] [CrossRef]
- Carpenedo, R.L.; Sargent, C.Y.; McDevitt, T.C. Rotary Suspension Culture Enhances the Efficiency, Yield, and Homogeneity of Embryoid Body Differentiation. Stem Cells 2007, 25, 2224–2234. [Google Scholar] [CrossRef]
- Brancato, V.; Comunanza, V.; Imparato, G.; Corà, D.; Urciuolo, F.; Noghero, A.; Bussolino, F.; Netti, P.A. Bioengineered tumoral microtissues recapitulate desmoplastic reaction of pancreatic cancer. Acta Biomater. 2017, 49, 152–166. [Google Scholar] [CrossRef]
- Kirstein, M.N.; Brundage, R.C.; Moore, M.M.; Williams, B.W.; Hillman, L.A.; Dagit, J.W.; Fisher, J.E.; Marker, P.H.; Kratzke, R.A.; Yee, D. Pharmacodynamic characterization of gemcitabine cytotoxicity in an in vitro cell culture bioreactor system. Cancer Chemother. Pharmacol. 2007, 61, 291–299. [Google Scholar] [CrossRef]
- Candini, O.; Grisendi, G.; Foppiani, E.M.; Brogli, M.; Aramini, B.; Masciale, V.; Spano, C.; Petrachi, T.; Veronesi, E.; Conte, P.; et al. A Novel 3D In Vitro Platform for Pre-Clinical Investigations in Drug Testing, Gene Therapy, and Immuno-oncology. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bjånes, T.; Kotopoulis, S.; Murvold, E.T.; Kamčeva, T.; Gjertsen, B.T.; Gilja, O.H.; Schjøtt, J.; Riedel, B.; McCormack, E. Ultrasound- and Microbubble-Assisted Gemcitabine Delivery to Pancreatic Cancer Cells. Pharmaceutics 2020, 12, 141. [Google Scholar] [CrossRef] [Green Version]
- Mandrycky, C.; Wang, Z.; Kim, K.; Kim, D.-H. 3D bioprinting for engineering complex tissues. Biotechnol. Adv. 2016, 34, 422–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asghar, W.; El Assal, R.; Shafiee, H.; Pitteri, S.; Paulmurugan, R.; Demirci, U. Engineering cancer microenvironments for in vitro 3-D tumor models. Mater. Today 2015, 18, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Langer, E.M.; Allen-Petersen, B.L.; King, S.M.; Kendsersky, N.D.; Turnidge, M.A.; Kuziel, G.M.; Riggers, R.; Samatham, R.; Amery, T.S.; Jacques, S.L.; et al. Modeling Tumor Phenotypes In Vitro with Three-Dimensional Bioprinting. Cell Rep. 2019, 26, 608–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakobyan, D.; Médina, C.; Dusserre, N.; Stachowicz, M.-L.; Handschin, C.; Fricain, J.-C.; Guillermet-Guibert, J.; Oliveira, H. Laser-assisted 3D bioprinting of exocrine pancreas spheroid models for cancer initiation study. Biofabrication 2020, 12, 035001. [Google Scholar] [CrossRef]
- Conchello, J.-A.; Lichtman, J.W. Optical sectioning microscopy. Nat. Methods 2005, 2, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J. Optical sectioning microscopy with planar or structured illumination. Nat. Methods 2011, 8, 811–819. [Google Scholar] [CrossRef]
- Reynaud, E.G.; Kržič, U.; Greger, K.; Stelzer, E.H.K. Light sheet-based fluorescence microscopy: More dimensions, more photons, and less photodamage. HFSP J. 2008, 2, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef]
- Beer, M.; Kuppalu, N.; Stefanini, M.; Becker, H.; Schulz, I.; Manoli, S.; Schuette, J.; Schmees, C.; Casazza, A.; Stelzle, M.; et al. A novel microfluidic 3D platform for culturing pancreatic ductal adenocarcinoma cells: Comparison with in vitro cultures and in vivo xenografts. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Bradney, M.J.; Venis, S.M.; Yang, Y.; Konieczny, S.F.; Han, B. A Biomimetic Tumor Model of Heterogeneous Invasion in Pancreatic Ductal Adenocarcinoma. Small 2020, 16, e1905500. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Shirure, V.S.; Liu, R.; Cunningham, C.; Ding, L.; Meacham, J.M.; Goedegebuure, S.P.; George, S.C.; Fields, R.C. Tumor-on-a-chip platform to interrogate the role of macrophages in tumor progression. Integr. Biol. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.-R.; Ozcelikkale, A.; Yang, Y.; Elzey, B.D.; Konieczny, S.F.; Han, B. An engineered pancreatic cancer model with intra-tumoral heterogeneity of driver mutations. Lab Chip 2020, 20, 3720–3732. [Google Scholar] [CrossRef]
- Kramer, B.; De Haan, L.; Vermeer, M.; Olivier, T.; Hankemeier, T.; Vulto, P.; Joore, J.; Lanz, H.L. Interstitial Flow Recapitulates Gemcitabine Chemoresistance in A 3D Microfluidic Pancreatic Ductal Adenocarcinoma Model by Induction of Multidrug Resistance Proteins. Int. J. Mol. Sci. 2019, 20, 4647. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.-H.T.; Lee, E.; Alimperti, S.; Norgard, R.J.; Wong, A.; Lee, J.J.-K.; Eyckmans, J.; Stanger, B.Z.; Chen, C.S. A biomimetic pancreatic cancer on-chip reveals endothelial ablation via ALK7 signaling. Sci. Adv. 2019, 5, eaav6789. [Google Scholar] [CrossRef] [Green Version]
- Siret, C.; Collignon, A.; Silvy, F.; Robert, S.; Cheyrol, T.; André, P.; Rigot, V.; Iovanna, J.; Van De Pavert, S.; Lombardo, D.; et al. Deciphering the Crosstalk Between Myeloid-Derived Suppressor Cells and Regulatory T Cells in Pancreatic Ductal Adenocarcinoma. Front. Immunol. 2020, 10, 3070. [Google Scholar] [CrossRef]
- Gresham, G.K.; A Wells, G.; Gill, S.; Cameron, C.; Jonker, D.J. Chemotherapy regimens for advanced pancreatic cancer: A systematic review and network meta-analysis. BMC Cancer 2014, 14, 471. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Hiroshima, Y.; Matsuyama, R.; Homma, Y.; Hoffman, R.M.; Endo, I. Role of the tumor microenvironment in pancreatic cancer. Ann. Gastroenterol. Surg. 2019, 3, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Seton-Rogers, S. Fibroblasts shape PDAC architecture. Nat. Rev. Cancer 2019, 19, 418. [Google Scholar] [CrossRef] [PubMed]
- Adiseshaiah, P.P.; Crist, R.M.; Hook, S.S.; McNeil, S.E. Nanomedicine strategies to overcome the pathophysiological barriers of pancreatic cancer. Nat. Rev. Clin. Oncol. 2016, 13, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Brachi, G.; Bussolino, F.; Ciardelli, G.; Mattu, C. Nanomedicine for Imaging and Therapy of Pancreatic Adenocarcinoma. Front. Bioeng. Biotechnol. 2019, 7, 307. [Google Scholar] [CrossRef] [PubMed]
- Van Der Meel, R.; Sulheim, E.; Shi, Y.; Kiessling, F.; Mulder, W.J.M.; Lammers, T. Smart cancer nanomedicine. Nat. Nanotechnol. 2019, 14, 1007–1017. [Google Scholar] [CrossRef]
- Jang, H.L.; Sengupta, S. Transcellular transfer of nanomedicine. Nat. Nanotechnol. 2019, 14, 731–732. [Google Scholar] [CrossRef]
- Al-Batran, S.-E.; Geissler, M.; Seufferlein, T.; Oettle, H. Nab-Paclitaxel for Metastatic Pancreatic Cancer: Clinical Outcomes and Potential Mechanisms of Action. Oncol. Res. Treat. 2014, 37, 128–134. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Hubner, R.A.; Siveke, J.T.; Von Hoff, D.D.; Belanger, B.; de Jong, F.A.; Mirakhur, B.; Chen, L.-T. NAPOLI-1 phase 3 study of liposomal irinotecan in metastatic pancreatic cancer: Final overall survival analysis and characteristics of long-term survivors. Eur. J. Cancer 2019, 108, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Jiang, T.; Shen, S.; She, X.; Tuo, Y.; Hu, Y.; Pang, Z.; Jiang, X. Cyclopamine disrupts tumor extracellular matrix and improves the distribution and efficacy of nanotherapeutics in pancreatic cancer. Biomaterials 2016, 103, 12–21. [Google Scholar] [CrossRef]
- Ray, P.; Confeld, M.; Borowicz, P.; Wang, T.; Mallik, S.; Quadir, M. PEG-b-poly (carbonate)-derived nanocarrier platform with pH-responsive properties for pancreatic cancer combination therapy. Colloids Surf. B Biointerfaces 2019, 174, 126–135. [Google Scholar] [CrossRef]
- Oluwasanmi, A.; Al-Shakarchi, W.; Manzur, A.; Aldebasi, M.H.; Elsini, R.S.; Albusair, M.K.; Haxton, K.J.; Curtis, A.D.; Hoskins, C. Diels Alder-mediated release of gemcitabine from hybrid nanoparticles for enhanced pancreatic cancer therapy. J. Control. Release 2017, 266, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wason, M.S.; Colon, J.; Das, S.; Seal, S.; Turkson, J.; Zhao, J.; Baker, C.H. Sensitization of pancreatic cancer cells to radiation by cerium oxide nanoparticle-induced ROS production. Nanomedicine Nanotechnol. Biol. Med. 2013, 9, 558–569. [Google Scholar] [CrossRef] [Green Version]
- Tummers, W.S.; Bs, S.E.M.; Teraphongphom, N.T.; Gomez, A.; Steinberg, I.; Huland, D.M.; Hong, S.; Kothapalli, S.-R.; Hasan, A.; Ertsey, R.; et al. Intraoperative Pancreatic Cancer Detection using Tumor-Specific Multimodality Molecular Imaging. Ann. Surg. Oncol. 2018, 25, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Hoogstins, C.E.S.; Boogerd, L.S.F.; Bsc, B.G.S.M.; Mieog, J.S.D.; Swijnenburg, R.J.; Van De Velde, C.J.H.; Sarasqueta, A.F.; Bonsing, B.A.; Framery, B.; Pèlegrin, A.; et al. Image-Guided Surgery in Patients with Pancreatic Cancer: First Results of a Clinical Trial Using SGM-101, a Novel Carcinoembryonic Antigen-Targeting, Near-Infrared Fluorescent Agent. Ann. Surg. Oncol. 2018, 25, 3350–3357. [Google Scholar] [CrossRef]
- Perlman, O.; Weitz, I.S.; Azhari, H. Copper oxide nanoparticles as contrast agents for MRI and ultrasound dual-modality imaging. Phys. Med. Biol. 2015, 60, 5767–5783. [Google Scholar] [CrossRef] [PubMed]
- Handgraaf, H.J.M.; Boonstra, M.C.; Van Erkel, A.R.; Bonsing, B.A.; Putter, H.; Van De Velde, C.J.H.; Vahrmeijer, A.L.; Mieog, J.S.D. Current and Future Intraoperative Imaging Strategies to Increase Radical Resection Rates in Pancreatic Cancer Surgery. BioMed Res. Int. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, X.; Wu, Z.; Sun, T.; Huang, W.; Wang, Z.; Ding, X.; Jiang, C.; Li, F. Theranostic nanoparticles enabling the release of phosphorylated gemcitabine for advanced pancreatic cancer therapy. J. Mater. Chem. B 2020, 8, 2410–2417. [Google Scholar] [CrossRef]
- Rosenberger, I.; Strauss, A.; Dobiasch, S.; Weiß, C.; Szanyi, S.; Gil-Iceta, L.; Alonso, E.; Esparza, M.G.; Gómez-Vallejo, V.; Szczupak, B.; et al. Targeted diagnostic magnetic nanoparticles for medical imaging of pancreatic cancer. J. Control. Release 2015, 214, 76–84. [Google Scholar] [CrossRef]
- Hilmi, M.; Bartholin, L.; Neuzillet, C. Immune therapies in pancreatic ductal adenocarcinoma: Where are we now? World J. Gastroenterol. 2018, 24, 2137–2151. [Google Scholar] [CrossRef]
- Nakamura, T.; Harashima, H. Integration of nano drug-delivery system with cancer immunotherapy. Ther. Deliv. 2017, 8, 987–1000. [Google Scholar] [CrossRef]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef]
- Dermani, F.K.; Samadi, P.; Rahmani, G.; Kohlan, A.K.; Najafi, R. PD-1/PD-L1 immune checkpoint: Potential target for cancer therapy. J. Cell. Physiol. 2019, 234, 1313–1325. [Google Scholar] [CrossRef]
- Miao, L.; Li, J.; Liu, Q.; Feng, R.; Das, M.; Lin, C.M.; Goodwin, T.J.; Dorosheva, O.; Liu, R.; Huang, L. Transient and Local Expression of Chemokine and Immune Checkpoint Traps To Treat Pancreatic Cancer. ACS Nano 2017, 11, 8690–8706. [Google Scholar] [CrossRef] [PubMed]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan–Kynurenine–Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Liu, X.; Liao, Y.-P.; Salazar, F.; Sun, B.; Jiang, W.; Chang, C.H.; Jiang, J.; Wang, X.; Wu, A.M.; et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, F.; Li, Y.; Wang, H.; Ren, H.; Chen, J.; Nie, G.; Hao, J. Co-delivery of HIF1α siRNA and gemcitabine via biocompatible lipid-polymer hybrid nanoparticles for effective treatment of pancreatic cancer. Biomaterials 2015, 46, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Ellah, N.H.A.; Abouelmagd, S.A. Surface functionalization of polymeric nanoparticles for tumor drug delivery: Approaches and challenges. Expert Opin. Drug Deliv. 2017, 14, 201–214. [Google Scholar] [CrossRef]
- Wu, C.Y.; Pu, Y.; Liu, G.; Shao, Y.; Ma, Q.S.; Zhang, X.M. MR imaging of human pancreatic cancer xenograft labeled with superparamagnetic iron oxide in nude mice. Contrast Media Mol. Imaging 2012, 7, 51–58. [Google Scholar] [CrossRef]
- Darrigues, E.; Dantuluri, V.; Nima, Z.A.; Vang-Dings, K.B.; Griffin, R.J.; Biris, A.R.; Ghosh, A. Raman spectroscopy using plasmonic and carbon-based nanoparticles for cancer detection, diagnosis, and treatment guidance. Part 2: Treatment. Drug Metab. Rev. 2017, 49, 253–283. [Google Scholar] [CrossRef]
- Managò, S.; Migliaccio, N.; Terracciano, M.; Napolitano, M.; Martucci, N.M.; De Stefano, L.; Rendina, I.; De Luca, A.C.; Lamberti, A.; Rea, I. Internalization kinetics and cytoplasmic localization of functionalized diatomite nanoparticles in cancer cells by Raman imaging. J. Biophotonics 2018, 11, e201700207. [Google Scholar] [CrossRef] [PubMed]
- Durr, N.J.; Larson, T.; Smith, D.K.; Korgel, B.A.; Sokolov, K.; Ben-Yakar, A. Two-Photon Luminescence Imaging of Cancer Cells Using Molecularly Targeted Gold Nanorods. Nano Lett. 2007, 7, 5. [Google Scholar] [CrossRef]
- Phan, T.T.V.; Bui, N.Q.; Cho, S.-W.; Bharathiraja, S.; Manivasagan, P.; Moorthy, M.S.; Mondal, S.; Kim, C.-S.; Oh, J. Photoacoustic Imaging-Guided Photothermal Therapy with Tumor-Targeting HA-FeOOH@PPy Nanorods. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lazzari, G.; Vinciguerra, D.; Balasso, A.; Nicolas, V.; Goudin, N.; Garfa-Traore, M.; Féher, A.; Dinnyés, A.; Nicolas, J.; Couvreur, P.; et al. Light sheet fluorescence microscopy versus confocal microscopy: In quest of a suitable tool to assess drug and nanomedicine penetration into multicellular tumor spheroids. Eur. J. Pharm. Biopharm. 2019, 142, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Darrigues, E.; Nima, Z.A.; Nedosekin, D.A.; Watanabe, F.; Alghazali, K.M.; Zharov, V.P.; Biris, A.S. Tracking Gold Nanorods’ Interaction with Large 3D Pancreatic-Stromal Tumor Spheroids by Multimodal Imaging: Fluorescence, Photoacoustic, and Photothermal Microscopies. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Advantages | Disadvantages |
|---|---|---|
| Monolayer cultures | Cost effective | Shape changed from original tissue (polarization lost) |
| a | Easy-to-use protocol | Lack vasculature |
| Scalable to different plate formats | Reduces cell-to-cell interactions | |
| Compliant with high-throughput screening (HTS) | Less biologically relevant models | |
| High reproducibility | ||
| Formed from primary cells and cell lines | ||
| Spheroids | Easy-to-use protocol | Simple architectures |
| Scalable to different plate formats | Cannot control uniformity (size, composition) | |
| Compliant with high-throughput screening (HTS) | Not all cell lines form spheroids | |
| Formed from primary cells and cell lines | Agglomeration | |
| Long-term culture | Necrotic cores | |
| Allows co-cultures | Lack vasculature | |
| Organoids | Patient-specific | Costly |
| Scalable to different plate formats | Not easy-to-use protocol | |
| Formed from primary cells | Cannot control uniformity (size, composition) | |
| Allows co-cultures | Less amenable to HTS | |
| In vivo-like complexity | May lack key cell types | |
| In vivo-like architecture | Lack vasculature | |
| Amenable for tissue engineering and transplantation | Require validation to identify outgrowth of unwanted cells | |
| Requires access to human samples | ||
| Scaffolds | Scalable to different plate formats | Costly |
| Compliant with high-throughput screening (HTS) | Not easy-to-use protocol | |
| High reproducibility | Simple architectures | |
| Formed from primary cells and cell lines | Batch-to-batch variability of natural matrixes | |
| Long term culture | Might require complex cell retrieval/imaging methods | |
| Allows co-cultures | ||
| In vivo-like complexity | ||
| In vivo-like architecture | ||
| Amenable for tissue engineering and transplantation | ||
| Naturally-derived ECM components of synthetic polymers | ||
| Resemble mechanical forces in tumors | ||
| Versatile | ||
| Tunable composition | ||
| Bioreactors | High density cell expansion | Costly |
| Controllable culture parameters | Requires optimization of cell parameters and biomaterial inclusion | |
| Hydrodynamic shear stress | ||
| Organ-on-a-chip | Compliant with high-throughput screening (HTS) | Costly |
| High reproducibility | Requires special equipment | |
| Formed from primary cells and cell lines | Difficult to scale up | |
| Allow co-cultures | ||
| In vivo-like complexity | ||
| In vivo-like architecture | ||
| Controllable culture parameters | ||
| Vascularized | ||
| 3D Bioprinting | High-throughput production | Costly |
| High reproducibility | Requires special equipment | |
| Formed from primary cells and cell lines | Challenges with cells/materials | |
| Allow co-cultures | Issues with tissue maturation | |
| In vivo-like complexity | Needs optimization | |
| In vivo-like architecture | ||
| Controllable culture parameters | ||
| Vascularized | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delle Cave, D.; Rizzo, R.; Sainz, B., Jr.; Gigli, G.; del Mercato, L.L.; Lonardo, E. The Revolutionary Roads to Study Cell–Cell Interactions in 3D In Vitro Pancreatic Cancer Models. Cancers 2021, 13, 930. https://doi.org/10.3390/cancers13040930
Delle Cave D, Rizzo R, Sainz B Jr., Gigli G, del Mercato LL, Lonardo E. The Revolutionary Roads to Study Cell–Cell Interactions in 3D In Vitro Pancreatic Cancer Models. Cancers. 2021; 13(4):930. https://doi.org/10.3390/cancers13040930
Chicago/Turabian StyleDelle Cave, Donatella, Riccardo Rizzo, Bruno Sainz, Jr., Giuseppe Gigli, Loretta L. del Mercato, and Enza Lonardo. 2021. "The Revolutionary Roads to Study Cell–Cell Interactions in 3D In Vitro Pancreatic Cancer Models" Cancers 13, no. 4: 930. https://doi.org/10.3390/cancers13040930
APA StyleDelle Cave, D., Rizzo, R., Sainz, B., Jr., Gigli, G., del Mercato, L. L., & Lonardo, E. (2021). The Revolutionary Roads to Study Cell–Cell Interactions in 3D In Vitro Pancreatic Cancer Models. Cancers, 13(4), 930. https://doi.org/10.3390/cancers13040930