
Bone Marrow Homeostasis Is Impaired via JAK/STAT and Glucocorticoid Signaling in Cancer Cachexia Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. LLC-Induced Cancer Cachexia in Mice
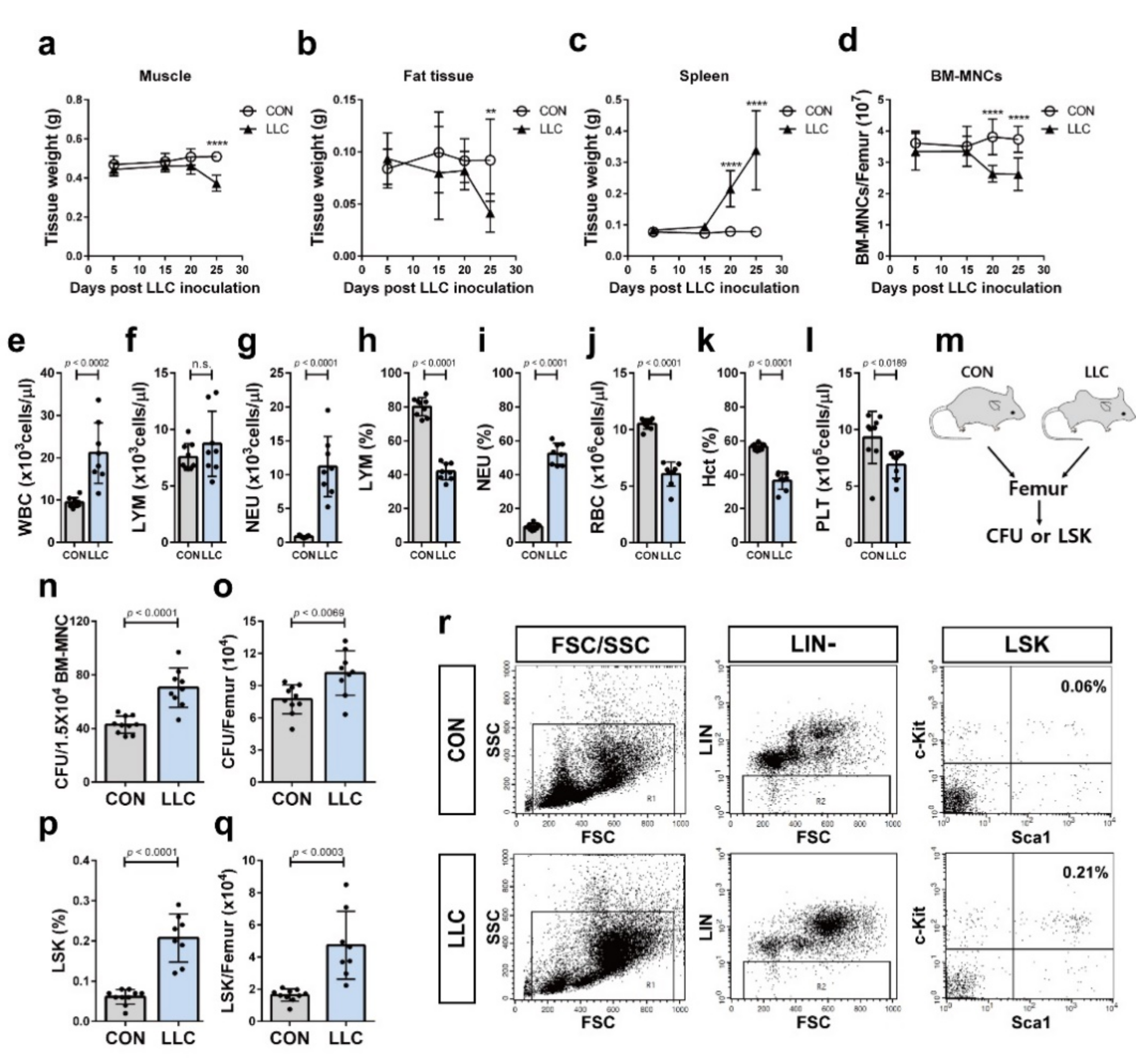
2.2. Cancer Cachexia Leads to Myeloid Skewing in the Circulation and Increases Hematopoietic Progenitors of the Bone Marrow
2.3. Cancer Cachexia-Induced Bone Loss Is Associated with the Decrease in the Population of Bone Marrow MSCs
2.4. Cancer Cachexia Increases Adipogenic Differentiation of the Bone Marrow MSCs via Activation of JAK/STAT as Well as Glucocorticoid Signaling and Decreases Bone Marrow MSCs in a Glucocorticoid-Dependent Manner
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Experimental Model of Cancer Cachexia
4.3. Enzyme-Linked Immunosorbent Assay (ELISA)
4.4. Protein Antibody Array
4.5. Serum Preparation for In Vitro Experiments
4.6. Flow Cytometry
4.7. CFU Assay
4.8. Ex Vivo Microcomputed Tomography (μ-CT) Imaging
4.9. Tartrate-Resistant Acid Phosphatase (TRAP) Staining
4.10. CFU-F Assay
4.11. Osteoclast Differentiation
4.12. Osteoblast Differentiation
4.13. Adipocyte Differentiation and Oil Red O Staining
4.14. Western Blot Analysis
4.15. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Argiles, J.M.; Busquets, S.; Stemmler, B.; Lopez-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- da Silva, S.P.; Santos, J.M.O.; Costa, E.S.M.P.; da Costa, R.M.G.; Medeiros, R. Cancer cachexia and its pathophysiology: Links with sarcopenia, anorexia and asthenia. J. Cachexia Sarcopenia Muscle 2020, 11, 619–635. [Google Scholar] [CrossRef]
- Tomasin, R.; Martin, A.; Cominetti, M.R. Metastasis and cachexia: Alongside in clinics, but not so in animal models. J. Cachexia Sarcopenia Muscle 2019, 10, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Shiono, M.; Huang, K.; Downey, R.J.; Consul, N.; Villanueva, N.; Beck, K.; Fenn, K.; Dietz, D.; Yamaguchi, T.; Kato, S.; et al. An analysis of the relationship between metastases and cachexia in lung cancer patients. Cancer Med. 2016, 5, 2641–2648. [Google Scholar] [CrossRef] [Green Version]
- Petruzzelli, M.; Wagner, E.F. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 2016, 30, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Argiles, J.M.; Lopez-Soriano, F.J. The role of cytokines in cancer cachexia. Med. Res. Rev. 1999, 19, 223–248. [Google Scholar] [CrossRef]
- Argiles, J.M.; Lopez-Soriano, F.J.; Toledo, M.; Betancourt, A.; Serpe, R.; Busquets, S. The cachexia score (CASCO): A new tool for staging cachectic cancer patients. J. Cachexia Sarcopenia Muscle 2011, 2, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Keshavarz-Fathi, M.; Baracos, V.; Arends, J.; Mahmoudi, M.; Rezaei, N. Cancer cachexia: Diagnosis, assessment, and treatment. Crit. Rev. Oncol. Hematol. 2018, 127, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Strassmann, G.; Fong, M.; Freter, C.E.; Windsor, S.; D’Alessandro, F.; Nordan, R.P. Suramin interferes with interleukin-6 receptor binding in vitro and inhibits colon-26-mediated experimental cancer cachexia in vivo. J. Clin. Investig. 1993, 92, 2152–2159. [Google Scholar] [CrossRef] [Green Version]
- Strassmann, G.; Kambayashi, T. Inhibition of experimental cancer cachexia by anti-cytokine and anti-cytokine-receptor therapy. Cytokines Mol. Ther. 1995, 1, 107–113. [Google Scholar]
- Gordon, J.N.; Trebble, T.M.; Ellis, R.D.; Duncan, H.D.; Johns, T.; Goggin, P.M. Thalidomide in the treatment of cancer cachexia: A randomised placebo controlled trial. Gut 2005, 54, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Braun, T.P.; Grossberg, A.J.; Krasnow, S.M.; Levasseur, P.R.; Szumowski, M.; Zhu, X.X.; Maxson, J.E.; Knoll, J.G.; Barnes, A.P.; Marks, D.L. Cancer- and endotoxin-induced cachexia require intact glucocorticoid signaling in skeletal muscle. FASEB J. 2013, 27, 3572–3582. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Young, M.R.; Newby, M.; Wepsic, H.T. Hematopoiesis and suppressor bone marrow cells in mice bearing large metastatic Lewis lung carcinoma tumors. Cancer Res. 1987, 47, 100–105. [Google Scholar]
- Ho, Y.H.; del Toro, R.; Rivera-Torres, J.; Rak, J.; Korn, C.; Garcia-Garcia, A.; Macias, D.; Gonzalez-Gomez, C.; del Monte, A.; Wittner, M.; et al. Remodeling of Bone Marrow Hematopoietic Stem Cell Niches Promotes Myeloid Cell Expansion during Premature or Physiological Aging. Cell Stem Cell 2019, 25, 407–418 e406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtonyuk, L.V.; Fritsch, K.; Feng, X.; Manz, M.G.; Takizawa, H. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Front. Immunol. 2016, 7, 502. [Google Scholar] [CrossRef] [Green Version]
- Cuenca, A.G.; Cuenca, A.L.; Winfield, R.D.; Joiner, D.N.; Gentile, L.; Delano, M.J.; Kelly-Scumpia, K.M.; Scumpia, P.O.; Matheny, M.K.; Scarpace, P.J.; et al. Novel role for tumor-induced expansion of myeloid-derived cells in cancer cachexia. J. Immunol. 2014, 192, 6111–6119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winfield, R.D.; Delano, M.J.; Pande, K.; Scumpia, P.O.; Laface, D.; Moldawer, L.L. Myeloid-derived suppressor cells in cancer cachexia syndrome: A new explanation for an old problem. JPEN J. Parenter. Enteral Nutr. 2008, 32, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Casbon, A.J.; Reynaud, D.; Park, C.; Khuc, E.; Gan, D.D.; Schepers, K.; Passegue, E.; Werb, Z. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl. Acad. Sci. USA 2015, 112, E566–E575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourgholaminejad, A.; Aghdami, N.; Baharvand, H.; Moazzeni, S.M. The effect of pro-inflammatory cytokines on immunophenotype, differentiation capacity and immunomodulatory functions of human mesenchymal stem cells. Cytokine 2016, 85, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, S.; James, A.W.; Blough, J.; Donneys, A.; Wang, S.C.; Cederna, P.S.; Buchman, S.R.; Levi, B. Reconciling the effects of inflammatory cytokines on mesenchymal cell osteogenic differentiation. J. Surg. Res. 2013, 185, 278–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacey, D.C.; Simmons, P.J.; Graves, S.E.; Hamilton, J.A. Proinflammatory cytokines inhibit osteogenic differentiation from stem cells: Implications for bone repair during inflammation. Osteoarthr. Cartil. 2009, 17, 735–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noronha, N.C.; Mizukami, A.; Caliari-Oliveira, C.; Cominal, J.G.; Rocha, J.L.M.; Covas, D.T.; Swiech, K.; Malmegrim, K.C.R. Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem. Cell Res. Ther. 2019, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Yuasa, M.; Yamada, T.; Taniyama, T.; Masaoka, T.; Xuetao, W.; Yoshii, T.; Horie, M.; Yasuda, H.; Uemura, T.; Okawa, A.; et al. Dexamethasone enhances osteogenic differentiation of bone marrow- and muscle-derived stromal cells and augments ectopic bone formation induced by bone morphogenetic protein-2. PLoS ONE 2015, 10, e0116462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Yi, W.; Suino-Powell, K.; Zhou, X.E.; Tolbert, W.D.; Tang, X.; Yang, J.; Yang, H.; Shi, J.; Hou, L.; et al. Structures and mechanism for the design of highly potent glucocorticoids. Cell Res. 2014, 24, 713–726. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, R.S. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol. Metab. Clin. N. Am. 2012, 41, 595–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernigou, P.; Beaujean, F.; Lambotte, J.C. Decrease in the mesenchymal stem-cell pool in the proximal femur in corticosteroid-induced osteonecrosis. J. Bone Joint Surg. Br. 1999, 81, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Oshina, H.; Sotome, S.; Yoshii, T.; Torigoe, I.; Sugata, Y.; Maehara, H.; Marukawa, E.; Omura, K.; Shinomiya, K. Effects of continuous dexamethasone treatment on differentiation capabilities of bone marrow-derived mesenchymal cells. Bone 2007, 41, 575–583. [Google Scholar] [CrossRef]
- Chen, T.L. Inhibition of growth and differentiation of osteoprogenitors in mouse bone marrow stromal cell cultures by increased donor age and glucocorticoid treatment. Bone 2004, 35, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Sproston, N.R.; Ashworth, J.J. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, F.A.; Rivera, N.I. The role of C-reactive protein as a prognostic indicator in advanced cancer. Curr. Oncol. Rep. 2002, 4, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, T.A.; Hentila, J.; Penna, F.; Lampinen, A.; Lautaoja, J.H.; Fachada, V.; Holopainen, T.; Ritvos, O.; Kivela, R.; Hulmi, J.J. Treating cachexia using soluble ACVR2B improves survival, alters mTOR localization, and attenuates liver and spleen responses. J. Cachexia Sarcopenia Muscle 2018, 9, 514–529. [Google Scholar] [CrossRef]
- Wu, C.; Hua, Q.; Zheng, L. Generation of Myeloid Cells in Cancer: The Spleen Matters. Front. Immunol. 2020, 11, 1126. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Xie, C.; Ma, H.; Zhang, S.; Liang, Y.; Shi, L.; Yu, D.; Feng, Y.; Zhang, T.; Wu, G. Gr-1+CD11b+ cells facilitate Lewis lung cancer recurrence by enhancing neovasculature after local irradiation. Sci. Rep. 2014, 4, 4833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giles, A.J.; Reid, C.M.; Evans, J.D.; Murgai, M.; Vicioso, Y.; Highfill, S.L.; Kasai, M.; Vahdat, L.; Mackall, C.L.; Lyden, D.; et al. Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression within the Pre-metastatic Niche. Cancer Res. 2016, 76, 1335–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras, E.M.; Reynaud, D.; Kang, Y.A.; Carlin, D.; Calero-Nieto, F.J.; Leavitt, A.D.; Stuart, J.M.; Gottgens, B.; Passegue, E. Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 2015, 17, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Okada, S.; Nakauchi, H.; Nagayoshi, K.; Nishikawa, S.; Miura, Y.; Suda, T. In vivo and in vitro stem cell function of c-kit- and Sca-1-positive murine hematopoietic cells. Blood 1992, 80, 3044–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetto, A.; Kays, J.K.; Parker, V.A.; Matthews, R.R.; Barreto, R.; Puppa, M.J.; Kang, K.S.; Carson, J.A.; Guise, T.A.; Mohammad, K.S.; et al. Differential Bone Loss in Mouse Models of Colon Cancer Cachexia. Front. Physiol. 2016, 7, 679. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone Res. 2013, 1, 11–26. [Google Scholar]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Harada, S.; Rodan, G.A. Control of osteoblast function and regulation of bone mass. Nature 2003, 423, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Stemmler, B.; Lopez-Soriano, F.J.; Busquets, S. Inter-tissue communication in cancer cachexia. Nat. Rev. Endocrinol. 2018, 15, 9–20. [Google Scholar] [CrossRef]
- Zhao, H.; Lazarenko, O.P.; Chen, J.R. Hippuric acid and 3-(3-hydroxyphenyl) propionic acid inhibit murine osteoclastogenesis through RANKL-RANK independent pathway. J. Cell Physiol. 2020, 235, 599–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Kim, N. Regulation of NFATc1 in Osteoclast Differentiation. J. Bone Metab. 2014, 21, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Shao, J.; Chen, W.; Li, Y.P. Osteoclast differentiation and gene regulation. Front. Biosci. 2007, 12, 2519–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engblom, C.; Pfirschke, C.; Zilionis, R.; Martins, J.d.; Bos, S.A.; Courties, G.; Rickelt, S.; Severe, N.; Baryawno, N.; Faget, J.; et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF(high) neutrophils. Science 2017, 358, eaal5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, N.; Haynesworth, S.E.; Caplan, A.I.; Bruder, S.P. Osteogenic differentiation of purified, culture-expanded human mesenchymal stem cells in vitro. J. Cell Biochem. 1997, 64, 295–312. [Google Scholar] [CrossRef]
- Hong, J.H.; Hwang, E.S.; McManus, M.T.; Amsterdam, A.; Tian, Y.; Kalmukova, R.; Mueller, E.; Benjamin, T.; Spiegelman, B.M.; Sharp, P.A.; et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 2005, 309, 1074–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Nagai, K.; Woo, J.T. Insulin-dependent adipogenesis in stromal ST2 cells derived from murine bone marrow. Biosci. Biotechnol. Biochem. 2003, 67, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Moresi, V.; Adamo, S.; Berghella, L. The JAK/STAT Pathway in Skeletal Muscle Pathophysiology. Front. Physiol. 2019, 10, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [Green Version]
- la Rosee, F.; Bremer, H.C.; Gehrke, I.; Kehr, A.; Hochhaus, A.; Birndt, S.; Fellhauer, M.; Henkes, M.; Kumle, B.; Russo, S.G.; et al. The Janus kinase 1/2 inhibitor ruxolitinib in COVID-19 with severe systemic hyperinflammation. Leukemia 2020, 34, 1805–1815. [Google Scholar] [CrossRef]
- Fleseriu, M.; Biller, B.M.; Findling, J.W.; Molitch, M.E.; Schteingart, D.E.; Gross, C.; SEISMIC Study Investigators and SEISMIC Study Investigators. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing’s syndrome. J. Clin. Endocrinol. Metab. 2012, 97, 2039–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyart, E.; Bindels, L.B.; Mina, E.; Menga, A.; Stanga, S.; Porporato, P.E. Cachexia, a Systemic Disease beyond Muscle Atrophy. Int. J. Mol. Sci. 2020, 21, 8592. [Google Scholar] [CrossRef]
- Fearon, K.C. The Sir David Cuthbertson Medal Lecture 1991. The mechanisms and treatment of weight loss in cancer. Proc. Nutr. Soc. 1992, 51, 251–265. [Google Scholar] [CrossRef] [Green Version]
- Cuminetti, V.; Arranz, L. Bone Marrow Adipocytes: The Enigmatic Components of the Hematopoietic Stem Cell Niche. J. Clin. Med. 2019, 8, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinngrebe, J.; Debatin, K.M.; Fischer-Posovszky, P. Adipocytes in hematopoiesis and acute leukemia: Friends, enemies, or innocent bystanders? Leukemia 2020, 34, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Naveiras, V.; Nardi, P.L.; Wenzel, P.V.; Hauschka, F.; Fahey, G.Q. Daley, Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 2009, 460, 259–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontula, K.; Paavonen, T.; Luukkainen, T.; Andersson, L.C. Binding of progestins to the glucocorticoid receptor. Correlation to their glucocorticoid-like effects on in vitro functions of human mononuclear leukocytes. Biochem. Pharmacol. 1983, 32, 1511–1518. [Google Scholar] [CrossRef]
- Yeh, S.S.; Schuster, M.W. Megestrol acetate in cachexia and anorexia. Int. J. Nanomed. 2006, 1, 411–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Choi, S.; Park, A.; Do, J.; Nam, D.; Kim, Y.; Noh, J.; Lee, K.Y.; Maeng, C.H.; Park, K.-S. Bone Marrow Homeostasis Is Impaired via JAK/STAT and Glucocorticoid Signaling in Cancer Cachexia Model. Cancers 2021, 13, 1059. https://doi.org/10.3390/cancers13051059
Yu J, Choi S, Park A, Do J, Nam D, Kim Y, Noh J, Lee KY, Maeng CH, Park K-S. Bone Marrow Homeostasis Is Impaired via JAK/STAT and Glucocorticoid Signaling in Cancer Cachexia Model. Cancers. 2021; 13(5):1059. https://doi.org/10.3390/cancers13051059
Chicago/Turabian StyleYu, Jinyeong, Sanghyuk Choi, Aran Park, Jungbeom Do, Donghyun Nam, Youngjae Kim, Jinok Noh, Kil Yeon Lee, Chi Hoon Maeng, and Ki-Sook Park. 2021. "Bone Marrow Homeostasis Is Impaired via JAK/STAT and Glucocorticoid Signaling in Cancer Cachexia Model" Cancers 13, no. 5: 1059. https://doi.org/10.3390/cancers13051059
APA StyleYu, J., Choi, S., Park, A., Do, J., Nam, D., Kim, Y., Noh, J., Lee, K. Y., Maeng, C. H., & Park, K. -S. (2021). Bone Marrow Homeostasis Is Impaired via JAK/STAT and Glucocorticoid Signaling in Cancer Cachexia Model. Cancers, 13(5), 1059. https://doi.org/10.3390/cancers13051059