
A Novel Bispecific Antibody Targeting EGFR and VEGFR2 Is Effective against Triple Negative Breast Cancer via Multiple Mechanisms of Action


Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
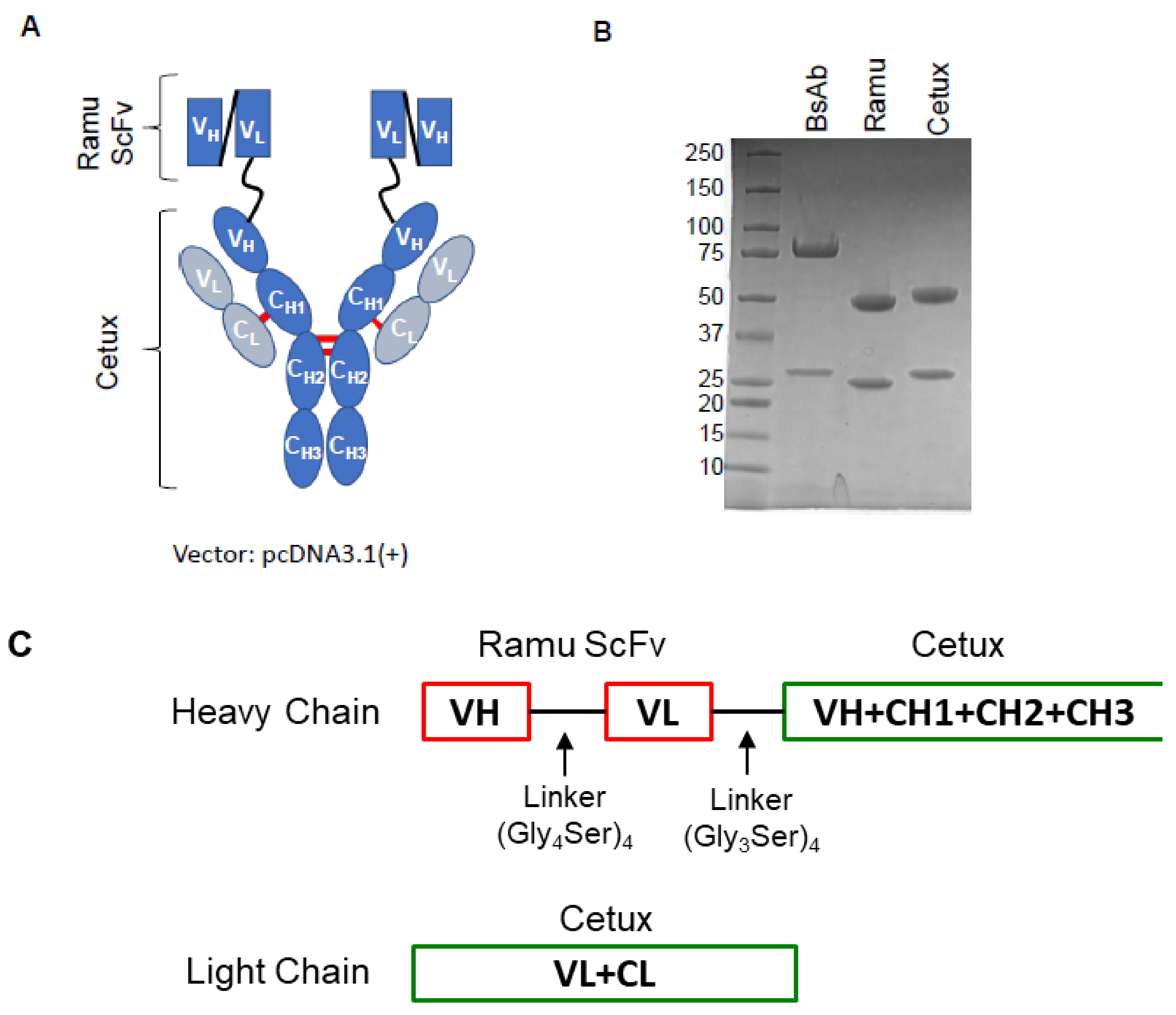
2.1. Construction, Production and Purification of BsAb
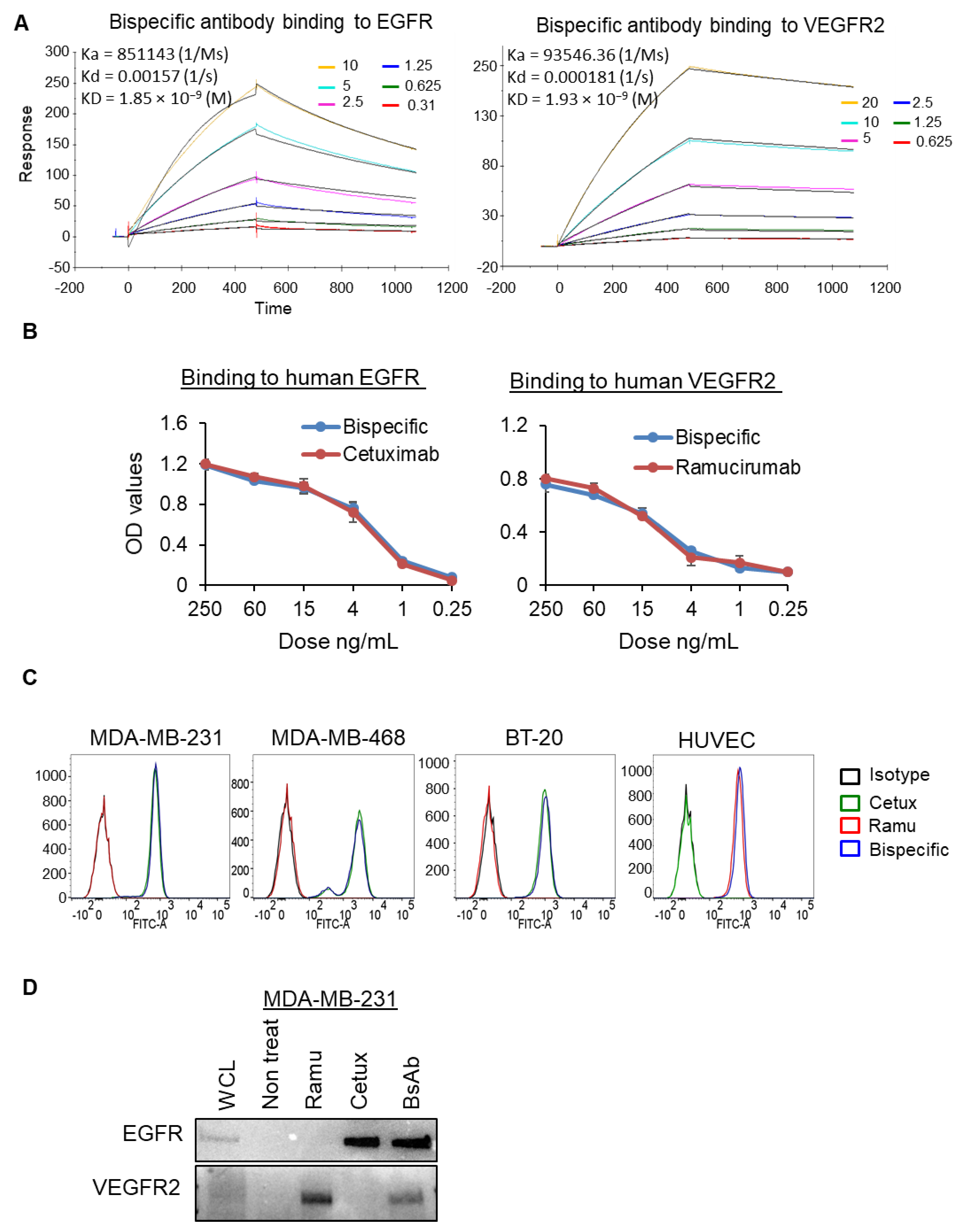
2.2. Binding Characterization of Anti-EGFR/VEGFR2 BsAb
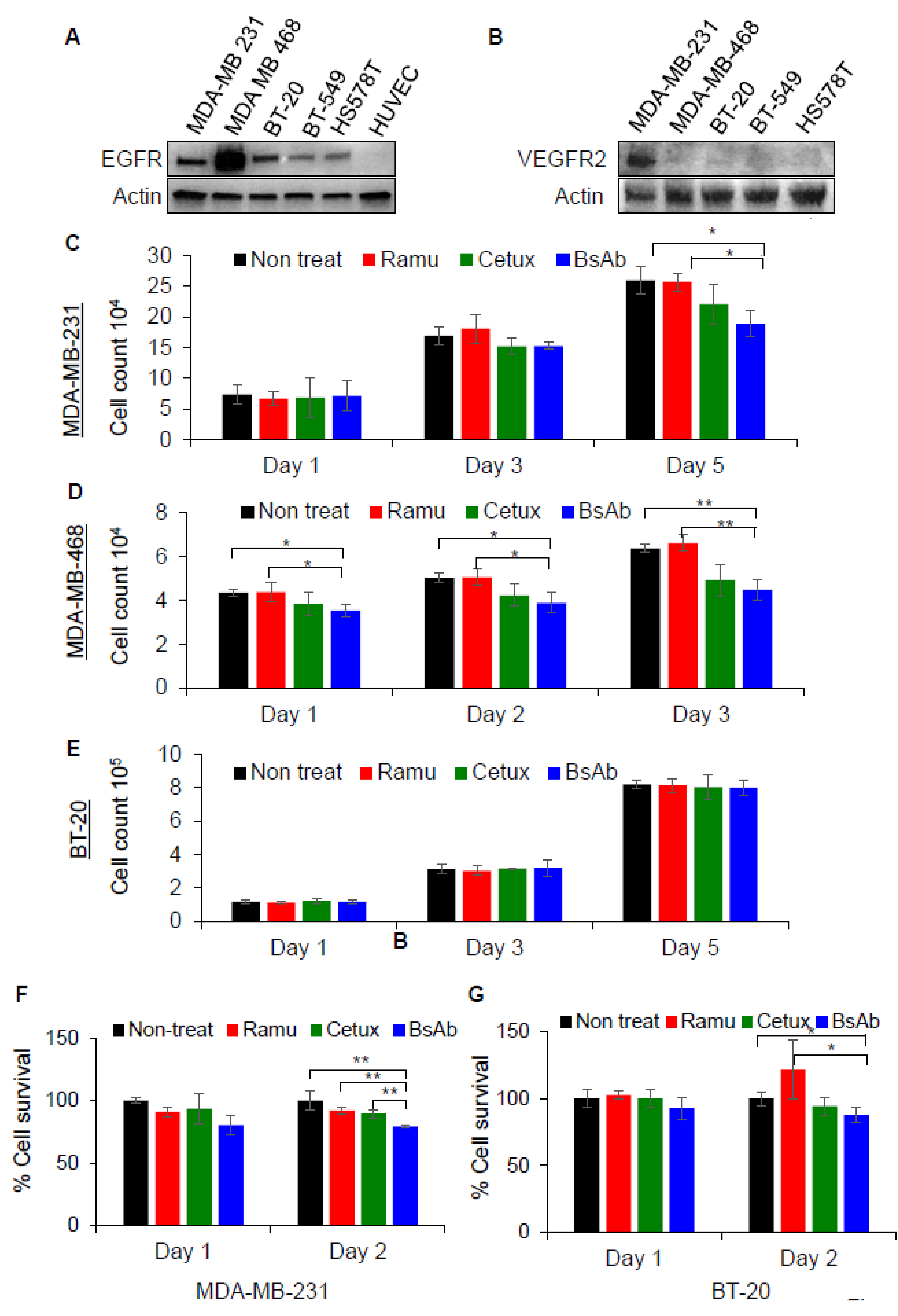
2.3. Anti-EGFR/VEGFR2 BsAb Effectively Inhibits Cellular Proliferation in TNBC Cells
2.4. Anti-EGFR/VEGFR2 BsAb Shows Antitumor Activity in Tumor Xenograft Model
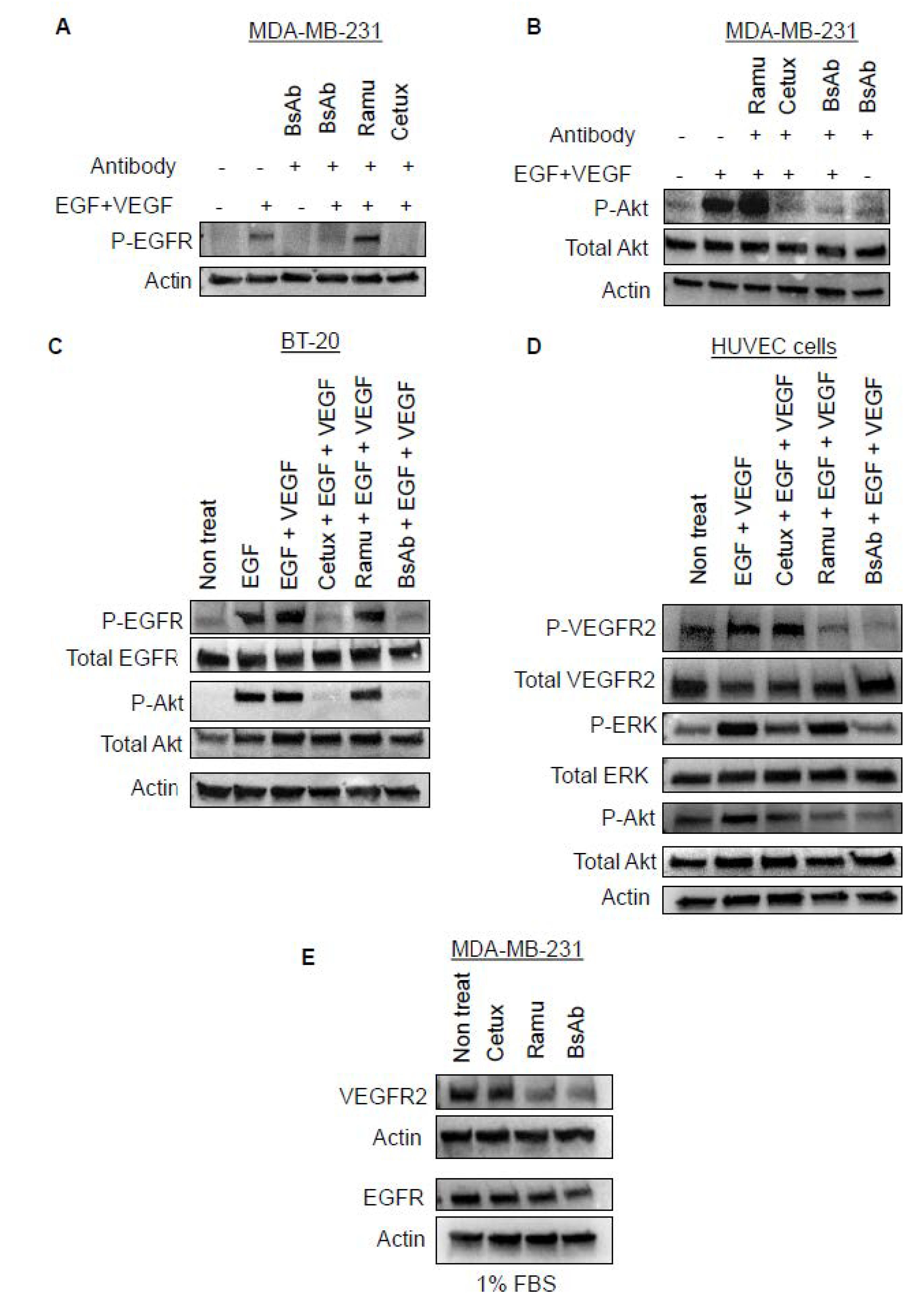
2.5. Anti-EGFR/VEGFR2 BsAb Inhibits Ligand-Induced EGFR and VEGFR2 Signaling
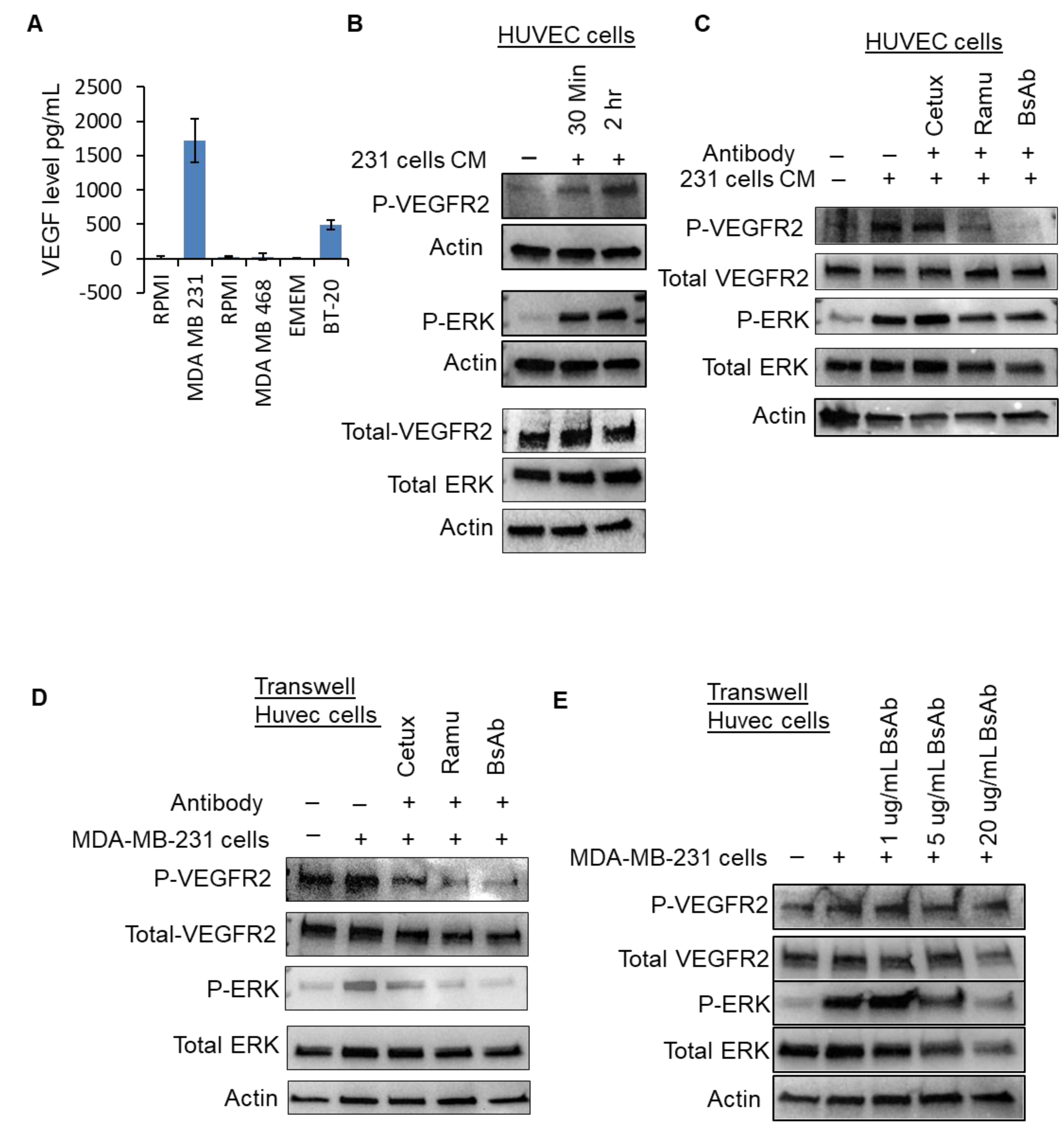
2.6. Anti-EGFR/VEGFR2 BsAb Impairs Paracrine VEGFR2 Signaling in HUVEC Cells Induced by Cancer Cell Media
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Construction, Expression and Purification of the Anti-EGFR/VEGFR2 BsAb
4.3. ELISA Assay
4.4. Flow Cytometry Binding Assay
4.5. Biacore Binding Kinetics Assay
4.6. Western Blot Analysis
4.7. Tumor Xenograft Model
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chames, P.; Daniel, B. Bispecific Antibodies for Cancer Therapy: The Light at the End of the Tunnel? MAbs 2009, 1, 539–547. [Google Scholar] [CrossRef]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies Drug Discovery Today 2015. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, U.; Kontermann, R.E. The Making of bispecfic antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Ghayur, T. Generation of Dual-Variable-Domain Immunoglobulin Molecules for Dual-Specific Targeting. Methods Enzymol. 2012, 502, 25–41. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Mohan, N.; Hosain, S.; Zhao, J.; Shen, Y.; Luo, X.; Jiang, J.; Endo, Y.; Wu, W.J. Atezolizumab potentiates Tcell-mediated cytotoxicity and coordinates with FAK to suppress cell invasion and motility in PD-L1+ triple negative breast cancer cells. OncoImmunology 2019, 8, e1624128. [Google Scholar] [CrossRef] [Green Version]
- Reis-Filho, J.S.; Tutt, A.N.J. Triple negative tumours: A critical review. Histopathol. 2007, 52, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Hung, M.-C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar]
- Rimawi, M.F.; Shetty, P.B.; Weiss, H.L.; Schiff, R.; Osborne, C.; Chamness, G.C.; Elledge, R.M. EGFR Expression in Breast Cancer Association with biologic phenotype and clinical outcomes. Cancer 2010, 116, 1234–1242. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Norton, L.; Masui, H.; Pandiella, A.; Coplan, K.; Miller, J.W.H.; Mendelsohn, J. Antitumor Effects of Doxorubicin in Combination With Anti-epidermal Growth Factor Receptor Monoclonal Antibodies. J. Natl. Cancer Inst. 1993, 85, 1327–1333. [Google Scholar] [CrossRef]
- Baselga, J.; Gómez, P.P.; Greil, R.R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.S.; A Pêgo, A.; A Chan, A.; et al. Randomized Phase II Study of the Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Cetuximab With Cisplatin Versus Cisplatin Alone in Patients With Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.; Liu, W.; Xu, J.; Wang, M.; Bi, Y. Tumor-derived vascular endothelial growth factor (VEGF)-A facilitates tumor metastasis through the VEGF-VEGFR1 signaling pathway. Int. J. Oncol. 2011, 39, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- A A Mohammed, R.; O Ellis, I.; Mahmmod, A.M.; Hawkes, E.C.; Green, A.R.; A Rakha, E.; Martin, S.G. Lymphatic and blood vessels in basal and triple-negative breast cancers: Characteristics and prognostic significance. Mod. Pathol. 2011, 24, 774–785. [Google Scholar] [CrossRef] [Green Version]
- Perrot-Applanat, M.; Di Benedetto, M. Autocrine functions of VEGF in breast tumor cells. Cell Adhes. Migr. 2012, 6, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linderholm, B.K.; Hellborg, H.; Johansson, U.; Elmberger, G.; Skoog, L.; Lehtiö, J.; Lewensohn, R. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann. Oncol. 2009, 20, 1639–1646. [Google Scholar] [CrossRef]
- Yardley, D.A.; Reeves, J.; Dees, E.C.; Osborne, C.; Paul, D.; Ademuyiwa, F.; Soliman, H.; Guthrie, T.; Andersen, J.; Krekow, L.; et al. Ramucirumab with Eribulin Versus Eribulin in Locally Recurrent or Metastatic Breast Cancer Previously Treated With Anthracycline and Taxane Therapy: A Multicenter, Randomized, Phase II Study. Clin. Breast Cancer 2016, 16, 471–479.e1. [Google Scholar] [CrossRef] [Green Version]
- Larsen, A.K.; Ouaret, D.; El Ouadrani, K.; Petitprez, A. Targeting EGFR and VEGF(R) pathway cross-talk in tumor survival and angiogenesis. Pharmacol. Ther. 2011, 131, 80–90. [Google Scholar] [CrossRef]
- Ohba, T.; Cates, J.M.; Cole, H.A.; Slosky, D.A.; Haro, H.; Ando, T.; Schwartz, H.S.; Schoenecker, J.G. Autocrine VEGF/VEGFR1 Signaling in a Subpopulation of Cells Associates with Aggressive Osteosarcoma. Mol. Cancer Res. 2014, 12, 1100–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigand, M.; Hantel, P.; Kreienberg, R.; Waltenberger, J. Autocrine vascular endothelial growth factor signalling in breast cancer. Evidence from cell lines and primary breast cancer cultures in vitro. Angiogenesis 2005, 8, 197–204. [Google Scholar] [CrossRef]
- Dong, J.; Sereno, A.; Aivazian, D.; Langley, E.; Miller, B.R.; Snyder, W.B.; Chan, E.; Cantele, M.; Morena, R.; Joseph, I.B.; et al. A stable IgG-like bispecific antibody targeting the epidermal growth factor receptor and the type I insulin-like growth factor receptor demonstrates superior anti-tumor activity. MAbs 2011, 3, 273–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Heukamp, L.C.; Siobal, M.; Schöttle, J.; Wieczorek, C.; Peifer, M.; Frasca, D.; Koker, M.; König, K.; Meder, L.; et al. Tumor VEGF:VEGFR2 autocrine feed-forward loop triggers angiogenesis in lung cancer. J. Clin. Investig. 2013, 123, 1732–1740. [Google Scholar] [CrossRef] [Green Version]
- Pal, H.C.; Sharma, S.; Strickland, L.R.; Agarwal, J.; Athar, M.; Elmets, C.A.; Afaq, F. Delphinidin Reduces Cell Proliferation and Induces Apoptosis of Non-Small-Cell Lung Cancer Cells by Targeting EGFR/VEGFR2 Signaling Pathways. PLoS ONE 2013, 8, e77270. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Xie, W.; Acheampong, D.O.; Xu, M.; He, H.; Yang, M.; Li, C.; Luo, C.; Wang, M.; Zhang, J. A human IgG-like bispecific antibody co-targeting epidermal growth factor receptor and the vascular endothelial growth factor receptor 2 for enhanced antitumor activity. Cancer Biol. Ther. 2016, 17, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Jin, H.; Chen, Z.; Xie, W.; Wang, Y.; Wang, Y.; Wang, M.; Zhang, J.; Acheampong, D.O. A novel bispecific diabody targeting both vascular endothelial growth factor receptor 2 and epidermal growth factor receptor for enhanced antitumor activity. Biotechnol. Prog. 2016, 32, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Amin, D.N.; Bielenberg, D.R.; Lifshits, E.; Heymach, J.V.; Klagsbrun, M. Targeting EGFR activity in blood vessels is sufficient to inhibit tumor growth and is accompanied by an increase in VEGFR-2 dependence in tumor endothelial cells. Microvasc. Res. 2008, 76, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; De Palma, R.; Orditura, M.; De Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin. Exp. Immunol. 2009, 158, 1–9. [Google Scholar] [CrossRef]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.-J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the Treatment of Colorectal Cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef] [Green Version]
- Chong, C.R.; A Jänne, P. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [Green Version]
- McKnight, B.N.; Kim, S.; Boerner, J.L.; Viola, N.T. Cetuximab PET delineated changes in cellular distribution of EGFR upon dasatinib treatment in triple negative breast cancer. Breast Cancer Res. 2020, 22, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Verdaguer, H.; Tabernero, J.; Macarulla, T. Ramucirumab in metastatic colorectal cancer: Evidence to date and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Prewett, M.; Huber, J.; Li, Y.; Santiago, A.; O’Connor, W.; King, K.; Overholser, J.; Hooper, A.; Pytowski, B.; Witte, L.; et al. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res. 1999, 59, 5209–5218. [Google Scholar] [PubMed]
- Vazquez-Lombardi, R.; Nevoltris, D.; Luthra, A.; Schofield, P.; Zimmermann, C.; Christ, D. Transient expression of human antibodies in mammalian cells. Nat. Protoc. 2018, 13, 99–117. [Google Scholar] [CrossRef]
- Franco, A.; Damdinsuren, B.; Ise, T.; Dement-Brown, J.; Li, H.; Nagata, S.; Tolnay, M. Human Fc Receptor–Like 5 Binds Intact IgG via Mechanisms Distinct from Those of Fc Receptors. J. Immunol. 2013, 190, 5739–5746. [Google Scholar] [CrossRef] [PubMed]
- Mohan, N.; Endo, Y.; Elzarrad, M.K.; Wu, W.J.; Shen, Y. Trastuzumab, but Not Pertuzumab, Dysregulates HER2 Signaling to Mediate Inhibition of Autophagy and Increase in Reactive Oxygen Species Production in Human Cardiomyocytes. Mol. Cancer Ther. 2016, 15, 1321–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, N.; Shen, Y.; Dokmanovic, M.; Endo, Y.; Hirsch, D.S.; Wu, W.J. VPS34 regulates TSC1/TSC2 heterodimer to mediate RheB and mTORC1/S6K1 activation and cellular transformation. Oncotarget 2016, 7, 52239–52254. [Google Scholar] [CrossRef] [Green Version]
- Faustino-Rocha, A.; Oliveira, P.A.; Pinho-Oliveira, J.; Teixeira-Guedes, C.; Soares-Maia, R.; da Costa, R.G.; Colaço, B.; Pires, M.J.; Colaço, J.; Ferreira, R.; et al. Estimation of Rat Mammary Tumor Volume Using Caliper and Ultrasonography Measurements. Lab. Anim. 2013, 42, 217–224. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatments | MDA MB 231 | BT-20 | MDA-MB-468 | HUVEC |
|---|---|---|---|---|
| Isotype control | 10 | 15.9 | 10.8 | 12.4 |
| Cetuximab | 670 | 958 | 3186 | 12.4 |
| Ramucirumab | 9.86 | 11.2 | 14.7 | 670 |
| Anti-EGFR/VEGFR2 BsAb | 725 | 1054 | 2930 | 769 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohan, N.; Luo, X.; Shen, Y.; Olson, Z.; Agrawal, A.; Endo, Y.; Rotstein, D.S.; Pelosof, L.C.; Wu, W.J. A Novel Bispecific Antibody Targeting EGFR and VEGFR2 Is Effective against Triple Negative Breast Cancer via Multiple Mechanisms of Action. Cancers 2021, 13, 1027. https://doi.org/10.3390/cancers13051027
Mohan N, Luo X, Shen Y, Olson Z, Agrawal A, Endo Y, Rotstein DS, Pelosof LC, Wu WJ. A Novel Bispecific Antibody Targeting EGFR and VEGFR2 Is Effective against Triple Negative Breast Cancer via Multiple Mechanisms of Action. Cancers. 2021; 13(5):1027. https://doi.org/10.3390/cancers13051027
Chicago/Turabian StyleMohan, Nishant, Xiao Luo, Yi Shen, Zachary Olson, Atul Agrawal, Yukinori Endo, David S. Rotstein, Lorraine C. Pelosof, and Wen Jin Wu. 2021. "A Novel Bispecific Antibody Targeting EGFR and VEGFR2 Is Effective against Triple Negative Breast Cancer via Multiple Mechanisms of Action" Cancers 13, no. 5: 1027. https://doi.org/10.3390/cancers13051027
APA StyleMohan, N., Luo, X., Shen, Y., Olson, Z., Agrawal, A., Endo, Y., Rotstein, D. S., Pelosof, L. C., & Wu, W. J. (2021). A Novel Bispecific Antibody Targeting EGFR and VEGFR2 Is Effective against Triple Negative Breast Cancer via Multiple Mechanisms of Action. Cancers, 13(5), 1027. https://doi.org/10.3390/cancers13051027